
Abstract
Hypophosphatasia (HPP) is an inherited systemic bone disease that is characterized by bone hypomineralization. HPP is classified into six forms according to the age of onset and severity as perinatal (lethal), perinatal benign, infantile, childhood, adult, and odontohypophosphatasia. The causative gene of the disease is the ALPL gene that encodes tissue-nonspecific alkaline phosphatase (TNAP). TNAP is expressed ubiquitously, and its physiological role is apparent in bone mineralization. A defect in bone mineralization can manifest in several ways, including rickets or osteomalacia in HPP patients. Patients with severe forms suffer from respiratory failure because of hypoplastic chest, which is the main cause of death. They sometimes present with seizures due to a defect in vitamin B6 metabolism resulting from the lack of alkaline phosphatase activity in neuronal cells, which is also lethal. Patients with a mild form of the disease exhibit rickets or osteomalacia and a functional defect of exercise. Odontohypophosphatasia shows only dental manifestations. To date, 302 mutations in the ALPL gene have been reported, mainly single-nucleotide substitutions, and the relationships between phenotype and genotype have been partially elucidated. An established treatment for HPP was not available until the recent development of enzyme replacement therapy. The first successful enzyme replacement therapy in model mice using a modified human TNAP protein (asfotase alfa) was reported in 2008, and subsequently success in patients with severe form of the disease was reported in 2012. In 2015, asfotase alfa was approved in Japan in July, followed by in the EU and Canada in August, and then by the US Food and Drug Administration in the USA in October. It is expected that therapy with asfotase alfa will drastically change treatments and prognosis of HPP.
Introduction
Hypophosphatasia (HPP; OMIM #241500, 241510, 146300), which was first reported by the Canadian pediatrician John Campbell Rathbun in 1948, is a systemic bone disease caused by the deficiency of tissue-nonspecific alkaline phosphatase (TNAP).Citation1,Citation2 HPP is classified into six forms: perinatal (lethal), perinatal benign, infantile, childhood, adult, and odontohypophosphatasia, according to the age of onset and severity.Citation2,Citation3 The perinatal form occurs in utero and has the most severe manifestations; it is lethal for patients, and these patients are usually stillborn or die during the early postnatal period. Recently, however, a group of patients with such early manifestation of HPP that have good prognosis was reported, and this group is now classified as a perinatal benign form.Citation4 The infantile form occurs before 6 months of age, and the phenotypes are generally less severe than the perinatal form. The childhood form occurs after 6 months of age and shows milder manifestations. Their prognosis for life is good, although the patients usually exhibit functional musculoskeletal disorder. The adult form occurs at middle age. Odontohypophosphatasia is only a dental disorder without skeletal manifestations. The symptoms of HPP vary depending on the form involved. Except for odontohypophosphatasia, a common feature is rickets or osteomalacia leading to bone deformity and disorder of musculoskeletal function.Citation2 These symptoms are due to systemic hypomineralization of bone. Severe forms (perinatal and infantile forms) also show respiratory failure as a result of hypoplasia of the chest. Patients with severe forms sometimes suffer from seizures because of a defect of γ-aminobutyric acid (GABA) in neuronal cells in the brain. Respiratory failure and seizures are the main causes of death. No established therapy has been available until the recent success of enzyme replacement therapy (ERT) using asfotase alfa.
Alkaline phosphatase
Tissue-nonspecific alkaline phosphatase
Alkaline phosphatases (orthophosphoric-monoester phosphohydrolase [alkaline optimum], EC 3.1.3.1) are membrane-bound ectoenzymes that hydrolyze monophosphate esters between pH 8 and 10.Citation5 Alkaline phosphatases exist in organisms ranging from bacteria to mammals, and DNA and amino acid sequences of these enzymes are fairly conserved:Citation5 ~57.8% of the amino acids that make up the human TNAP sequence are conserved among mammalian alkaline phosphatases.Citation6 Human TNAP is one of four isoenzymes of human alkaline phosphatase. The other three isozymes, namely, intestinal, placental, and germ cell (placental-like) alkaline phosphatases, are tissue-specific, whereas TNAP is expressed ubiquitously, with an especially marked expression reported in the liver, bone, kidney, neuronal cells, and neutrophils.Citation5,Citation7 TNAP is, therefore, also known as the liver/bone/kidney (LBK) alkaline phosphatase, although no precise physiological roles in the liver and kidney are known. TNAP is encoded by a gene on the short arm of chromosome 1 (1p36.1–34) and is spread over 50 kb, whereas the genes for the tissue-specific alkaline phosphatases are clustered on the long arm of chromosome 2 (2q34–37).Citation8–Citation13 The TNAP gene (ALPL) is ~1.5 kb in length and consists of 12 exons, of which exons 2–12 are coding exons, and there are also two alternative noncoding exons 1 (1B and 1L).Citation9,Citation14 Functional TNAP enzyme is considered to be existed as a homodimer.Citation5 The molecular weight of each monomer is ~80 kDa and is linked to the outer membrane of the cells via a glycosylphosphatidylinositol (GPI) anchor.Citation5 After the TNAP peptide is synthesized as a native protein with a molecular weight of 66 kDa, carbohydrate chains are added as O- and N-linked sugar chains in the endoplasmic reticulum, and the modified protein is then processed in the Golgi apparatus and is eventually localized on the outer membrane via a GPI anchor.Citation5,Citation15 There are also isoforms of TNAP itself that are tissue-specific (liver-specific, bone-specific, etc). These isoforms have different sugar chains, mainly because of different O-linked glycosylation, although they all have the same peptide sequence.Citation5,Citation16 TNAP isoforms and isoenzymes can be distinguished by through electrophoresis and antibody, and this can be used for diagnosis to determine the tissue origin of the enzyme, which can suggest defective organs.
