Abstract
An understanding of the risks, benefits, and relative value of glatiramer acetate (GA) in multiple sclerosis (MS) has been evolving based on recently completed head-to-head studies: REGARD (REbif vs Glatiramer Acetate in Relapsing MS Disease); BEYOND (Betaseron Efficacy Yielding Outcomes of a New Dose); and BECOME (BEtaseron vs COpaxone in Multiple Sclerosis with Triple-Dose Gadolinium and 3-Tesla MRI Endpoints). Outcomes in the primary endpoints of these trials showed no significant differences between GA and high-dose beta-interferons (IFNβs). Results of the PreCISe (Early GA Treatment in Delaying Conversion to Clinically Definite Multiple Sclerosis [CDMS] in Subjects Presenting With a Clinically Isolated Syndrome [CIS]) trial led to the US Food and Drug Administration approval of GA in patients with a CIS. Furthermore, the ongoing follow-up study to the original pivotal GA trial, now extending beyond 15 years, continues to support the safety of GA. Currently, GA and IFNβs are no longer the only immunomodulators available for MS. Introduction of the monoclonal antibody, natalizumab (Tysabri®; Biogen Idec, Inc., Cambridge, MA, USA) provides an alternative immunomodulator for MS and has changed the therapeutic landscape dramatically. However, the rare but serious cases of progressive multifocal leukoencephalopathy that have occurred with natalizumab have raised concerns among clinicians and patients about using this agent and some of the emerging agents. The potential risks and benefits of the emerging therapies (cladribine, alemtuzumab, rituximab, fingolimod, laquinimod, teriflunomide, and dimethyl fumarate) based on phase II/III trials, as well as their use for indications other than MS, will be presented. This review provides available data on GA, natalizumab, and the emerging agents to support new developments in our understanding of GA and how its long-standing role as a first-line therapy in MS will evolve within the increasingly complex MS therapeutic landscape.
Introduction
Glatiramer acetate (GA) (Copaxone®; Teva Neuroscience, Kansas City, MO, USA) has been available as a first-line therapy for relapsing-remitting multiple sclerosis (RRMS) since 1996. Our understanding of the therapeutic value of GA has evolved since then based on a number of important studies, which were recently reviewed by Johnson and Due.Citation1 Until recently, GA and the beta-interferons (IFNβs) were the only immunomodulatory therapies available for treatment of RRMS; however, with the introduction of the monoclonal antibody, natalizumab (Tysabri®; Biogen Idec, Inc., Cambridge, MA, USA), the MS therapeutic landscape has changed dramatically. Many new considerations have contributed to therapeutic decisions being made in MS. In patients with continued disease activity despite treatment with disease-modifying agents, natalizumab may be used as second-line therapy.Citation2 However, use of natalizumab has led to rare but serious cases of progressive multifocal leukoencephalopathy (PML). There are many concerns regarding natalizumab as new cases of PML and other infections continue to occur in association with its use.Citation3 Recently, a 40-year-old male with RRMS developed a primary central nervous system lymphoma (PCNSL) following 21 doses of natalizumab monotherapy. Because this patient was not otherwise immunocompromised and there is no increased prevalence of PCNSL in MS, the association between natalizumab therapy and PCNSL was conceivable.Citation4 These findings of rare opportunistic infections and malignancies have raised a cautionary note among clinicians and patients who may be contemplating the use of this agent and other therapies that will be emerging in the near future. Also, recent reports show that the appearance of high-titered (>1:100)Citation5 neutralizing antibodies (NABs) totally blocks the biologic activity of IFNβs. The development of NABsCitation6 may occur in up to 35% of IFNβ-treated patients, and this phenomenon may reduce or completely eliminate IFNβ bioactivity, depending upon the level of titers present.Citation2 This information is changing treatment alternatives, especially in cases of suboptimal response.Citation2
With the emergence of multiple novel therapies currently in development, the MS therapeutic landscape is about to undergo more significant changes. Although some of these agents could be more efficacious than existing therapies (at least in the short term), risk-vs-benefit analyses must also take into account long-term safety and tolerability. Based on their use in clinical trials and experiences in therapeutic areas outside of MS, there is evidence that use of some of these emerging therapies may pose risks of developing opportunistic and community-acquired infections, malignancies, autoimmune disorders,Citation3 and complications affecting body systems other than the neurologic system. They may, therefore, require ongoing complex monitoring. The scope, as well as the duration of these effects, may not be completely predictable, complicating attempts to make reasonable and safe therapeutic decisions for MS patients.
With the current use of natalizumab and future use of several novel therapies, risk-vs-benefit analyses promise to be very different from what therapists faced when first-line MS regimens included solely GA and IFNs. The field of MS therapy is changing, and until the phase III results of trials for the emerging therapies are available, and the US Food and Drug Administration makes a decision regarding the approval of these novel agents, it will be difficult to determine their role in the armamentarium of MS. Understanding of how GA will be used in the future will depend not only on the risk-vs-benefit analysis of GA, but also on the risks and benefits of these emerging therapies. At the present time, our vision must be forward-looking rather than entirely evidence-based. This review will discuss the new developments in our understanding of GA, and the role GA may play in this new, evolving, and complex therapeutic landscape in MS.
Glatiramer acetate
GA, a member of the glatiramoid class of compounds,Citation7 is composed of a mixture of synthetic polypeptides derived from 4 amino acids that was originally designed to create a multiple peptide analog of myelin basic protein.Citation8 It was synthesized in the 1960s and studied first in many animal models and then in human patients with RRMS over a period of 30 years, culminating in its approval in the United States in 1996.Citation8 The mechanism of action (MOA) of GA is immunomodulatory. GA appears to modulate inflammation and act in a neuroprotective capacity ().Citation9 GA may shift T-lymphocyte populations in the systemic circulation via the induction of reactive Th2 immunoregulatory cellsCitation10 that have been demonstrated in experimental autoimmune encephalomyelitis (EAE) models to cross into the central nervous system (CNS) to release antiinflammatory cytokines and growth factors.Citation11 In addition, GA stimulates the secretion of neurotrophins that protect axons and may promote repair to damaged neurons.Citation10
Figure 1 Mechanisms of action of glatiramer acetate (GA) in MS.
Reprinted from Autoimmun Rev, Vol. 6, Schrempf W, Ziemssen T, Glatiramer acetate: mechanisms of action in multiple sclerosis, 469–475.Citation9 Copyright © 2007, with permission from Elsevier. 1. GA exhibits competitive binding at the MHC-II complex and TCRantagonism. In addition, GA is able to displace MBP from the binding site on MHC-II molecules. Treatment with GA leads to the induction of antigen specific TH2 T cells in the periphery. 2. In addition CD8+ and CD4+ CD25+ regulatory T cells are induced by GA therapy. 3. The constant activation seems to have an important impact on the induction and maintenance of the regulatory/suppressive immune cells. 4. Because of the daily activation, GA T cells are believed to be able to cross the blood–brain barrier. 5. Inside the CNS, some GA-specific T cells cross-react with products of local myelin turnover presented by local APCs. 6. In response anti-inflammatory cytokines are secreted which dampen the local inflammatory process (bystander suppression). 7. Furthermore GA specific T cells secrete neurotrophic factors which might favor remyelination and axonal protection.
The safety and efficacy of GA have been demonstrated over many years, starting in 1995, with the findings provided by a pivotal, placebo-controlled trial,Citation12 and more recently by head-to-head trials comparing GA with high-dose IFNβs. Also, prospective, follow-up studies of over a decade using GA as monotherapy have provided additional confirmation of safety.Citation13
Head-to-head trials
Three head-to-head trials have been completed that compared the efficacy and safety of high-dose IFNs with GA. Although it is difficult to compare results across different studies due to the lack of a placebo arm, these 3 trials found that there were no significant differences between high-dose IFN-based therapies and GA in primary endpoints evaluating reductions in relapse rates.Citation14–Citation16
The REGARD (REbif vs Glatiramer Acetate in Relapsing MS Disease) study, a randomized, comparative, parallel-group, open-label trial, included 764 patients with RRMS (diagnosed by McDonald criteria) who had experienced at least 1 relapse in the past year. The subjects received either 44 μg of IFNβ-1a subcutaneous (SC) (Rebif®; EMD Serono, Rockland, MA, USA and Pfizer, Inc., New York, NY, USA) 3 times a week or 20 mg of GA SC once a day for 96 weeks – 65 patients discontinued IFN and 49 patients discontinued GA, with 82% of 764 patients completing the trial. Time to first relapse (primary outcome) and magnetic resonance imaging (MRI) benchmarks (T2-weighted and gadolinium-enhancing [GdE] lesion number and brain volume) were studied. After 96 weeks, there was no significant difference between groups in the time to first relapse () (hazard ratio [HR] 0.94, 95% confidence interval [CI], 0.74–1.21; P = 0.64), and both groups had lower than predicted relapse rates.Citation14
Figure 2 REGARD trial: Kaplan–Meier plot of time to first relapse.Citation14
Reprinted with permission from Mikol DD, Barkhof F, Chang P, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol. 2008;7(10):903–914. Citation14 Copyright © 2009, with permission from Elsevier.
![Figure 2 REGARD trial: Kaplan–Meier plot of time to first relapse.Citation14Reprinted with permission from Mikol DD, Barkhof F, Chang P, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol. 2008;7(10):903–914. Citation14 Copyright © 2009, with permission from Elsevier.](/cms/asset/9c4aba3c-0ff6-43a4-8f13-85238e2ed91d/dtcr_a_6743_f0002_b.jpg)
Annualized relapse rates (ARRs) were not different between the groups (). In terms of MRI outcomes, there were no significant differences in the number and change in volume of T2 active lesions. Patients treated with IFNβ-1a SC had significantly fewer GdE lesions (0.24 vs 0.41 lesions per patient per scan; P = 0.0002) and patients treated with GA experienced significantly less brain atrophy (P = 0.018).Citation14
Figure 3 ARRs before and during treatment in the REGARD and BEYOND trials.