Structure of alkaline phosphatase
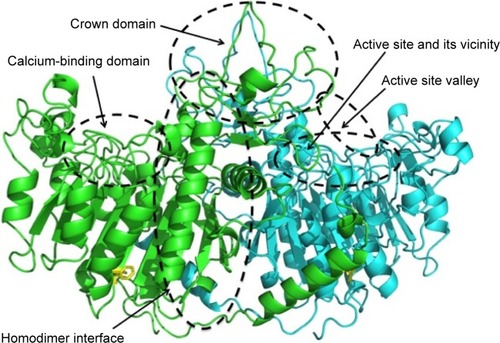
To express enzymatic activity, TNAP needs two Zn2+, one Mg2+, and one Ca2+ ion as cofactors.Citation5,Citation17 The amino acid sequence of the human TNAP molecule is 57% identical and 74% homologous with the human placental alkaline phosphatase (PLAP) molecule. The three-dimensional (3D) structure of TNAP based on the crystallography of the human TNAP protein has not been elucidated. However, the 3D structure of human TNAP can be obtained using a simulation modelCitation18 () based on human PLAPCitation19 and rat-intestinal alkaline phosphatase,Citation20 whose 3D structures have been solved. The core structure of the PLAP protein consists of an extended β-sheet and flanking α-helices. PLAP also has an N-terminal α-helix. The active site comprises a catalytic serine residue (S92) and metal ion-binding sites (two Zn2+-binding sites and a Mg2+-binding site).Citation19 Mammalian alkaline phosphatases have a characteristic crown domain of an interfacial flexible loop consisting of 60 residues, which may interact with extracellular proteins, as well as a long N-terminal α-helix and a Ca2+-binding domain.Citation18–Citation21 Although basic structures of the alkaline phosphatase proteins are well conserved, the crown domain is lacking in bacterial (Escherichia coli) alkaline phosphatase.Citation22 In the C-terminus of the PLAP protein, 29 hydrophobic amino acid residues are removed after translation in the endoplasmic reticulum and the GPI anchor is then added to an aspartate residue (D484).Citation23 The GPI anchor consists of an ethanolamine phosphate, three residues of mannose, a glucosamine, and a phosphatidylinositol.
Figure 1 Structure and mutation sites of TNAP.
Abbreviations: 3D, three-dimensional; PLAP, placental alkaline phosphatase; TNAP, tissue-nonspecific alkaline phosphatase.
Nomenclature of the ALPL gene and protein
Nucleotides are numbered by reference to the first nucleotide (+1), which corresponds to the A of the ATG initiation codon,Citation9,Citation24 and are represented by a c.number. The standardized nomenclature of amino acid number follows the HGVS recommendations, in which the first codon is the ATG initiation codon, and amino acid numbers are represented by a p.number.Citation25,Citation26
Alkaline phosphatase and mineralization
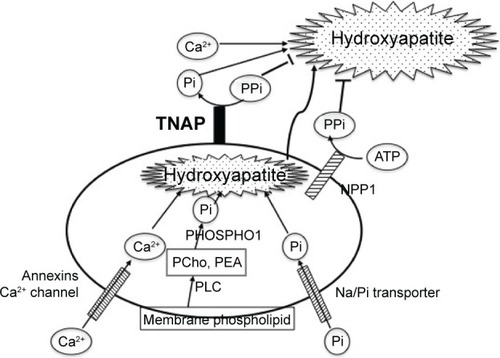
TNAP is essential for tissue biomineralization ().Citation27 Mineralization takes place in two distinct processes.Citation28 Hypertrophic chondrocytes, osteoblasts, and odontoblasts bud matrix vesicles when mineralization begins. Matrix vesicles are membrane-invested vesicles of 50–200 nm in diameter and are rich in annexins A2, A5, and A6, and in Ca2+-ATPase, TNAP, nucleotide pyrophosphate phosphodiesterase 1 (NPP1, formerly known as PC-1), Pit1 (a sodium–phosphate [Na/Pi] cotransporter), and PHOSPHO1.Citation28 The first step of the mineralization process occurs within the matrix vesicles, in which hydroxyapatite (Ca10(PO4)6(OH)2) crystals are formed.Citation28 Phosphate is derived from membrane phospholipids, which are hydrolyzed by phospholipase C to produce phosphocholine and phosphoethanolamine.Citation29 These phosphocompounds are hydrolyzed by PHOSPHO1, a cytosolic phosphatase that is abundant in the matrix vesicles, to yield inorganic phosphate (Pi).Citation30,Citation31 Another source of Pi in the matrix vesicles is Pi that is transported through the Na/Pi cotransporter Pit1 that is also abundant on the matrix vesicle membrane.Citation28 Calcium is incorporated into the matrix vesicles through annexin Ca2+ channels that consist of annexins A2, A5, and A6.Citation28 Developing hydroxyapatite crystals then penetrate the matrix vesicle membrane, are elongated in the extracellular space, and eventually deposit in the spaces between collagen fibrils to complete extracellular matrix mineralization.Citation28 The concentration ratio of Pi to inorganic pyrophosphate (PPi) in the extracellular matrix is crucial in the second step of mineralization because PPi is an inhibitor of hydroxyapatite formation.Citation32 Two mechanisms are used for PPi formation.Citation27,Citation33 PPi is formed in the extracellular matrix from ATP by the matrix vesicle membrane enzyme NPP1.Citation34 PPi is also provided through the PPi transporter ANKH (a homologue of the mouse progressive ankylosis gene product) from the cytoplasm, in which PPi is routinely formed by cellular metabolism.Citation35 ANKH is distributed on the plasma membrane of hypertrophic chondrocytes and osteoblasts.Citation35 TNAP on the membrane of the matrix vesicles hydrolyzes PPi and yields Pi, thereby reducing the levels of the PPi inhibitor and promoting hydroxyapatite formation. The importance of the Pi/PPi ratio has been proved by knockout (KO) mice of the respective genes as described in the “KO mice as models of HPP” section. TNAP KO mice (Akp2−/−) show hypomineralization (as will be described). NPP1 KO mice (Enpp1−/−) and naturally generated mutant mice (tiptoe walking mice; ttw/ttw) show calcification of articular cartilage and aorta.Citation36,Citation37 The human ENPP1 gene mutation causes idiopathic infantile arterial calcification.Citation38 Naturally occurring progressive ankylosis (ank/ank) mice present hypercalcification of vertebrae and joints, leading to progressive ankylosis.Citation39 Double KO animals of TNAP (Akp2−/−) and NPP1 (Enpp1−/−) or ANK (ank/ank) show a seemingly normal skeleton, indicating that TNAP, NPP1, and ANK(H) regulate mineralization in concert.Citation40,Citation41
Figure 2 Mineralization in and surrounding a matrix vesicle.
Abbreviations: NPP1, nucleotide pyrophosphate phosphodiesterase 1; Pi, inorganic phosphate; PPi, inorganic pyrophosphate; TNAP, tissue-nonspecific alkaline phosphatase; PCho, phosphocholine; PEA, phosphoethanolamine; Na/Pi transporter, sodium-phosphate transporter; PLC, phospholipase C.
Pathophysiology of HPP
Bone mineralization
The pathophysiology of HPP is derived from the defective function of TNAP. TNAP deficiency causes not only defective enzymatic activity but also defective mineralizing activity. The common feature of HPP is hypomineralization of bones and teeth. In HPP patients, extracellular hydroxyapatite crystals are reduced and mineralization is impaired.Citation42 However, mineralization does occur within the matrix vesicles formed in these patientsCitation42 because PHOSPHO1 is expressed normally in the patients’ matrix vesicles. This has been proven using double KO mice of TNAP and PHOSPHO1, which show little formation of hydroxyapatite either in the matrix vesicles or in the extracellular space.Citation43 HPP patients exhibit an increase in osteoid tissue that contains abundant nonmineralized bone extracellular matrix without hydroxyapatite crystals, leading to rickets and osteomalacia.Citation2 Mineralization can be assessed by relatively easy procedures. An expression plasmid is introduced into U2OS osteoblastic cells that express trace levels of TNAP enzymatic activity, and then the cells are cultured with a mineralization medium that contains β-glycerophosphate as an artificial TNAP substrate with or without ascorbic acid. The cells in which the wild-type TNAP cDNA is introduced mineralize around the cells within 5 days, whereas cells in which mutant cDNAs are introduced show less mineralization.Citation44 Mineralizing ability parallels TNAP enzymatic activity in this assay, and therefore the level of TNAP activity can reflect phenotype, especially with regard to bone mineralization.Citation44
Central nervous system and seizures
Another physiological role of TNAP is associated with neurotransmitter synthesis in the central nervous system.Citation45 One of the physiological substrates of TNAP is pyridoxal 5′-phosphate, a derivative of vitamin B6, which is necessary for the biosynthesis of GABA, a cofactor of glutamic acid carboxylase in neuronal cells.Citation45,Citation46 GABA is known to act as a repressive neurotransmitter.Citation46 For pyridoxal 5′-phosphate entry into cells, the phosphate moiety has to be first released, and then pyridoxal is incorporated into the cells, followed by rephosphorylation within the cells.Citation47 It has been confirmed in fibroblasts from normal and HPP patients that TNAP hydrolyzes pyridoxal 5′-phosphate to release phosphate outside the cell membrane,Citation47 and therefore a similar mechanism is presumed to operate in the neuronal cell membrane.Citation45 Failure of this role of TNAP leads to epileptic seizures in HPP patients because reduced GABA in the neuronal cells results in hyperactivity of the central nervous system.Citation48,Citation49 However, although almost all patients with seizures are pyridoxine responsive, the etiology of seizures other than deteriorated vitamin B6 metabolism may be considered, such as high intracranial pressure because of craniosynostosis, hypercalcinemia, and hypoxia.Citation2
Inheritance of HPP
Although HPP is inherited as an autosomal recessive trait, autosomal dominant cases have been reported in patients with milder HPP.Citation50,Citation51 The dominant negative effect of severe alleles accounts for almost all autosomal dominant cases.Citation52 Severe forms are mostly compound heterozygotes of severe alleles, and residual activity is important to decide clinical forms.Citation53 Infrequently, however, siblings with different phenotypes have been reported;Citation54,Citation55 this phenomena may raise the suspicion of epigenetic factors being involved in the expression of phenotypes.
Mutations in alkaline phosphatase
The first patient reported by Rathbun was suffering from an infantile form of HPP. Fifty years later, his mutations were identified using the surviving parents’ DNA as a compound heterozygote of p.A114T (c.340G > A) and p.D294A (c.881A > C).Citation56 Mutations found in HPP patients are scattered throughout the molecule, and 302 mutations, mainly missense mutations, have been described to date in the ALPL gene.Citation57 In addition, although some deletions, insertions, and splice mutations have been reported, no large deletion or insertion of the ALPL gene area was reported.Citation57 Regarding the relationships between genotype and phenotype, mutation sites are classified by the protein regions in which they occur.Citation58 Mutations located in the active site and in its vicinity, the homodimer interface, the crown domain, and the calcium-binding domain are mainly associated with a severe phenotype, whereas mutations in the active site valley show less severe phenotypes (). The phenotypic severity of mutations of specific residues depends on the type of mutation at each residue. For example, although p.F327 is a frequently mutated site, it is a moderate allele,Citation59 but p.F327del is a severe allele.Citation60 The difference in the severity of the phenotype probably depends on the stability of the molecule. As the phenylalanine residue at 327 is in a β-sheet that consists of 10 core β-sheets near the Mg2+-binding site, its deletion may result in deterioration of the structure, resulting in a severe phenotype. Mutations in the C-terminus also result in a severe phenotype. Deletion of nucleotide 1559 (c.1559del) causes a frameshift mutation, which adds 80 amino acids to the C-terminus, resulting in the failure of GPI-anchor formation.Citation61 The mutant protein cannot be processed properly in the cells, aggregates in the Golgi apparatus, and is then treated in the proteasome; therefore, this deletion results in almost no enzymatic activity and is a severe allele.Citation62 Nevertheless, most HPP patients are compound heterozygotes, indicating that interaction of the two mutated alleles may determine the phenotype.Citation63,Citation64 The relationships between genotype and phenotype have been previously discussed.Citation65 Generally, phenotypes are related to residual enzymatic activity and the mineralizing ability of the cells that express the mutant enzymes.Citation44 However, the relationship between genotype and phenotype is not so simple, and more research is needed to clarify this relationship.