A) REGARD trial: ARRs before and during treatment.Citation14 ARR for the 2 years before and during the 96 weeks of the study in the intention-to-treat population.a Number of relapses during 96 weeks was analyzed using a Poisson regression model with factors for treatment and center. The log of time on study was used as the offset variable. B) BEYOND trial: ARRs 1 year before and during treatment. (Assessed by hazard ratios derived from generalized linear Poisson regression).Citation15 Reprinted with permission from Mikol DD, Barkhof F, Chang P, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol. 2008;7(10):903–914.Citation14 Copyright © 2009, with permission from Elsevier.
![Figure 3 ARRs before and during treatment in the REGARD and BEYOND trials.A) REGARD trial: ARRs before and during treatment.Citation14 ARR for the 2 years before and during the 96 weeks of the study in the intention-to-treat population.a Number of relapses during 96 weeks was analyzed using a Poisson regression model with factors for treatment and center. The log of time on study was used as the offset variable. B) BEYOND trial: ARRs 1 year before and during treatment. (Assessed by hazard ratios derived from generalized linear Poisson regression).Citation15 Reprinted with permission from Mikol DD, Barkhof F, Chang P, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol. 2008;7(10):903–914.Citation14 Copyright © 2009, with permission from Elsevier.](/cms/asset/1f5c07dd-5607-4195-8a89-81da3dfba50c/dtcr_a_6743_f0003_b.jpg)
The BEYOND (Betaseron Efficacy Yielding Outcomes of a New Dose) study compared 3 treatment groups (N = 2244 in a 2:2:1 randomization): 250 μg of IFNβ-1b SC (Betaseron®, Bayer HealthCare Pharmaceuticals, Montville, NJ, USA) dosed every other day; IFNβ-1b 500 μg SC dosed every other day; GA 20 mg SC daily over ≥2 years.Citation15 The primary endpoint for this study was the risk of relapses; however, the study also examined the time to confirmed Expanded Disability Status Scale (EDSS) progression and MRI parameters. Annualized relapse rates were 0.33, 0.36, and 0.34 for IFNβ-1b 500 μg, IFNβ-1b 250 μg, and GA, respectively, and did not differ (). No significant differences were found in time to first relapse, overall relapse rates during the study period, and proportion of patients who remained relapse free.Citation15 For MRI data, there was no significant difference in GdE lesions, T1 lesions, and normalized brain volume.
There were significant differences in the cumulative number of new T2 lesions between the 3.3 lesions in patients on IFNβ-1b 500 μg and the 3.3 lesions in patients on IFNβ-1b 250 μg compared with the 4.6 lesions in patients treated with GA (P = 0.0009 and P = 0.011, respectively). T2 lesion volume increased for all 3 treatment groups: IFNβ-1b 500 μg, IFNβ-1b 250 μg, and GA 20 mg, by 12%, 10%, and 17%, respectively, with a significant difference between GA and the high and low IFN doses, P = 0.0008 and P = 0.0001, respectively.Citation15 Among 2244 patients, 1934 (86%) completed the study. The highest number of dropouts (161 patients) was observed among those patients receiving IFNβ-1b 500 μg, followed by patients receiving IFNβ-1b 250 μg (104 patients), with 71 patients receiving GA dropping out.
The BECOME (BEtaseron vs COpaxone in Multiple Sclerosis with Triple-Dose Gadolinium and 3-Tesla MRI Endpoints) study was a head-to-head study conducted to determine the efficacy of treatment with IFNβ-1b or GA as assessed by monthly brain MRI.Citation16 A total of 75 patients with RRMS or clinically isolated syndrome (CIS) were enrolled in the study; 4 patients in each group discontinued the study medication. They were randomized to receive either IFNβ-1b 250 μg SC every other day or GA 20 mg SC daily and underwent enhanced MRI scans for up to 2 years. Investigators used a specialized protocol with triple-dose Gd and delayed imaging post-injection, utilizing a 3-Tesla MRI scanner – all of which were intended to maximize detection of combined active lesions (CALs; CAL refers to the total number of contrast-enhancing lesions plus new nonenhancing lesions on long repetition time scans that have appeared since the most recent examination).Citation16 There were similar median (75th percentile) CALs per patient per scan for Months 1 to 12 – 0.63 (2.76) for IFNβ-1b and 0.58 (2.45) for GA. In addition, there were no significant differences in the effects of the medications on relapse rates. The ARR for IFNβ-1b changed from 1.8 to 0.37, with a reduction of 79% compared with a difference of 1.9 to 0.33 for GA, representing an 83% reduction in ARR from baseline with treatment for each agent, respectively.Citation16
When examining the data from these 3 head-to-head trials, it is important to note that GA performed similarly to the high-dose IFN-based therapies in efficacy on the primary endpoints, including relapse rate reductions and onset of action. Overall, reductions in relapse rates were much greater in each of these head-to-head trials than those reported during the pivotal trial for GA in 1995.Citation12 Furthermore, the ARR achieved with GA (range, 0.29 to 0.34)Citation14–Citation16 was much improved compared with that achieved in the pivotal trial (0.59).Citation12 In the pivotal trial, there was a 29% reduction in favor of GA over placebo (P = 0.007), with an ARR of 0.59 for GA and 0.84 for placebo.Citation12 A difference in patient populations was the most likely explanation for this trend. Currently, earlier diagnosis of MS and earlier treatment initiation are possible through the use of imaging-based McDonald criteria rather than clinically based criteria, and the patient populations of these trials reflect this shift. Results from the more recent trialsCitation14–Citation16 are probably more indicative of the effectiveness that should be expected with initiation of GA therapy in a newly diagnosed RRMS patient who is more likely to resemble patients in these newer studies rather than those in the pivotal trial.Citation12
Indication for CIS
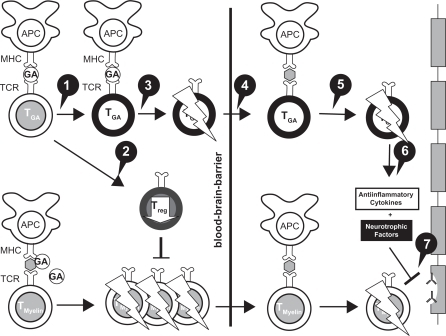
The PreCISe (Early GA Treatment in Delaying Conversion to Clinically Definite Multiple Sclerosis [CDMS] in Subjects Presenting With a Clinically Isolated Syndrome [CIS]) trial evaluated the effect of GA on delaying conversion of patients with CIS to CDMS. Data from this trial, including results that showed GA delays conversion of CIS to CDMS, led to the recent US Food and Drug Administration (FDA) approval of the use of GA for the treatment of patients with CIS.Citation17 The primary endpoint of the study was the time to CDMS. A total of 481 patients who had experienced a single, unifocal, clinical attack participated in the study. Those in the active treatment arm received GA 20 mg SC per day. The Drug Monitoring Committee (DMC) conducted an interim analysis (mean exposure was 2.3 [SD 0.65] years, 81% of the targeted 3-year study) and based on the results, the DMC recommended the placebo arm of the trial be stopped, and all placebo patients be given the opportunity to receive active treatment with GA for 2 years.Citation17 The trial data demonstrated that there was a significant delay of 386 days in conversion to CDMS: 722 days for the GA-treated group compared with 336 days for the untreated patients (HR = 0.55, 95% CI, 0.40–0.77; P = 0.0005) (). Patients who converted to CDMS then continued on active treatment for an additional 2 years.Citation17
Figure 4 Time to conversion to CDMS in the placebo-controlled phase. Kaplan–Meier curves with Cox’s proportional hazard ratio were used to model the amount of time to conversion to CDMS for patients assigned to GA and placebo. A delay of 386 days (115%) in conversion to CDMS was noted for first quartile of patients receiving GA compared with those receiving placebo. Reprinted from The Lancet, Comi G, Martinelli V, Rodegher M, et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet. 2009;374:1503–1511.Citation17 Copyright 2009, with permission from Elsevier.