Clinical features of HPP
Clinical features of HPP vary depending on the form.Citation3 The perinatal (lethal) form is the most severe form, which shows shortened and deformed limbs during gestation and at birth.Citation2 The cranium presents as membranous and the ribs are hypomineralized and deformed, resulting in respiratory failure after birth. Epileptic seizures sometimes occur. Respiratory failure and/or seizures precipitate a lethal course of the disease. The infantile form also displays rickets and deformity of limbs and ribs because of hypomineralization of bones. In addition, craniosynostosis often occurs. Patients fail to thrive and often need respiratory aid, and the main cause of death is respiratory failure; the elongation of lifespan recently observed can be accounted for by developments in respiratory management. Hypercalcinemia and hypercalciuria are often seen, and the latter causes nephrocalcinosis. Patients with childhood form of the disease exhibit a wide range of clinical findings,Citation66 including deformity of limbs, delayed walking, and waddling gait. Radiography shows characteristic focal bone defect at the ends of long bones as a tongue-like radiolucent projection from the rachitic growth plate into the metaphysis.Citation2 These patients also show premature loss of deciduous teeth because of disturbed cementum formation.Citation67 Craniosynostosis occurs in some cases, leading to raised intracranial pressure.Citation68 Patients also exhibit problems in doing exercise because of not only rickets but also muscle weakness and functional muscular failure. The adult form occurs during middle age. Although the natural history of the adult form has not been well characterized,Citation69 patients sometimes have a history of rickets and/or premature exfoliation of deciduous teeth. In the adult form, osteomalacia develops with pain associated with often recurrent metatarsal stress fractures.Citation2 In some patients, calcium pyrophosphate dehydrate crystals are deposited on articular cartilage due to an increase in endogenous levels of PPi.Citation2 Odontohypophosphatasia affects only dental tissue without any manifestations of rickets or osteomalacia, but should present with a low serum alkaline phosphatase level. The perinatal benign form is a newly recognized form in which patients manifest symptoms in utero, but this form is nonlethal.Citation4
HPP can be diagnosed using ultrasonography and radiography in utero for the perinatal form.Citation2,Citation3 In any case, serum alkaline phosphatase activity is strong evidence of the disease. In some cases, carriers show subnormal enzymatic activity. In addition, some natural TNAP substrates have diagnostic value. Alkaline phosphatase activity is measured in the clinical laboratory using the artificial substrate p-nitrophenylphosphate. There is some controversy regarding natural substrates of TNAP;Citation2 PPi and pyridoxal 5′-phosphate are natural substrates (as already mentioned). The status of phosphoethanolamine is controversial; it is probably not a natural TNAP substrate, but it does have diagnostic value because the measurement of urine phosphoethanolamine is easier, and so is more widely used than that of serum PPi.Citation2 HPP patients show higher serum PPi and urine phosphoethanolamine; low alkaline phosphatase activity with high PPi or phosphoethanolamine indicates strong evidence of HPP. In some milder cases, however, an increase in phosphoethanolamine is not shown, and, in some cases, phosphoethanolamine is slightly elevated in carriers.Citation70
Prevalence of HPP
The prevalence of HPP was estimated to be 1 in 100,000 live births in the Toronto area in Canada.Citation71 It is known to be as high as 1 in 2,500 for Mennonite families in Manitoba, Canada.Citation72 For severe forms, prevalence has been estimated at 1 in 300,000 in Europe.Citation73 A recent report from Japan estimated a prevalence of 1 in 450,000 for patients who have the particular mutant allele c.1559delT,Citation70 which is possessed by 46.8% of Japanese patients with HPP.Citation74
A possible role of asfotase alfa
KO mice as models of HPP
KO mice of the murine TNAP gene (Akp2) were createdCitation45,Citation75 and were confirmed as a suitable model of HPP.Citation76 The KO mice show hypomineralization of bone, rickets and osteomalacia, failure to thrive, abnormal dental manifestations,Citation77 and seizures.Citation45 The latter finding is essential, and they die within 20 days after birth. Although these mice mimic the manifestations of severe forms of HPP patients, one difference between them is the presence of epileptic seizures, which HPP patients rarely exhibit but almost all KO mice show.
History of treatment of HPP
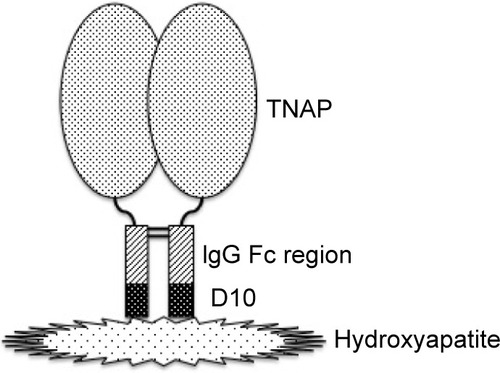
Trials of treatment of HPP have been performed and have failed.Citation5 Artificial respiration is used for respiratory failure. Other symptomatic treatments including dietary therapy (calcium restriction, vitamin D supplementation, etc) are also used.Citation2,Citation3 As a fundamental therapy, ERT has been attempted. Symptoms of female patients with mild form of the disease are known to improve when they are pregnant because of the activity of PLAP.Citation5 Therefore, infusion of PLAP into an infant with severe form of HPP was attempted, but no improvement was observed.Citation5 TNAP infusion was also attempted using the sera of patients with Paget disease who exhibit hyperphosphatemia, but almost no improvement was observed.Citation78 On the other hand, mesenchymal stem-cell transplantation has shown slight improvement.Citation79 Those results suggested that cells expressing TNAP on the cell surface is necessary for successful treatment. In 2008, successful treatment of TNAP null mice (Akp2−/−) with human TNAP bioengineered with the C-terminus extended by the Fc region of human IgG and a deca-aspartate sequence was reported.Citation80 In this experiment, TNAP was cut at the C-terminal membrane-bound region to yield the soluble type but without a loss of enzymatic activity. The Fc region was added in order to purify the enzyme by a one-step procedure. Given that deca-aspartate was proven to bind to hydroxyapatite with high affinity,Citation81 it was used for bone targeting (). The modified enzyme also prevents dental defects in Akp2−/− mice.Citation82 As Akp2−/− is a good model for perinatal form HPP,Citation76 this report gave hope of ERT to the patients. A clinical trial was conducted, and the first results of the trials in the USA were reported in 2012.Citation83 The recruited eleven patients (five perinatal and six infantile) were treated with 2 mg/kg of ENB-0040 (asfotase alfa; 40 mg/mL) provided by Enobia Pharma, Inc. (San Carlos, CA, USA) as a single intravenous infusion followed by subcutaneous injections three times per week at a dose of 1 mg/kg for 48 weeks. With the exception of one case who died of respiratory failure that was unrelated to asfotase alfa, the recruited patients showed great improvement in rickets, developmental milestones, and pulmonary function.Citation83 Asfotase alfa is the same bioengineered enzyme that was used in the animal experiment described earlier.Citation80 A successful case of ERT of the perinatal lethal form with asfotase alfa was also reported from Japan.Citation84 In July 2015, asfotase alfa (Strensiq™; Alexion Pharmaceuticals, Inc., New Haven, CT, USA) was approved in Japan, followed by approval in the EU and Canada in August and then by the US Food and Drug Administration in the USA in October 2015.
Figure 3 Structure of asfotase alfa.
Abbreviations: D10, deca-aspartate; TNAP, tissue-nonspecific alkaline phosphatase.
Usage and evaluation of asfotase alfa
Symptomatic and radiographic evaluation, motor and cognitive development, and laboratory findings should confirm the outcome of the treatment. The recommended administration method of asfotase alfa is subcutaneous injection six times a week at a dose of 1 mg/kg or three times a week at 2 mg/kg, and the maximal volume of injection is 1 mL.Citation85 In the report of the first trial, the authors employed laboratory data (serum alkaline phosphatase activity, plasma pyridoxal phosphate level, and serum PTH level), radiography (to check the severity of rickets, craniosynostosis, and nephrocalcinosis), respiratory state, feeding state, and gross motor function to evaluate the outcome of the treatment.Citation83 Gross motor function was assessed with the use of the Bayley Scales of Infant and Toddler Development, third edition (Bayley-III) instrument. The main adverse event of the trial directly associated with the administration was transient erythema at the injection site, although some patients showed serious respiratory distress, craniosynostosis, and conductive hearing loss, which were considered possibly related to the study treatment.Citation83 Regarding immunogenicity, about half of the patients showed the presence of neutralizing antibody, but as yet no clinical adverse effects have been observed.Citation83 Common adverse reactions were hypersensitivity reactions, localized lipodystrophy at the injection site, ectopic calcifications of the eye, including the cornea and conjunctiva, and nephrocalcinosis.Citation85 Although calcification of blood vessels was not reported,Citation83 since medial artery calcifications are the most important pathological state associated with elevated alkaline phosphatase expression,Citation86 cautious follow-up is necessary.
Future perspective
The targets of clinical trials of asfotase alfa were perinatal and infantile forms of HPP.Citation83,Citation84 For milder forms, namely, the childhood form, adult form, and odontohypophosphatasia, feasibility and safety have not been established. The current indication of asfotase alfa is therefore for perinatal/infantile-and juvenile-onset HPP.Citation85 Furthermore, the natural course of the adult form of the disease is not well understoodCitation68 and should be elucidated, and more evidence regarding the feasibility of asfotase alfa for the milder form is needed. In addition, there are some problems with administration methods of asfotase alfa. Asfotase alfa must be administered at least three times per week, which is a big burden for the growing patients. The end point of the drug is also not known. It is reasonable that TNAP is necessary for growing children, but the enzyme is also required for bone remodeling in adulthood. It is still unclear whether asfotase alfa should be administered to help bone remodeling in patients who reach middle age. Other problems with administration are the frequency and dosage. The half-life of asfotase alfa following subcutaneous administration is 5 days.Citation85 Modification of the drug can be considered to increase the interval of the injection. Otherwise, combination treatment can also be considered; asfotase alfa could be combined with stem-cell transplantation. Bone marrow stem-cell transplantation has been attempted, but the outcome was not very effective.Citation87,Citation88 Currently, the combination treatment is being attempted. In addition, a recent successful attempt of ERT in Akp2−/− mice using a soluble chimeric alkaline phosphatase, which is a chimeric molecule of human intestinal alkaline phosphatase with the crown domain of human PLAP,Citation89 showed therapeutic benefit of the nonmineral-targeting recombinant enzyme.Citation90 Another method to reduce the injection burden is gene therapy, especially ex vivo gene therapy into bone marrow stem cells. Gene therapy was successfully attempted in KO mice.Citation91,Citation92 Although administration of a viral vector containing the TNAP gene into blood is effective, the effect is transient. Administration into muscular tissue or combination with stem-cell transplantation (ex vivo gene therapy)Citation93 may be effective with a one-shot administration. Nevertheless, those experiments were performed in the KO mouse model, and it is necessary to prove the safety of vector administration for application to human patients. Furthermore, it is necessary to investigate the optimal vector that has the highest transduction efficacy.
Conclusion
HPP is an inherited systemic bone disease characterized by bone hypomineralization. The responsible gene ALPL encodes TNAP, which is a GPI-anchored membrane-bound ectoenzyme expressed ubiquitously and plays an essential role in bone mineralization. Since the 3D structure of human TNAP has not been solved, a simulation model based on human PLAP is used to discuss the relationship between TNAP structure and mutations seen in the disease. The symptoms of HPP vary depending on the clinical form. Patients with severe forms suffer from respiratory failure, which is the main cause of death. An established treatment for HPP was not available until the recent development of ERT with a modified human TNAP (asfotase alfa). Treatment for HPP has changed drastically since the advent of asfotase alfa. However, there is still not enough data regarding the outcome of its administration, especially regarding the end points. In addition, daily or thrice-weekly injection of asfotase alfa may be a burden for patients. Further improvement of the therapy for HPP patients as well as more knowledge of the pathophysiology is needed.