During the PreCISe trial, it was noted that GA also had an impact on MRI and magnetic resonance spectroscopy (MRS) parameters.Citation18,Citation19 Specifically, patients receiving GA experienced a 58% reduction in new T2-weighted lesions (P < 0.0001) and exposure-adjusted T2-weighted lesion volume (P = 0.0002).Citation18 GA also reduced the number of new T1 GdE and new T1 hypointense lesions (P = 0.0001).Citation18 In addition, evidence exists for axonal protection with use of GA in patients with CIS. MRS techniques demonstrated a significant difference in paired changes of N-acetylaspartate (NAA)/creatine ratios between patients treated with GA vs those treated with placebo, which remained for up to 24 months.Citation19 Finally, GA was well tolerated among the patients with CIS, with a side-effect profile similar to that seen in patients treated with GA for RRMS.Citation17
Long-term data
Despite the chronic nature of MS and the growing trend to recommend initiation of disease-modifying therapies (DMTs) early in the disease course (eg, upon diagnosis of CIS), there are scant data on the efficacy and safety of DMTs beyond 5 years. Ford et alCitation13 conducted a prospective, open-label, extension study to evaluate the neurologic status and EDSS scores in patients from the original pivotal trial of GACitation12 who had used GA for up to 15 years.Citation13 As of November 2003, 108 (47%) patients from the 232 originally enrolled in the US Glatiramer Acetate Pivotal Trial who had taken GA for an average of 10 years remained in the open-label extension phase. Of the 124 who withdrew from the study, 50 were available for long-term follow-up (LTFU) and 74 were not, resulting in a total of 158 (68%) patients from the original study who were evaluable at 10 years.Citation13 Investigators found that while on GA, relapse rates declined from 1.18 per year pre-study to approximately 1 relapse every 5 years – a decline of >80%.Citation13 The median time to ≥1-point increase in EDSS score was 8.8 years; 58% of patients had stable or improved EDSS scores,Citation13 while 38%,18%, and 3% reached EDSS scores of 4, 6, and 8, respectively (among patients who had received at least 1 GA dose since study inception). Citation20 While receiving GA, nearly all patients (with a mean disease duration of 15 years) were ambulatory, with EDSS scores of <6.Citation13 A LTFU visit of patients who had withdrawn from the pivotal trial showed that these patients had greater disability than patients who had continued on GA, with slightly higher EDSS scores (3.00 ± 1.59 [SD] vs 2.569 ± 1.35, respectively; P = 0.03).Citation13
The safety profile of GA was favorable over the long term, with the most common adverse events (AEs) related to GA use being local injection-site reactions (erythema, pain, mass, edema) and self-limited immediate post-injection reactions. No other immune-mediated disorders, infections, or malignancies have been associated with long-term use of GA.Citation13 The mean duration of GA treatment was 4.269 ± 3.13 [SD] years (range: 0.2–11.5) in the Withdrawn Total cohort. The GA exposure was 4.479 ± 2.95 years (range: 0.2–10.4) in the Withdrawn with LTFU cohort, and 4.139 ± 3.26 years (range: 0.2–11.5) in the Withdrawn without LTFU cohort, and there were no statistical differences in demographic or disease characteristics between withdrawn patients who returned for LTFU and withdrawn patients who did not return at the time of GA initiation. Because no objective neurologic evaluations were conducted on the patients at the time they withdrew, subjective data about patients who withdrew from the study due to their own perception that their disease was worsening cannot be supported by objective data. In addition, patients who withdrew from this study did so for various reasons, not only based on the perception of worsening disease. For example, patients withdrew for non-MS-related issues, such as lack of transportation to the study site or pregnancy.Citation13
Suboptimal responses to DMTs
The relative frequency of suboptimal responses to MS therapy has been noted to be as high as 30% in 3 years post-initiation of first-line therapies.Citation21 Criteria for defining what constitutes a suboptimal response vary, however. Typical criteria include relapse rates greater than 1 per year or unchanged from pretreatment rates; incomplete recovery from relapses; new brainstem or spinal cord lesions; or progression of disability or cognitive impairment that leads to a disruption in activities of daily living.Citation22 A significant cause of suboptimal response in the case of IFN therapy is the development of NABs (this has been recently reviewed by Rudick and Polman).Citation2 Reports indicate how NABs reduce or abolish IFNβ bioactivity in a titer-dependent manner.Citation2,Citation23 The appearance of high-titered (>1:100)Citation5 NABs has been shown to block the biologic activity of IFNβs. NABs may occur in up to 35% of IFNβ-treated patients, with several studies suggesting that IFNβ-1b is most immunogenic (35% of patients NAB-positive) followed by subcutaneous IFNβ-1a (23.7% of patients NAB-positive) and lastly intramuscular IFNβ-1a (7% of patients NAB-positive), as indicated in one review by Rudick.Citation2
Another potential cause of suboptimal responses to DMTs is a lack of adherence to therapies over the long term, because 4 of 6 currently available therapies require self-injection, and all have side effects that may range from influenza-like symptoms to injection-site reactions. Barriers to adherence thus include needle phobia and coping with the aforementioned AEs as well as others, but also include additional issues, such as not taking medication because of forgetfulness, complacency, treatment fatigue, changes in socioeconomic circumstances, and perceived lack of efficacy.Citation24 Specifically related to GA, a study by Haas and Firzlaff of 308 patients with RRMS conducted over 24 months indicated a significantly (P < 0.001) lower discontinuation rate for GA compared with the 3 IFN-based therapies.Citation25 However, regardless of whether patients are treated with IFNβ or GA, adherence rates for 2 to 5 years range from 60% to 76% overall, Citation24 and 49% discontinue treatment within the first 2 years.Citation25 Adverse events that most commonly resulted in treatment discontinuation (affecting approximately 5% of 563 patients) for GA during the clinical trials included injection-site reactions, dyspnea, urticaria, vasodilatation, and hypersensitivity. Citation26 The most common adverse reactions overall were: injection-site reactions, vasodilatation, rash, dyspnea, and chest pain.Citation24,Citation26 Localized lipoatrophy is another AE that may lead to nonadherence. This occurred in approximately 2% of patients participating in the clinical trials.Citation26 Evidence suggests that healthcare providers can have an impact on patient adherence by assuring a close relationship with their patients, providing education and support, reinforcing the need to continue treatment, and managing treatment expectations, as well as side effects.Citation24
Although there is minimal class I or class II evidence on the impact of switching DMTs to improve patient response or eliminate AEs, 3 open-label studies have been conducted to investigate whether changing from an IFN-based therapy to GA improves outcomes in patients who have experienced suboptimal responses to IFNs.Citation27–Citation29
Caon et alCitation27 evaluated the clinical course of 85 patients with RRMS who had received weekly doses of IFNβ-1a intramuscular (IM) (Avonex®, Biogen, Inc, Cambridge, MA, USA) for ≥18 months, but who were switched to GA therapy due to either persistent suboptimal efficacy (n = 62) or persistently unacceptable toxicity (n = 23).Citation27 Treatment with IFNβ-1a IM reduced the ARR from 1.41 to 1.23 after a mean duration of 19.7 months. In the patients with a persistent suboptimal efficacy who were switched to GA, the mean ARR was reduced from 1.32 to 0.52 (P = 0.0001).Citation27 GA therapy was effective in a group of patients who switched due to persistent toxicity associated with IFN therapy. Within this group, therapeutic efficacy was maintained after the switch to GA: the ARR decreased slightly from 0.61 to 0.47.Citation27 In addition, in each group, EDSS scores improved by approximately 0.5 points in each group after the switch to GA.Citation27 There were 5 (8%) patients who, due to suboptimal efficacy, were switched to GA, but failed to respond to this therapy as well. Two patients who had been switched to GA due to intolerable toxicity from IFNβ-1a IM also did not demonstrate a reduction in ARR. Excluding these patients from the analysis, however, did not affect the results. Six patients were lost to follow-up, and no data are available on whether patients taking GA subsequently discontinued this therapy.Citation27
Similarly, Carrá et alCitation28 found that switching to GA following IFN therapy improved effectiveness and stabilized disability progression when there had been a suboptimal response to IFN therapy. This was a prospective longitudinal observational study of 114 patients with RRMS who switched treatments after 3 years. Patients were switched either from low-dose to high-dose IFNβ (n = 31), from IFNβ to GA (n = 52) or mitoxantrone (n = 13), or from GA to IFNβ (n = 16).Citation28 Patients who had failed treatment were candidates for a switch in therapies. Criteria for treatment failure included either inadequate efficacy or the occurrence of AEs. In the case of the treatment group of 18 patients originally treated with GA, 14 (77.8%) were switched due to inadequate efficacy. In the group of patients switched from IFNβ to GA, the ARR was reduced from 0.63 to 0.14, a decrease of 77%, with 68% of those who had been switched due to efficacy concerns not experiencing a relapse over the entire treatment period of 3 years (an improvement from 16% who were relapse free in the period before the switch to GA).Citation28 Furthermore, patients who switched from IFNβ to either GA or mitoxantrone did not experience a significant progression in their EDSS scores. This was in contrast to patients who switched from low-dose IFN-based therapy to a high-dose IFN-based therapy and continued to increase their disability scores.Citation28 All groups (IFNβ to GA, low-dose IFNβ to high-dose IFNβ, IFNβ to mitoxantrone, and GA to IFNβ) in this study benefited from switching therapies.Citation28 The best outcomes, as reflected by improvements in ARR and stabilization of EDSS scores, were in those patients switching to GA or mitoxantrone.Citation28
A third study, by ZwibelCitation29 compared clinical outcomes between 558 treatment-naive patients initiating treatment with GA and 247 patients who were switched from IFNβ-1b SC primarily due to occurrence of AEs, such as flu-like symptoms or injection-site reactions (36% stopped treatment with IFNβ-1b due to perceived lack of efficacy).Citation29 Patients in the switch group were older and demonstrated more advanced disease, with higher baseline EDSS scores.Citation29 For example, 22% of those previously treated with IFNβ-1b had an EDSS score of 6.0% vs 10% of treatment-naïve patients. Following a switch, mean ARR in the switched patients decreased by 75%, which was the same decrease noted in treatment-naive patients initially treated with GA.Citation29 A total of 107 patients who had previously been on IFNβ-1b and 247 patients in the previously treatment-naive group discontinued GA before study completion. Both groups cited similar reasons for discontinuing GA – most commonly “patient decision” ([n = 147; 18.3%], which was not further explained in the study), followed by “adverse experience” (n = 88; 10.9% of each cohort). Of the latter group, 30 patients discontinued due to an injection-site reaction; however, more than 97% of injection-site reactions overall were rated as mild or moderate in severity.Citation29
In summary, these studies clearly demonstrate improvements in ARRs and disability (as reflected by EDSS scores) when patients are switched from an IFN to GA.