Acknowledgments
The author thanks Dr Tomohiro Matsumura, Division of Metabolism and Nutrition, Department of Biochemistry and Molecular Biology, Nippon Medical School, for arranging the 3D structure model of TNAP.
Disclosure
The author received honoraria from Alexion Pharmaceuticals, Inc. The author reports no other conflicts of interest in this work.
References
- RathbunJC“Hypophosphatasia”: a new developmental anomalyAm J Dis Child19487582283118110134
- WhyteMPHypophosphatasia: nature’s window on alkaline phosphatase function in manBilezikianJPRaiszLGRodanGAPrinciples of Bone Biology2nd edSan Diego, CAAcademic Press200212291248
- BianchiMLHypophosphatasia: an overview of the disease and its treatmentOsteoporos Int20152627422757
- WenkertDMcAlisterWHCoburnSPHypophosphatasia: nonlethal disease despite skeletal presentation in utero (17 new cases and literature review)J Bone Miner Res2011262389239821713987
- MillánJLMammalian Alkaline Phosphatases: From Biology to Applications in Medicine and BiotechnologyWeinheim, GermanyWiley-VCH Verlag GmbH & Co2006
- SilventJGasseBMornetESireJYMolecular evolution of the tissue-nonspecific alkaline phosphatase allows prediction and validation of missense mutations responsible for hypophosphatasiaJ Biol Chem2014289241682417925023282
- HarrisHThe human alkaline phosphatases: what we know and what we don’t knowClin Chim Acta19891861331502178806
- SmithMWeissMJGriffinCARegional assignment of the gene for human liver/bone/kidney alkaline phosphatase to chromosome 1p36.1-34Genomics198821391433410475
- WeissMJRayKHenthornPSLambBKadeschTHarrisHStructure of the human liver/bone/kidney alkaline phosphatase geneJ Biol Chem198826312002120103165380
- HenthornPSRaduchaMKadeschTWeissMJHarrisHSequence and characterization of the human intestinal alkaline phosphatase geneJ Biol Chem198826312011120192841341
- KnollBJRothblumKNLongleyMNucleotide sequence of the human placental alkaline phosphatase gene: evolution of the 5′ flanking region by deletion/substitutionJ Biol Chem198826312020120273042787
- MillánJLManesTSeminoma-derived Nagao isozyme is encoded by a germ-cell alkaline phosphatase geneProc Natl Acad Sci U S A19888531243028
- GriffinCASmithMHenthornPSHuman placental and intestinal alkaline phosphatase genes map to 2q34–q37Am J Hum Genet198741102510343687940
- MatsuuraSKishiFKajiiTCharacterization of a 5′-flanking region of the human liver/bone/kidney alkaline phosphatase gene: two kinds of mRNA from a single geneBiochem Biophys Res Commun199016899310002346496
- ShibataHFukushiMIgarashiADefective intracellular transport of tissue-nonspecific alkaline phosphatase with an Ala162 → Thr mutation associated with lethal hypophosphatasiaJ Biochem19981239689779562633
- NosjeanOKoyamaIGosekiMRouxBKomodaTHuman tissue-nonspecific alkaline phosphatases: sugar-moiety-induced enzymic and antigenic modulation and genetic aspectsBiochem J19973212973039020858
- HallSLDimaiHPFarleyJREffects of zinc on human skeletal alkaline phosphatase activity in vitroCalcif Tissue Int1999641631729914326
- Le DuMHMillánJLStructural evidence of functional divergence in human alkaline phosphatasesJ Biol Chem2002277498084981412372831
- Le DuMHStigbrandTTaussigMJMénezASturaEACrystal structure of alkaline phosphatase from human placenta at 1.8 Å resolutionJ Biol Chem20012769158916511124260
- GhoshKTagoreDMAnumulaRCrystal structure of rat intestinal alkaline phosphatase – role of crown domain in mammalian alkaline phosphatasesJ Struct Biol201318418219224076154
- LlinasPSturaEAMénezAStructural studies of human placental alkaline phosphatase in complex with functional ligandsJ Mol Biol200535044145115946677
- SowadskiJMHandschumacherMDMurthyHMFosterBAWyckoffHWRefined structure of alkaline phosphatase from Escherichia coli at 2.8 Å resolutionJ Mol Biol19851864174333910843
- MicanovicRGerberLDBergerJKodukulaKUdenfriendSSelectivity of the cleavage/attachment site of phosphatidylinositol-glycan-anchored membrane proteins determined by site-specific mutagenesis at Asp-484 of placental alkaline phosphataseProc Natl Acad Sci U S A1990871571612153284
- AntonarakisSEthe Nomenclature Working GroupRecommendation for a nomenclature system for human gene mutationsHum Mutat199811139450896
- den DunnenJAntonarakisSEMutation nomenclature extensions and suggestions to describe complex mutations: a discussionHum Mutat20001571210612815
- den DunnenJNomenclature for the description of sequence variantsMelbourne, AustraliaHGVS Available from: http://www.hgvs.org/mutnomen/Accessed April 18, 2016
- OrimoHThe mechanism of mineralization and the role of alkaline phosphatase in health and diseaseJ Nippon Med Sch20107741220154452
- AndersonHCThe role of matrix vesicles in physiological and pathological calcificationCurr Opin Orthop200718428433
- MebarekSAbousalhamAMagneDPhospholipases of mineralization competent cells and matrix vesicles: roles in physiological and pathological mineralizationsInt J Mol Sci2013145036512923455471
- RobertsSJStewartAJSadlerPJFarquharsonCHuman PHOSPHO1 exhibits high specific phosphoethanolamine and phosphocholine phosphatase activitiesBiochem J2004382596515175005
- RobertsSNarisawaSHarmeyDMillánJLFarquharsonCFunctional involvement of PHOSPHO1 in matrix vesicle-mediated skeletal mineralizationJ Bone Miner Res20072261762717227223
- AddisonWNAzariFSørensenESKaartinenMTMcKeeMDPyrophosphate inhibits mineralization of osteoblast cultures by binding to mineral, upregulating osteopontin, and inhibiting alkaline phosphatase activityJ Biol Chem2007282158721588317383965
- MillánJLThe role of phosphatases in the initiation of skeletal mineralizationCalcif Tissue Int20139329930623183786
- JohnsonKMoffaAChenYPritzkerKGodingJTerkeltaubRMatrix vesicle plasma cell membrane glycoprotein-1 regulates mineralization by murine osteoblastic MC3T3 cellsJ Bone Miner Res19991488389210352096
- WangWXuJDuBKirschTRole of the progressive ankylosis gene (ank) in cartilage mineralizationMol Cell Biol20052531232315601852
- JohnsonKGodingJVan EttenDLinked deficiencies in extracellular PPi and osteopontin mediate pathologic calcification associated with defective PC-1 and ANK expressionJ Bone Miner Res200318994100412817751
- OkawaANakamuraIGotoSMoriyaHNakamuraYIkegawaSMutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spineNat Genet1998192712739662402
- RutschFRufNVaingankarSMutations in ENPP1 are associated with “idiopathic” infantile arterial calcificationNat Genet20033437938112881724
- HoAMJohnsonMDKingsleyDMRole of the mouse ank gene in control of tissue calcification and arthritisScience200028926527010894769
- HessleLJohnsonKAAndersonHCTissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralizationProc Natl Acad Sci U S A2002999445944912082181
- HarmeyDHessleLNarisawaSJohnsonKATerkeltaubRMillánJLConcerted regulation of inorganic pyrophosphate and osteopontin by Akp2, Enpp1, and AnkAm J Pathol20041641199120915039209
- AndersonHCHsuHHMorrisDCFeddeKNWhyteMPMatrix vesicles in osteomalacic hypophosphatasia bone contain apatite-like mineral crystalsAm J Pathol1997151155515619403706
- YadavMSimãoAMNarisawaSLoss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function: a unified model of the mechanisms of initiation of skeletal calcificationJ Bone Miner Res20112628629720684022
- OrimoHGoseki-SoneMHosoiTShimadaTFunctional assay of the mutant tissue-nonspecific alkaline phosphatase gene using U2OS osteoblast-like cellsMol Genet Metab20089437538118455459
- WaymireKGMahunenJDJajeJMGuilarteTRCoburnSPMacGregorGRMice lacking tissue nonspecific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6Nat Genet19951145517550313
- GuilarteTRRegional changes in the concentrations of glutamate, glycine, taurine, and GABA in the vitamin B-6 deficient developing rat brain: association with neonatal seizuresNeurochem Res1989148898972574423
- FeddeKNWhyteMPAlkaline phosphatase (tissue-nonspecific isoenzyme) is a phosphoethanolamine and pyridoxal-5′-phosphate ectophosphatase: normal and hypophosphatasia fibroblast studyAm J Hum Genet1990477677752220817
- WhyteMPMahunenJDFeddeKNColeSMcCabeERCoburnSPPerinatal hypophosphataia: tissue levels of vitamin B6 are unremarkable despite markedly increased circulating concentrations of pyridoxal-5′-phosphateJ Clin Invest198881123412393350970
- Baumgartner-SiglSHaberlandtEMummSPyridoxine-responsive seizures as the first symptom of infantile hypophosphatasia caused by two novel missense mutations (c.677T > C, p.M226T; c.1112C > T, p.T371I) of the tissue-nonspecific alkaline phosphatase geneBone2007401655166117395561
- SilvermanJLApparent dominant inheritance of hypophosphatasiaArch Int Med196211019119813912932
- MooreCACurryCJHenthornPMild autosomal dominant hypophosphatasia: in utero presentation in two familiesAm J Med Genet19998641041510508980
- FauvertDBrun-HeathILia-BaldiniASMild forms of hypophosphatasia mostly result from dominant negative effect of severe alleles or from compound heterozygosity for severe and moderate allelesBMC Med Genet2009105119500388
- MornetEHypophosphatasiaOrphanet J Rare Dis200724017916236
- MacfarlaneJDKroonHMvan den HartenJJPhenotypically dissimilar hypophosphatasia in two sibshipsAm J Med Genet1992421171211308350
- PeachCAZhangYWordsworthBPMutations of the tissue-nonspecific alkaline phosphatase gene (TNAP) causing a non-lethal case of perinatal hypophosphatasiaRheumatology2007461037104017409132
- MummSJonesJFinneganPWhyteMPHypophosphatasia: molecular diagnosis of Rathbun’s original caseJ Bone Miner Res2001161724172711547844
- MornetEThe tissue nonspecific alkaline phosphatase gene mutations database Available from: http://www.sesep.uvsq.fr/03_hypo_mutations.phpAccessed April 18, 2016
- MornetESturaELia-BaldiniASStigbrandTMénezALe DuMHStructural evidence for a functional role of human tissue-nonspecific alkaline phosphatase in bone mineralizationJ Biol Chem2001276311713117811395499
- CaiGMichigamiTYamamotoTAnalysis of localization of mutated tissue-nonspecific alkaline phosphatase proteins associated with neonatal hypophosphatasia using green fluorescent protein chimerasJ Clin Endocrinol Metab199883393639429814472
- OrimoHGoseki-SoneMSatoSShimadaTDetection of deletion 1154–1156 hypophosphatasia mutation using TNSALP exon amplificationGenomics1997423643669192863
- Goseki-SoneMOrimoHIimuraTExpression of the mutant (1735T-DEL) tissue-nonspecific alkaline phosphatase gene from hypophosphatasia patientsJ Bone Miner Res199813182718349844100
- KomaruKIshidaYAmayaYGoseki-SoneMOrimoHOdaKNovel aggregate formation of a frame-shift mutant protein of tissue-nonspecific alkaline phosphatase is ascribed to three cysteine residues in the C-terminal extension: retarded secretion and proteasomal degradationFEBS J20052721704171715794757