Effect of GA on fatigue, depression, and work absenteeism
Although most commonly regarded as a progressive neurologic disease that causes physical disability, MS is also associated with psychological symptoms that affect quality of life.Citation30 These include fatigue and depression, which have an effect on such key functions as work performance.Citation31 Fatigue is highly prevalent among patients with MS,Citation31,Citation32 reported by approximately 75% of patientsCitation33,Citation34 and noted to be “one of the most distressing symptoms” of MS by >50% of patients.Citation32,Citation33 As a result, fatigue has a major impact on productivity. Among individuals with MS forced to work part-time, 90% reported fatigue as the cause.Citation31,Citation35 Zeimssen et alCitation33 conducted a prospective, observational, noninterventional study of patients with RRMS (by McDonald criteria) to determine the impact of initiating GA treatment on fatigue and absenteeism. All patients were diagnosed with RRMS (n = 291), treatment naive, and evaluated every 3 months for 1 year with neurologic assessments. Patients also were assessed for disability with EDSS; fatigue was assessed by a visual analog scale (VAS) and the Modified Fatigue Impact Scale (MFIS).Citation33
Use of GA resulted in a significant reduction in fatigue and lost work days. MFIS scores decreased by 7.6 ± 16.4 from 34.6 to 27.0 (P ≤ 0.001) and fatigue symptoms using a VAS decreased by 1.04 ± 2.88 cm from 4.47 cm to 3.43 cm (P ≤ 0.001). The proportion of patients absent from work at least once was reduced by a factor of 2 from 65.1% to 30.1% (P ≤ 0.001). These reductions in fatigue and absenteeism corresponded with improvements in disability and relapses. The mean EDSS score at the end of treatment was 2.45, representing a mean, statistically significant decrease from baseline of 0.55 points (P < 0.05; Wilcoxon rank test). Furthermore, GA was found to be well tolerated. AEs, most frequently local injection-site reactions, were reported in 15.1% of patients.Citation33
While the pathogenesis of fatigue in MS is not clearly understood, it is thought to be either a nonspecific outcome of overall disease improvement or due to an impact of treatment on the pathophysiology of MS fatigue.Citation33 High levels of proinflammatory cytokines, for example, have been associated with exacerbation of fatigue.Citation36 Therefore, the reduction in fatigue associated with GA may be due to the drug’s attenuation of secretion and activity of these cytokines within the CNS.Citation37
Likewise, depression has been identified by several studies to be prevalent among patients with MS.Citation30 For example, a Canadian Community Health Survey found that the 12-month prevalence of major depression in patients with MS was 15.7% compared with 4.0% of patients without a chronic illness, but was also higher vs individuals with other chronic conditions in whom prevalence of depression was 9.1%.Citation30,Citation38 MS patients experience approximately a 50% risk of major depression over their lifetime,Citation39 and about 25% have suicidal ideation.Citation30,Citation40
GA does not appear to exacerbate depression, and may stimulate production of brain-derived neurotrophic factor (BDNF) that may have a positive effect on depression.Citation30,Citation41–Citation43 Depression has been associated with decreased levels of BDNF as well as with an inflammatory process in the brain.Citation41 Studies have demonstrated that GA administration can enhance central BDNF activity or increase serum BDNF levels. When administered to animals, GA also has a central anti-inflammatory effect through the release of interleukin-10.Citation41 Finally, peripheral administration of GA has been found to augment neurogenesis in rodents.Citation41 Therefore, evidence from preclinical and clinical studies indicates that GA could have antidepressant effects by increasing central BDNF, decreasing inflammation, and stimulating neurogenesis.Citation41
Compounds related to currently approved GA
Other related compounds – of higher molecular weight (MW) and of higher dosage than currently approved GA – have also been explored for the treatment of MS. Like GA, protiramer is a glatiramoid but with a higher MW.Citation7 Preliminary data from 2 pilot studies in patients with RRMS (n = 62 for both studies) showed that protiramer, 15 mg/week, had no significant effect in reducing GdE lesions, while 30 mg/week significantly reduced the mean number of GdE lesions and new T2 W lesions (P = 0.0013 and P = 0.002, respectively) during a 36-week treatment phase.Citation7 However, animal studies involving chronic use of protiramer (>3 months) subsequently demonstrated serious toxicities that have not been shown with GA. For example, severe injection-site reactions (including disseminated necrosis and inflammation of dermal muscles, nerves, and blood vessels) in addition to kidney and liver lesions were observed in monkeys and rats.Citation44 Furthermore, alterations in a number of hematologic and chemistry parameters were noted, and 2 deaths occurred in the group of monkeys being treated with high-dose protiramer for ≥24 weeks.Citation44 As a result, further development of protiramer has not been pursued.Citation44
A phase II study to evaluate the safety, efficacy, and tolerability of GA at 40 mg/day (n = 46) vs the approved 20 mg/day (n = 44) in patients with RRMS showed that the decrease in mean GdE lesions was significantly greater with the 40-mg dose vs the 20-mg dose (65% vs 75%, respectively [P < 0.0001]). Furthermore, the higher dose was found to be safe and well tolerated, although some aspects of the injection-site reactions (eg, pain) were more common in the higher dose group.Citation45 However, results from a phase III randomized, double-blind, parallel-group study to evaluate the safety, tolerability, and efficacy of GA 40 mg compared with 20 mg showed that the current dose of 20 mg and the higher dose of 40 mg were equally effective in reducing clinical relapses and MRI activity.Citation46 In this study, 1155 patients with definite MS (revised McDonald criteria) and at least one documented relapse in the 12 months prior to screening, or at least 2 documented relapses in the 24 months prior to screening, and EDSS score 0–5.5, were enrolled. The primary endpoint was the rate of confirmed relapses. The patients were randomized to GA 20 mg (n = 586) or 40 mg (n = 569). The primary efficacy endpoint was similar in both groups (relative risk = 1.07, P = 0.4859) with mean ARR of 0.33 for the 20 mg and 0.35 for the 40 mg group, and 0.27 for those who completed 1 year of treatment (ARR in the last year prior to study was 1.498). Among both groups, 77% were relapse free, and both demonstrated a reduction in GdE lesions and new T2 lesions.Citation46
Summarizing recent developments with use of GA
Data now support the use of GA in a continuum of therapy from initiation of first-line therapy upon diagnosis with CISCitation17 or RRMS through its long-term use in patients with RRMS. Results from head-to-head trials with IFN therapies demonstrate similar potency and speed of onset for the first 1 to 2 years after initiating therapy.Citation14–Citation16 In addition, ARR reductions with use of GA in numerous recent trialsCitation14–Citation16 are much greater than those seen in its pivotal trial.Citation12 The 70% to 83% reduction in relapse rates (from that prior to initiating therapy) observed during these recent investigations Citation14–Citation16 is probably much more indicative of the results that should be expected when initiating treatment with an early RRMS patient today. These reductions in ARRs are similar to those found with use of natalizumab in the AFFIRM (Natalizumab Safety and Efficacy in Relapsing Remitting Multiple Sclerosis) trial.Citation47 Furthermore, the use of GA in cases of suboptimal response to IFNs appears in most cases to improve effectiveness.Citation27–Citation29
With regard to risks, long-term data (up to 15 years),Citation13 from the head-to-head trials,Citation14–Citation16 and data from the PreCISe trialCitation17 indicate that GA is generally safe. Opportunistic infections, malignancies, and the development of autoimmune diseases are not risks associated with GA, although tolerability problems, especially related to injections and injection-site reactions (including lipoatrophy), continue to be an issue.Citation13 These new findings – particularly those that reflect on its long-term safety, its use earlier in the disease, and a better understanding of its efficacy relative to high-dose IFNs, and its effectiveness in patients after stopping IFNs – are reshaping the risk-vs-benefit analysis for GA.
In an ever-evolving therapeutic landscape that will probably include several additions in the near future, GA will remain a viable option for patients and clinicians given these new data indicating its continued safety, even after years of use, along with efficacy comparable to other currently available therapies. Still, the future role of GA in MS therapy is dependent not only on the analysis of its own minor risks and considerable benefits, but also on the analysis of the risks and benefits associated with the emerging therapies for MS. Use will depend on the continuing risk-vs-benefit analysis of other drugs and, in the future, this will consist of multiple drugs currently under consideration, including those adapted based on use in other diseases; novel biologic agents; and – most eagerly anticipated by patients – the first oral therapies to be developed for the treatment of MS.
Risks and benefits of emerging therapies
Numerous new agents are in the final stages of development, particularly for the management of RRMS, and there is strong demand for oral therapies. With many of these agents, balancing the risks and benefits will be difficult. Robust preliminary efficacy data from initial clinical trials will have to be viewed against a backdrop of serious risks observed during the clinical trials; the protracted effects of some of these agents; and the limited data available, particularly long-term data. Several of the novel therapies are being developed by adapting oncology products, such as alemtuzumab (Campath®; Berlex Laboratories, Richmond, CA, USA), cladribine (Leustatin®; Ortho Biotech Products, Raritan, NJ, USA), and rituximab (Rituxan®; Genentech, Inc, South San Francisco, CA, USA), or those originally targeted for use in patients undergoing transplantation (fingolimod). However, the risk-vs-benefit considerations are very different in these therapeutic areas compared with those for MS. Much more risk may be acceptable when treating cancers and suppressing transplanted organ rejection. In these cases, the immediate morbidity and mortality are generally greater and the evaluations are made over the short term. In contrast, MS is a chronic, severely disabling, but not generally fatal disease, lasting decades. Also, MS often follows an unpredictable and clinically variable course. In this scenario, patients may be eager to prevent or minimize physical and cognitive disability; however, they may risk undergoing treatment with an agent for which a fairly complete understanding of its risks and benefits may not be available for several years, or even a decade or longer.
Several of the novel therapies alter host immune responses in a variety of ways and have strong immunosuppressive properties. Immunosuppressive therapies have been used much more extensively for treatment of conditions, such as in organ transplantation and for many neoplasms. Experience derived from use in transplantation reveals that the major long-term toxicities limiting the use of immunosuppressive regimens are opportunistic infections and malignancies. Citation48 With the opportunistic infections observed with use of natalizumab, Citation3 along with opportunistic infections and malignancies observed in several of the recently completed trials with use of some of the emerging agents,Citation49,Citation50 these same concerns are now becoming part of the challenges associated with managing MS.Citation48,Citation51 Patients undergoing transplantation and the physicians providing their care face a continually moving target as they provide antimicrobial prophylaxis for known threats, such as Pneumocystis carinii and cytomegalovirus (CMV) wherein a new organism emerges, causing a new host of challenges. Citation48,Citation51 The same or similar issues may be a concern with the use of these immunosuppressive agents in MS.