- OrimoHGoseki-SoneMInoueMTsubakioYSakiyamaTShimadaTImportance of deletion of T at nucleotide 1559 in the tissue-nonspecific alkaline phosphatase gene in Japanese patients with hypophosphatasiaJ Bone Miner Metab200220283311810413
- MichigamiTUchihashiTSuzukiATachikwaKNakajimaSOzonoKCommon mutation F310L and T1559del in the tissue-nonspecific alkaline phosphatase gene are related to distinct phenotypes in Japanese patients with hypophosphatasiaEur J Pediatr200516427728215660230
- ZurutuzaLMullerFGibratJFCorrelations of genotype and phenotype in hypophosphatasiaHum Mol Genet199981039104610332035
- WhyteMPZhangFWenkertDHypophosphatasia: validation and expansion of the clinical nosology for children from 25 years experience with 173 pediatric patientsBone20157522923925731960
- van den BosTHandokoGNiehofACementum and dentin in hypophosphatasiaJ Dent Res2005841021102516246934
- CollmannHMornetEGattenlöhnerSBeckCGirschickHNeurosurgical aspects of childhood hypophosphatasiaChilds Nerv Syst20092521722318769927
- BerksethKETebbenPJDrakeMTHefferranTEJewisonDEWermersRAClinical spectrum of hypophosphatasia diagnosed in adultsBone201354212723352924
- WatanabeAKarasugiTSawaiHPrevalence of c.1559delT in ALPL, a common mutation resulting in the perinatal (lethal) form of hypophosphatasia in Japanese and effects of the mutation on heterozygous carriersJ Hum Genet20115616616821179104
- FraserDHypophosphatasiaAm J Med19572273074613410963
- GreenbergCRTaylorCLHaworthJCA homoallelic Gly317 → Asp mutation in ALPL causes the perinatal (lethal) form of hypophosphatasia in Canadian mennonitesGenomics1990172152178406453
- MornetEYvardATaillandierAFauvertDSimon-BouyBA molecular-based estimation of the prevalence of hypophosphatasia in the European populationAnn Hum Genet20117543944521488855
- TaketaniTOnigataKKobayashiHMushimotoYFukudaSYamaguchiSClinical and genetic aspects of hypophosphatasia in Japanese patientsArch Dis Child20149921121524276437
- NarisawaSFröhlanderNMillánJLInactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasiaDev Dyn19972084324469056646
- FeddeKNBlairLSilversteinJAlkaline phosphatase knockout mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasiaJ Bone Miner Res1999142015202610620060
- BeertsenWVandenBosTEvertsVRoot development on mice lacking functional tissue non-specific alkaline phosphatase gene: inhibition of acellular cementum formationJ Dent Res1999781221122910371245
- WhyteMPMcAlisterWHPattonLSEnzyme replacement therapy for infantile hypophosphatasia attempted by intravenous infusions of alkaline phosphatase-rich Paget plasma: results in three additional patientsJ Pediatr19841059269336502342
- TadokoroMKanaiRTaketaniTUchinoYYamaguchiSOhgushiHNew bone formation by allogenic mesenchymal stem cell transplantation in a patient with perinatal hypophosphatasiaJ Pediatr200915492493019446101
- MillánJLNarisawaSLemireIEnzyme replacement therapy for murine hypophosphatasiaJ Bone Miner Res20082377778718086009
- NishiokaTTomatsuSGuitierrezMAEnhancement of drug delivery to bone: characterization of human tissue-nonspecific alkaline phosphatase tagged with an acidic oligopeptideMol Genet Metab20068824425516616566
- McKeeMDNakanoYMasicaDLEnzyme replacement therapy prevents dental defects in a model of hypophosphatasiaJ Dent Res20119047047621212313
- WhyteMPGreenbergCRSalmanNJEnzyme-replacement therapy in life-threatening hypophosphatasiaN Engl J Med201236690491322397652
- OkazakiYKitajimaHMochizukiNKitaokaTMichigamiTOzonoKLethal hypophosphatasia successfully treated with enzyme replacement from day 1 after birthEur J Pediatr201617543343726459154
- STRENSIQ™ (asfotase alfa) for Strensiq [prescribing information]Cheshire, CTAlexion Pharmaceuticals, Inc2015 Available from: http://strensiq.com/images/pi.pdfAccessed April 18, 2016
- SheenCRKussPNarisawaSPathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcificationJ Bone Miner Res20153082483625428889
- WhyteMPKurtzbergJMcAlisterWHMarrow cell transplantation for infantile hypophosphatasiaJ Bone Miner Res20031862463612674323
- TaketaniTOyamaCMiharaAEx vivo expanded allogeneic mesenchymal stem cells with bone marrow transplantation improved osteogenesis in infants with severe hypophosphatasiaCell Transplant2015241931194325396326
- SasajimaYKohamaYKojima-MisaizuMSimultaneous retension of thermostability and specific activity in chimeric human alkaline phosphatasesMol Biotechnol20145695396124906817
- GasqueKCFosterBLKussPImprovement of the skeletal and dental hypophosphatasia phenotype in Alpl−/− mice by administration of soluble (non-targeted) chimeric alkaline phosphataseBone20157213714725433339
- YamamotoSOrimoHMatsumotoTProlonged survival and phenotypic correction of Akp2−/− hypophosphatasia mice by lentiviral gene therapyJ Bone Miner Res20112613514220687159
- MatsumotoTMiyakeKYamamotoSRescue of severe infantile hypophosphatasia mice by AAV-mediated sustained expression of soluble alkaline phosphataseHum Gene Ther2011221355136421388343
- IijimaOMiyakeKWatanabeAPrevention of lethal murine hypophosphatasia by neonatal ex vivo gene therapy using lentivirally transduced bone marrow cellsHum Gene Ther20152680181226467745