The risks and benefits of 6 agents that are currently in or have just completed phase III trials will be discussed, including cladribine, alemtuzumab, rituximab, fingolimod, laquinimod, and dimethyl fumarate. The risks and benefits of the B-cell-targeted monoclonal antibodies, rituximab, and its humanized form, ocrelizumab, will be discussed due to the off-label use of rituximab.
Oncology agents in development for the treatment of MS
Cladribine
Cladribine was developed as a selective lymphocytotoxic agent for the treatment of lymphoid malignancies, including hairy-cell leukemia,Citation52 and is being investigated for treatment of chronic lymphocytic leukemia (CLL)Citation53,Citation54 and acute myelogenous leukemia.Citation55 It is a purine nucleoside analog that is converted intracellularly to its active nucleotides in cells with a high level of deoxycytidine kinase (phosphorylating enzyme) and low 5′-nucleotidase (dephosphorylating enzyme) activity.Citation52 The active nucleotides interrupt cellular purine metabolism, damage DNA (cause double-strand DNA breaks), and ultimately lead to cell death.Citation56 Lymphocytes have a high ratio of deoxycytidine kinase to 5′-nucleotidase, and resting and proliferating lymphocytes are selectively killed by cladribine.Citation57 Some T-cell classes are depleted for more than 1 year by a single weekly cycle of cladribine treatment, and may affect several other immune cell types for 4 to 12 months, an indication of the long duration of its actions.Citation58 Myelosuppression is the principal cause of toxicity, with life-threatening infections as an immunosuppressive complication of cladribine therapy for cancer.Citation59
Initial attempts at developing cladribine for the treatment of MS utilized parenteral formulations and involved 6 different trials (reviewed by Leist and Vermersch).Citation60 Rice et alCitation61 conducted a randomized, placebo-controlled trial of 159 patients with progressive MS and with 6.0 as their median baseline EDSS score. Participants were randomized to receive placebo or cladribine (a dosing cycle is 0.07 mg/kg/day SC for 5 consecutive days every 4 weeks), for either 2 or 6 cycles followed by placebo for a cumulative dose of 0.7 mg/kg or 2.1 mg/kg respectively.Citation61 The study included a 1-year, double-blind phase and a 6-year extension phase. During the 1-year phase, cladribine had no significant effects on progression of disability in patients with progressive MS.Citation61 However, efficacy was demonstrated based on MRI parameters, as well as reduced relapse rates and disability progression among 229 patients with RRMS and progressive forms of MS who were treated in 3 trials conducted at the Scripps Research Institute.Citation60 These patients received cumulative parenteral doses of cladribine, ranging from 0.7 mg/kg to 2.8 mg/kg administered over 4 to 6 months, using a monthly regimen of 5- to 7-day courses.Citation60
Cladribine is now being developed as an oral formulation based on its oral bioavailability of 37% to 51%.Citation62 Because the side effects of cladribine are generally dose dependent, development of oral cladribine is being pursued at doses that should give equivalent exposure to the lower doses used in the earlier parenteral studies (cumulative parenteral doses in the range of 0.7 mg/kg to 1.4 mg/kg).Citation60,Citation63,Citation64 A phase III clinical trial of an oral formulation of cladribine, the CLARITY (Cladribine Tablets in Treating MS Orally) trial, was recently completed.Citation65,Citation66 Based on this phase III trial, experience in the treatment of cancers, and the earlier parenteral studies, Merck Serono has submitted a New Drug Application for cladribine to the FDA for approval as a new therapy for treatment of RRMS.Citation67 It may be one of the first of the emerging oral agents to reach the market. The CLARITY trial, a phase III, 96-week, randomized, double-blind, placebo-controlled investigation (N = 1326) with oral cladribine (1 of 2 cumulative doses [3.5 mg/kg or 5.25 mg/kg] given in 2 or 4 short courses over the first 48 weeks with an additional 2 short courses starting at Week 48 and Week 52) reported reductions in ARRs by 58% (ARR 0.14; P < 0.001) and 55% (ARR 0.15; P < 0.001) in the cladribine low-dose and high-dose groups, respectively, compared with placebo (ARR 0.33). Secondary endpoints showed improvement in EDSS score and fewer T1 GdE lesions compared with placebo.Citation65,Citation66 With regard to safety issues, in the parenteral dosing MS trials involving cladribine, serious AEs (SAEs) similar to placebo were observed. However, there was a dose-related increase in infections, including 8 cases of herpes zoster, 1 case of Salmonella and 1 case of fatal, fulminating, newly acquired hepatitis B that was thought not to be connected to the administration of cladribine.Citation58,Citation68 AEs in the CLARITY trial were similar to those reported in the parenteral dosing studies. However, in addition, there were 4 malignancies reported in the cladribine treatment groups (none in the placebo group) during the 96-week trial (1 case each of cervical, ovarian, and pancreatic cancer, as well as a case of malignant melanoma).Citation49 A fifth case of cancer was reported in a woman who developed a choriocarcinoma when she became pregnant 6 months after study completion.Citation49,Citation69 Dermatomal herpes zoster cases also occurred in 1.9% of patients. Although cladribine offers a distinct advantage as a convenient oral therapy, requiring as little as 2 cycles of 5 consecutive days each at the beginning of the first and second month of treatment for the first full year of therapy, the malignancies and infections appearing in the CLARITY trial coupled with the long-lasting effects of cladribine on the lymphocyte populations leave the assessment of risks involved with cladribine therapy unclear. The need for long-term monitoring is also an unresolved issue.
Alemtuzumab
Alemtuzumab was originally developed, and is approved, for the treatment of B-cell CLL. This agent is a humanized monoclonal antibody that targets the CD52 antigen, a cell surface glycoprotein that is present on >95% of T lymphocytes, B lymphocytes, monocytes, and eosinophils, as well as tissues of the male reproductive system.Citation70,Citation71 Binding of alemtuzumab to CD52 results in the rapid depletion of targeted cells by complement-dependent cytotoxic and antibody-dependent cellular cytotoxic mechanisms. Citation71 Prolonged lymphocyte depletion results from a single dose that lasts for years. For example, CD4+ T cells require a median period of 61 months to recover; CD8+ lymphocytes recover in approximately 30 months (median recovery period); and monocytes and B lymphocytes return to baseline levels in 3 months.Citation70 Depletion of these cell types should suppress the immune responses involved in the pathophysiology of MS, especially the early inflammatory events. Early studies of alemtuzumab in the treatment of secondary progressive MS (SPMS) suggested efficacy in the treatment of relapses, however, not in the prevention of disability progression.Citation72
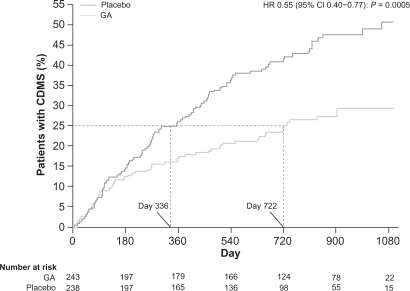
The CAMMS223 study was a phase II, randomized trial with 334 treatment-naive patients with RRMS who were selected to receive either IFNβ-1a SC (44 μg 3 times per week) or annual intravenous (IV) cycles of alemtuzumab (12 mg or 24 mg per day for 5 consecutive days per year) for 36 months.Citation73 The trial was designed to test the hypothesis that immunotherapy could influence long-term disability only if administered early in the disease course. Therefore, the eligibility criteria for this study limited the study to patients with an onset of symptoms of no more than 36 months and an EDSS score of 3 or less.Citation73 Alemtuzumab significantly reduced the rate of sustained accumulation of disability (9.0% for alemtuzumab vs 26.2% for IFNβ-1a SC; P < 0.001) and reduced the ARR (0.10 for alemtuzumab vs 0.36 for IFNβ-1a SC; P < 0.001) compared with IFNβ-1a SC (). Alemtuzumab also reduced MRI lesion burden; brain volume increased in the alemtuzumab group but decreased in the IFNβ-1a SC group.Citation73 Four-year pooled data recently released demonstrated that ARRs were 0.10 (95% CI: 0.08, 0.12) for alemtuzumab and 0.33 (95% CI: 0.28, 0.41) for IFNβ-1a SC, representing a 72% risk reduction for relapse (P < 0.0001) for alemtuzumab.Citation74 The results apparently demonstrate the superiority of alemtuzumab with regard to efficacy compared with IFNβ-1a SC.Citation73
Figure 5 Primary endpoints of the CAMMS223 trial – sustained disability and relapse.
A) Kaplan–Meier curves for patients who reached the criteria for sustained accumulation of disability. B) Cumulative number of relapses. Reprinted with permission from CAMMS223 Trial Investigators, Coles AJ, Compston DA, et al. Alemtuzumab vs interferon beta-1a in early multiple sclerosis. N Engl J Med. 2008;359(17):1786–1801.Citation73 Copyright ® 2009 Massachusetts Medical Society. All rights reserved.
There were SAEs during this trial, requiring the suspension of treatment with alemtuzumab after 3 patients developed immune thrombocytopenic purpura (ITP), one of whom died. The total number of patients with ITP included 1 patient receiving IFNβ-1a, and 6 patients receiving alemtuzumab. Citation73 Autoimmune disorders, such as ITP, have occurred at a high rate with the treatment of MS with alemtuzumab.Citation70,Citation73 These also include thyroid disorders (23% in the treatment group vs 3% in the placebo group in the CAMMS223 phase II trial) that occurred up to 30 months after the last dose of alemtuzumab was administered. Citation73 Among the affected patients, 96% developed thyroid autoantibodies. Of the 32 patients who developed hyperthyroidism, 3 experienced serious hyperthyroid events (eg, Graves’ ophthalmopathy) and 25 had sustained hyperthyroidism. Primary hypothyroidism developed in 10 patients, with 18 patients requiring long-term thyroid replacement therapy.Citation73 In an earlier trial, Grave’s disease developed in 27% of patients, and 1 of 58 patients developed autoimmune hypothyroidism. Citation70 Additionally, there was 1 case of renal failure due to Goodpasture’s syndrome that emerged 10 months after therapy.Citation70 Aside from autoimmune disorders, Coles et alCitation70 reported the occurrence of opportunistic infections, including CMV and herpes. There were 3 cases of cancer reported during the CAMMS223 trial (non-Epstein-Barr virus-associated Burkitt’s lymphoma, breast cancer, and cervical cancer in situ) among patients receiving alemtuzumab; 1 case of colon cancer was reported among patients receiving IFNβ-1a SC.Citation73 The occurrence of acute cytokine release syndrome necessitates the use of high-dose steroids to ameliorate side effects during delivery of alemtuzumab doses. Serious infusion reactions developed in 1.4% of patients during the CAMMS223 trial.Citation73
Based on current reports, risk-vs-benefit evaluations are difficult with alemtuzumab. The efficacy demonstrated in the CAMMS223 trial (superiority vs IFNβ-1a SC) suggests this may be one of the most potent agents available for use in treating MS (at least when dosed very early in the disease course). Although there may be very good efficacy when dosed early, there are major safety concerns, particularly those in association with autoimmunity, but also including opportunistic infections and malignancies. Many questions remain about the use of alemtuzumab, including how to use it over the long term. To date, alemtuzumab has been administered for 2 years (2 cycles). Experience from prior trials reveals that some safety issues have arisen years after the last dose was taken, yet no long-term data exist for this agent and the duration of effects of even a single 1-week cycle of alemtuzumab on the immune system is very long (>5 years). Issues and questions that still need to be addressed include the need for additional doses; the number of doses that may be safely administered; the effects of prolonged depletion of CD4+ lymphocytes; and how to proceed in cases of suboptimal response. There are very significant risks associated with alemtuzumab and a great number of unknowns. Yet, to achieve maximum efficacy with this agent, it may be best to use it as early as possible in the disease course. Long-term, frequent monitoring probably will be necessary to ultimately define risk vs benefit.
Rituximab/ocrelizumab
Unlike several of the emerging agents that target T cells, rituximab and its humanized form, ocrelizumab, are monoclonal antibodies that target B lymphocytes. Rituximab is approved in the United States for the treatment of non-Hodgkin’s lymphoma and as a therapy for rheumatoid arthritis (RA) in combination with methotrexate in patients who have an inadequate response to one or more tumor necrosis factor antagonist therapies.Citation75 Rituximab binds to the CD20 surface antigen on B lymphocytes, and directs the immune system to attack and kill the marked circulating CD20+ B lymphocytes. This depletion of B cells lasts more than 6 months.Citation71,Citation76 Affected cells include immature B cells, naive B cells, activated B cells, and memory B cells; however, stem cells, pro-B cells, and plasma cells are spared.Citation77
Hauser et alCitation78 conducted a double-blind, placebo-controlled, phase II study of rituximab 1000 mg administered intravenously in 2 infusions 15 days apart. The primary endpoint for this trial was total number of GdE lesions at several time points over the 48-week study. Investigators found that compared with placebo, there was a decrease in the number of GdE lesions of 91% (P < 0.001) and a decrease in new GdE lesions of 96% (P < 0.001), suggesting that this agent causes a strong suppression of inflammation. Although not powered to demonstrate a difference in ARR, compared with placebo, the ARR was reduced by 56% (P = 0.04) at 24 weeks and by 49% (P = 0.08) at 48 weeks.Citation78
In this study, 50 of the 54 patients in the rituximab group (92.6%) reported “mild to moderate” infusion-associated AEs. More patients in the rituximab group (78.3%) than in the placebo group (40.0%) had infusion-associated AEs within 24 hours after the first infusion, and a total of 5.7% of patients in the placebo group and 4.3% of patients in the rituximab group withdrew from the study due to AEs.Citation78 Experience with rituximab in non-MS patients reveals that most acute AEs (fever, chills, rigors, and flu-like symptoms) are related to infusions.Citation77 Additional less common AEs include nausea; pruritus; angioedema; asthenia; hypotension; acute respiratory distress syndrome; myocardial infarction; cardiogenic and anaphylactic shock; and mucocutaneous reactions.Citation71,Citation77 Tumor lysis syndrome, a rare and serious complication of rituximab infusion, has occurred in patients with cancer treated with high levels of rituximab.Citation71 Rituximab contains a boxed warning for fatal infusion reactions, tumor lysis syndrome, severe mucocutaneous reactions, and the occurrence of PML.Citation75 Carson et alCitation79 reports that there are 57 reported cases of PML after rituximab therapy in human immunodeficiency virus-negative patients. In general, these cases of PML are associated with the use of rituximab in patients receiving or having received other immunosuppressive therapies.Citation79,Citation77 Ocrelizumab, the humanized form of rituximab, is currently in development (phase II trials are underway), and the risks and benefits of this novel therapy remain an open question pending phase III trial results.
Other emerging agents
Fingolimod
Fingolimod (FTY720) is an oral, once-daily agent that was first pursued for use in transplantation patients; however, it was not found to offer any advantage over the standard of care, while having some increased risks, including macular edema.Citation80 It is a prodrug that is phosphorylated in vivo to FTY720-phosphate (FTY720-P), which acts as an agonist of 4 out of 5 members of the sphingosine-1-phosphate (S1P) receptor family.Citation81 Agonism of S1P receptor 1 on lymphocytes by FTY720-P leads to sequestration of lymphocytes in secondary lymphoid tissues, such as lymph nodes and Peyer’s patches,Citation81 lowering circulating lymphocytes by up to 85%.Citation82 Fingolimod, therefore, suppresses the immune response as T cells must migrate from the lymph nodes to the site of the pathogen or other foreign antigen to execute an immune response. Agonism of S1P receptors in the brain has been postulated to provide a mechanism by which fingolimod may be neuroprotective.Citation83 Agonism of S1P receptor 3 by FTY720-P is the likely cause of bradycardia commonly observed with use of fingolimod.Citation84
A phase II clinical trial of patients with RRMS revealed that fingolimod reduces GdE lesions, T2 lesions, and ARRs at daily doses of 1.25 mg (n = 83) and 5 mg (n = 77) compared with placebo (n = 81).Citation85,Citation86 ARRs were reduced by 55% (P = 0.009) and 53% (P = 0.01) compared with placebo with the 1.25-mg and 5-mg doses, respectively.Citation85 Patients (N = 189) were treated for 24 months in an extension trial. They continued to exhibit a low level of disease activity with a high proportion (>70%) of patients free of relapses (ARRs for Months 7 to 24: 0.12 to 0.26), and with low levels of inflammatory activity on MRI (79% to 91% demonstrated no GdE lesions).Citation86
Serious safety concerns, however, arose during the phase II clinical trial and extension. All patients receiving the 5-mg dose of fingolimod were switched to the 1.25-mg dose during the extension phase due to a less favorable safety profile at the higher dose.Citation87 Kappos et alCitation85 reported bradycardia (mostly with the initial dose), transient second-degree Wenckebach atrioventricular block, and decreased pulmonary function (forced expiratory volume in 1 second [FEVCitation1]).Citation85,Citation86 In addition, opportunistic infections and malignancies (7 cases of skin cancer) were reported.Citation87
TRANSFORMS (TRial Assessing injectable interferoN vs FTY720 Oral in RrMS), a 1-year, randomized, double-dummy, double-blind, parallel-group, phase III, active comparator investigation of fingolimod at 1.25 mg and 0.5 mg once daily with IFNβ-1a IM in patients with RRMS (n = 1292), was recently completed. The ARR was reduced by fingolimod to a range of 0.16 to 0.20 vs 0.33 by IFNβ-1a IM, correlating with a relative reduction of 38% to 52%.Citation88 However, during this trial, there were 2 deaths from opportunistic infections (1 death from herpes encephalitis and 1 death from disseminated varicella infection). In addition, there were 8 skin cancers (including 3 malignant melanomas) and 4 breast cancer cases in the fingolimod groups vs 2 skin cancers in the IFNβ-1a IM group. Macular edema was also reported in <1% of FTY20-treated patients.Citation50 During TRANSFORMS, the safety profile of the 0.5-mg dose appeared to be better than the 1.25-mg dose, including lower rates of infection and bradycardia, but not of cancer.Citation89 The occurrence of malignancies and opportunistic infections with fingolimod therapy is indicative of the potent immunosuppressive properties of this agent.
Two other phase III trials of fingolimod in MS include the placebo-controlled FREEDOMS (FTY720 Research Evaluating Effects of Daily Oral therapy in MS) and FREEDOMS II trial, which is underway. Results from the 2-year, phase III FREEDOMS study were recently reported: FTY720 reduced the relapse rate by 54% for the 0.5-mg dose and 60% for the 1.25-mg dose compared with placebo (P < 0.001, respectively).Citation90 In addition, FTY720 reduced the progression of disability over 2 years by 30% for patients on 0.5 mg and 32% for those on 1.25 mg compared with placebo (P = 0.02, respectively).Citation90 Both fingolimod doses were superior to placebo regarding MRI measures (P < 0.001). The proportions of patients in the 3 study groups who experienced AEs were similar at 93% to 94%.Citation90 The majority of AEs were mild to moderate in all 3 treatment groups, however, the authors concluded that AEs may be less frequent with the 0.5-mg dose than with the 1.25-mg dose.Citation90 AEs related to fingolimod and resulting in study discontinuation were most common in the 1.25-mg dose group, occurring in 14.2% of patients. AEs included bradycardia and atrioventricular heart block at the time of fingolimod initiation, as well as macular edema, elevated liver enzymes, and mild elevations in blood pressure.Citation90 The most common serious AEs were bradycardia, MS relapse, and basal cell carcinoma. A total of 7 patients taking fingolimod and 1 on placebo experienced bradycardia. Likewise, 7 patients in the fingolimod arms and 1 from the placebo arm experienced an MS relapse, whereas 5 patients in the treatment groups and 3 in the placebo arm developed basal cell carcinoma.Citation90
Any assessment of the risks versus the benefits for oral fingolimod must consider the dose-related risks identified during the phase II and phase III trials. To reduce the possibility of AEs, the high dose (5.0 mg) was eliminated during the extension trial,Citation86 and lower doses have been pursued (0.5-mg and 1.25-mg doses) for the phase III investigations. The phase III TRANSFORMS study demonstrated clear superiority in efficacy of fingolimod over IFNβ-1a IM;Citation88 however, efficacy also may be dose dependent: reductions in the numbers of GdE lesions and T2 lesions during the phase II trial were substantially less at the 1.25-mg dose than at the 5.0-mg dose,Citation85 and the induction of lymphopenia has been shown to be dose dependent as well.Citation82 Although patients and clinicians will be attracted to the convenience of an oral agent, this initial enthusiasm may be tempered by the need for extensive monitoring. For example, monitoring during the phase II fingolimod trial included serial pulmonary function testing, pulse rate monitoring for 6 hours after the first dose, and repeated ophthalmologic examinations.Citation86
Teriflunomide
Like fingolimod, teriflunomide is an oral agent with once-daily dosing. Teriflunomide is the active metabolite of leflunomide, a pyrimidine synthesis inhibitor used in the treatment of RA. It appears that the therapeutic effects of teriflunomide are primarily associated with its inhibition of dihydro-orate dehydrogenase, which is a key cellular enzyme involved in de novo pyrimidine synthesis. Teriflunomide has a cytostatic effect on cells that rely on a de novo pathway of pyrimidine synthesis rather than the salvage pathway, such as T and B lymphocytes, preventing their proliferation during an immune response. This appears to play a role in its therapeutic effect in MS.Citation91
A phase II study has been conducted among 179 patients with RRMS or SPMS to evaluate its safety and efficacy. The patients were randomized to receive teriflunomide 7 mg/day, 14 mg/day, or placebo for 36 weeks. Teriflunomide-treated patients had a significant reduction in T1- and T2-GdE-enhancing lesions with both doses of drug compared with placebo.Citation92 Although teriflunomide is generally well tolerated, several SAEs were reported during this phase II trial, including hepatic dysfunction, rhabdomyolysis, neutropenia/myelosuppression, and trigeminal neuralgia.Citation92
In a second study, 116 patients being treated with an IFN were randomized to receive a daily dose of either teriflunomide 7 mg (n = 37), teriflunomide 14 mg (n = 38), or placebo (n = 41), concurrently over 24 weeks. The objective of the study was to evaluate safety and efficacy of teriflunomide as an add-on therapy to IFN-based DMTs. This study revealed that compared with placebo, the number of T1-GdE lesions was significantly reduced (7 mg, 56%; 14 mg, 81%; all P < 0.001) and the percentage of patients who were free of T1-GdE lesions was higher in both groups treated with teriflunomide (placebo, 58%; 7 mg, 70%; 14 mg, 82%). In addition, fewer relapses were reported among patients receiving the higher dose of teriflunomide (14 mg) vs placebo. However, each study group experienced treatment-emergent AEs, causing 1 patient in each group to discontinue participation in the trial. The proportion of patients with abnormal liver function tests (increased alanine transaminase) was higher in those receiving 14 mg (28.9%) versus those receiving 7 mg (13.5%), or among placebo patients (12.2%). The percentage of patients with AEs potentially associated with immunosuppression (including low white blood cell counts, infections, and infestations) was also higher in the teriflunomide-treated groups (placebo, 32%; 7 mg, 49%; 14 mg, 47%).Citation93
Leflunomide, used for the treatment of RA, is rapidly and almost entirely converted to teriflunomide following oral ingestion.Citation91 After a higher loading dose, the standard daily dose of leflunomide is 20 mg/day,Citation94 while teriflunomide is being studied for use in MS at 7 and 14 mg/day.Citation91 The contraindications and warnings for leflunomide include a black box warning stating that it is contraindicated in pregnant women or women of childbearing potential who are not using reliable contraception, and the label includes other warnings regarding its immunosuppressive potential (bone marrow suppression), and rare but severe cases of liver injury.Citation94 Warnings for teriflunomide will not be determined until its approval.
Laquinimod
Laquinimod is a once-daily oral immunomodulatory agent being developed by targeted drug modification of another compound, roquinimex. Roquinimex had shown promising efficacy in MS clinical trials, but its development was discontinued in the late 1990s due to the occurrence of SAEs (myocardial infarction, pericarditis, and serositis).Citation95–Citation98 Laquinimod was derived from roquinimex by modifying the quinoline ring combined with chain elongation of the amidic methyl group, to create a new compound that is pharmacologically and chemically distinct from its origins.Citation98 Sixty compounds with structural relationships to roquinimex were studied, and laquinimod was found to be more potent than roquinimex and to have a better safety profile.Citation98,Citation99 During development of laquinimod from roquinimex, activity in animal models was enhanced approximately 20-fold, while toxicity was decreased.Citation98,Citation99 The MOA of laquinimod is believed to be immunomodulatory, not immunosuppressive,Citation98 and to be associated with a Th1/Th2 shift and inhibition of the migration of inflammatory cells into the CNS. Suppressing Th1 cytokines while enhancing Th2 cytokines appears to reduce subsequent demyelination of nerve tissue in animal models.Citation100
An initial double-blind, phase II study of laquinimod randomized 209 patients with RRMS to receive 0.1 mg or 0.3 mg of laquinimod or placebo as oral therapy for 24 weeks.Citation101 GdE MRI scans of the brain were performed at baseline, every 8 weeks, and 8 weeks post-treatment. The investigators found that laquinimod 0.3 mg reduced the mean cumulative number of active lesions by 44% compared with placebo. The reduction was slightly greater, at 52%, in the subgroup of patients with at least 1 active lesion at baseline.Citation101
A second double-blind, phase II study randomized 306 patients with RRMS and at least 1 enhancing lesion at screening to receive 0.3 mg or 0.6 mg of laquinimod or placebo for 36 weeks.Citation102 Monthly brain MRI scans, starting at Week 12, were performed during this trial. This was followed by a 36-week extension, during which the patients originally treated with laquinimod continued laquinimod treatment at the original dose, and patients receiving placebo were randomized to either the 0.3-mg or the 0.6-mg doses of laquinimod.Citation103 Compared with placebo, treatment with laquinimod 0.6 mg demonstrated a 40% reduction (P = 0.0048) in the mean cumulative number of GdE lesions on the last 4 scans of the first 36-week study and a 51% reduction (P = 0.0001) in the mean cumulative number of GdE lesions on the last 7 scans.Citation102
Although the trial was not powered to study relapse rates, there was a trend toward reduction with the 0.6-mg dose, with a 33% reduction (P = 0.09) in ARRs.Citation102 Following the 36-week, double-blind extension, an open-label extension was initiated. Results are now available for 155 of the 209 patients who entered the extension phase and are still undergoing treatment but have reached 24 months in the open-label phase. The mean ARR during the entire follow-up of 42 months for all patients was 0.46 compared with 0.53 in the period of 0 to 18 months. The ARR was 0.45 for the original 0.6-mg laquinimod group, 0.50 for the original 0.3-mg laquinimod group, and 0.42 for the original placebo group. During the 24-month extension, 10.5% of patients entering the extension phase met confirmed disability progression on EDSS compared with 14.8% of laquinimod and placebo groups during the first 18 months. On MRI parameters, 61% of patients were free of GdE lesions at Month 42. Laquinimod continued to be safe and well tolerated, with the only side effects reported to be nasopharyngitis, back pain, and headache.Citation104
Two phase III trials of laquinimod are currently underway, with recruitment complete: ALLEGRO (N<1000), which will compare laquinimod with placebo and is scheduled to be completed in November 2010; and BRAVO (N<1200), which includes a treatment arm with IFNβ-1a IM and is scheduled to be completed late in 2011. In evaluating the risks versus benefits of laquinimod, two principal questions remain to be resolved: 1) Will the clinical efficacy of laquinimod be robust? 2) Will laquinimod continue in its development without the appearance of the safety issues related to roquinimex (as it has thus far)?
Dimethyl fumarate (BG00012)
First introduced in the 1950’s, dimethyl fumarate has been in use as a treatment for psoriasis (an autoimmune skin disorder) in Europe for more than 30 years, although its formulation has evolved several times during that period. Dimethyl fumarate is administered 2 or 3 times daily. Its MOA is not clearly understood, although it is generally described as an immunomodulator. Citation105,Citation106 More specifically, it may reduce the secretion of proinflammatory cytokines, inhibit the expression of adhesion molecules involved in the transmigration of immune cells into the CNS, and cause a Th1 to Th2 shift.Citation106
A 12-month study of 10 psoriasis patients suggests that fumaric acid esters may have immunosuppressive activities as part of their MOA, as lymphocytes were decreased by about 60%.Citation107 In vitro studies by Treumer et alCitation108 could provide an explanation of the MOA for this effect, as dimethyl fumarate was reported to be a potent inducer of apoptosis in human activated T cells. The effect of dimethyl fumarate on T cells has not been directly studied in patients with MS; however, lymphopenia was reported as an AE by Schimrigk et alCitation109 in 3 of 10 patients with MS. Gold et alCitation110 reported that approximately 30% of patients with MS treated with dimethyl fumarate in the 24-week phase II trial had total white blood cell counts below the lower limit of the normal range.Citation110
Dimethyl fumarate was studied in a phase II clinical trial, in which 257 patients with RRMS were randomized to receive either dimethyl fumarate 120 mg once daily, 120 mg 3 times daily, 240 mg 3 times daily, or placebo for 24 weeks.Citation111 This was followed by a 24-week extension trial to assess safety. Patients taking placebo were switched during the extension phase of the trial to receive 240 mg of dimethyl fumarate 3 times daily. Compared with placebo, the highest dose (240 mg 3 times daily [720 mg total]) resulted in a significant reduction (69%; P < 0.0001) in the total number of new GdE lesions from Week 12 to 24. Lower doses, 120 mg once daily and 120 mg 3 times daily, did not cause a significant decrease in new GdE lesions, the primary endpoint of the study.Citation109,Citation111
Dimethyl fumarate is generally safe; however, AEs that relate to tolerability are common. These include gastrointestinal effects (upper abdominal pain, nausea, diarrhea) and flushing.Citation109,Citation111 In balancing the risks vs the benefits for this emerging therapy, efficacy was modest in the phase II trial and only significant with the highest dose. There are potentially significant tolerability and adherence issues with the highest dose (requiring administration 3 times a day). On the other hand, the long-term safety record established from its use in psoriasis suggests that the risk side of the equation may not be limiting.
Summary
The risk-vs-benefit analysis of GA is being reshaped by the recent findings on GA that reflect its long-term safety and efficacy in MS, its use earlier in the disease, and the realization of its greater efficacy than may have been perceived before the rigorous head-to-head trials. There is no evidence of NABs, which have reduced the effectiveness of all IFNβs. In an ever-evolving therapeutic landscape that will soon have several additional agents, the future role of GA in MS therapy will be dependent not only on the analysis of its own risks and benefits, but also on the analysis of the risks and benefits associated with the emerging therapies for MS. Like natalizumab, some of the emerging therapies are going to require the MS practitioner to consider new risks and to pay close attention to monitoring for potential AEs.
The risks and benefits of the emerging therapies vary widely. The therapies that are emerging from the fields of oncology and transplantation (cladribine, alemtuzumab, rituximab, and fingolimod) have been associated with, to various degrees, the risks associated with immunosuppressive therapies – serious opportunistic infections and/or malignancies. With alemtuzumab, the greatest risk appears to be the development of autoimmune syndromes. The effects of cladribine, alemtuzumab, and rituximab on the immune system are of a long duration that must be measured in years rather than in days or weeks. This makes the long-term risks associated with these treatments and how to use these treatments over the long term of particular concern. Long-term monitoring will probably be mandatory. The efficacy of alemtuzumab was clearly superior to that of IFNβ-1a SC in the CAMMS223 trial, and fingolimod was apparently superior to IFNβ-1a IM in the TRANSFORMS study. Rituximab, fingolimod, and cladribine all have impressive positive MRI data. The risks and benefits of these emerging therapies will be difficult to assess even at the end of their phase III trials due to the lack of long-term data and the long duration of biological activity of these agents.
With laquinimod, dimethyl fumarate, and teriflunomide, the life-threatening risks associated with the emerging therapies discussed above have not occurred. At this point in their development, they appear to be safer. However, questions remain about how robust the efficacy of these therapies will be. The phase II trials were not designed and/or powered to show efficacy on the important parameters of relapses and disability progression, and the risk-vs-benefit analyses will be incomplete until the end of phase III trials. Although their MOAs are not well understood, laquinimod and dimethyl fumarate appear to be largely immunomodulatory, whereas teriflunomide, by preventing lymphocyte proliferation, is largely immunosuppressive. If these therapies do prove to be relatively safe and effective, because they are oral therapies, they may significantly impact the use of GA.
Looking toward the future, there will be many interesting avenues to pursue in the treatment of MS as clinicians and their patients eagerly anticipate the approval of novel therapies that will be effective, convenient, and safe. Clearly, balancing the risks and benefits of selected therapies will become more challenging, and in the case of GA, more defined. Patients with MS, who are predominately young, active women, will look toward practitioners to guide them in making the best management decisions possible to ensure the best outcomes, with a minimum of physical and cognitive disability, and the most positive and improved quality of life possible.
Acknowledgements
The author acknowledges the assistance of Judith Greif, RN, MS, APNC, and James D. Bergstrom, PhD, of Advanced Clinical Concepts of Bridgewater, NJ, and Pippa Loupe, PhD, of Teva Neuroscience, Kansas City, MO, with the preparation of this manuscript.
Disclosures
Kenneth P Johnson, MD, is a consultant for Biogen Idec Inc. and Teva Neuroscience, and is a member of a Speakers’ Bureau for EMD Serono, Inc. He has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- JohnsonKPDueDLBenefits of glatiramer acetate in the treatment of relapsing-remitting multiple sclerosisExpert Rev Pharmacoecon Outcomes Res20099320521419527092
- RudickRAPolmanCHCurrent approaches to the identification and management of breakthrough disease in patients with multiple sclerosisLancet Neurol20098654555919446274
- BergerJRHouffSOpportunistic infections and other risks with newer multiple sclerosis therapiesAnn Neurol200965436737719399841
- SchweikertAKremerMRingelFPrimary central nervous system lymphoma in a patient treated with natalizumabAnn Neurol200966340340619798640
- SørensenPSDeisenhammerFDudaPGuidelines on use of anti-IFN-β antibody measurements in multiple sclerosis: report of an EFNS Task Force on IFN-β antibodies in multiple sclerosisEur J Neurol2005121181782716241970
- HesseDSellebjergFSorensenPSAbsence of MxA induction by interferon beta in patients with MS reflects complete loss of bioactivityNeurology200973537237719652141
- De StefanoNFilippiMConfavreuxCThe results of two multicenter, open-label studies assessing efficacy, tolerability and safety of protiramer, a high molecular weight synthetic copolymeric mixture, in patients with relapsing-remitting multiple sclerosisMult Scler200915223824318987103
- ArnonRThe development of Cop 1 (Copaxone®), an innovative drug for the treatment of multiple sclerosis: Personal reflectionsImmunol Lett1996501–21158793553
- SchrempfWZiemssenTGlatiramer acetate: Mechanisms of action in multiple sclerosisAutoimmun Rev20076746947517643935
- BlanchetteFNeuhausOGlatiramer acetate: evidence for a dual mechanism of actionJ Neurol20082551 Suppl263618317674
- AharoniRKayhanBEilamRSelaMArnonRGlatiramer acetate-specific T cells in the brain express T helper 2/3 cytokines and brain-derived neurotrophic factor in situProc Natl Acad Sci U S A20031002 Suppl141571416214614135
- JohnsonKPBrooksBRCohenJACopolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind, placebo-controlled trialNeurology1995457126812767617181
- FordCCJohnsonKPLisakRPA prospective open-label study of glatiramer acetate: over a decade of continuous use in multiple sclerosis patientsMult Scler200612330932016764344
- MikolDDBarkhofFChangPComparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trialLancet Neurol200871090391418789766
- O’ConnorPFilippiMArnasonB250 ug or 500 ug interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: a prospective, randomised, multicentre studyLancet Neurol200981088989719729344
- CadavidDWolanskyLJSkurnickJEfficacy of treatment of MS with IFNB-1b or glatiramer acetate by monthly brain MRI in the BECOME studyNeurol200919761983
- ComiGMartinelliVRodegherMEffect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trialLancet20093741503151119815268
- RoccaMAComiGCarraATreatment with glatiramer acetate reduces T1 and T2-weighted magnetic resonance imaging activity in patients with clinically isolated syndrome suggestive of multiple sclerosisMult Scler200814S29S293 [P501].
- ArnoldDLNarayananSAntelSTreatment with glatiramer acetate protects axons in patients with clinically isolated syndromes: evidence from the PreCISe trialMult Scler2008141S5S27
- FordCJohnsonKKachuckNContinuous long-term immunomodulatory therapy in relapsing multiple sclerosis: results from the 15-year analysis of the US prospective open-label study of glatiramer acetateMult Scler200814S29S293 [P44].
- GauthierSAGlanzBIMandelMIncidence and factors associated with treatment failure in the CLIMB multiple sclerosis cohort studyJ Neurol Sci20092841–211611919428028
- CohenBAKhanOJefferyDRIdentifying and treating patients with suboptimal responsesNeurology20046312 Suppl 6S33S4015623669
- FarrellRAGiovannoniGMeasuring and management of anti-interferon beta antibodies in subjects with multiple sclerosisMult Scler200713556757717548434
- CostelloKKennedyPScanzilloJRecognizing nonadherence in patients with multiple sclerosis and maintaining treatment adherence in the long termMedscape J Med200810922519008986
- HaasJFirzlaffMTwenty-four-month comparison of immunomodulatory treatments – A retrospective open label study in 308 RRMS patients treated with beta interferons or glatiramer acetate (Copaxone®)Eur J Neurol200512642543115885045
- Copaxone [package insert]Teva NeuroscienceKansas City, MO U S A 64131, 2009
- CaonCDinMChingWTselisALisakRKhanOClinical course after change of immunomodulating therapy in relapsing-remitting multiple sclerosisEur J Neurol200613547147416722971
- CarráAOnahaPLueticGTherapeutic outcome 3 years after switching of immunomodulatory therapies in patients with relapsingremitting multiple sclerosis in ArgentinaEur J Neurol200815438639318353125
- ZwibelHLGlatiramer acetate in treatment-naïve and prior interferon-β-1b-treated multiple sclerosis patientsActa Neurol Scand2006113637838616674604
- ZiemssenTMultiple sclerosis beyond EDSS: depression and fatigueJ Neurol Sci20092771 SupplS37S4119200865
- DennettSLCastelli-HaleyJOleen-BurkeyMThe Impact of Multiple Sclerosis on Patient Employment: a Review of the Medical LiteratureJ Health Productivity2007101218
- FiskJDPontefractARitvoPGArchibaldCJMurrayTJThe impact of fatigue on patients with multiple sclerosisCan J Neurol Sci19942119148180914
- ZiemssenTHoffmanJApfelRKernSEffects of glatiramer acetate on fatigue and days of absence from work in first-time treated relapsing-remitting multiple sclerosisHealth Qual Life Outcomes200866718775064
- FrealJEKraftGHCoryellJKSymptomatic fatigue in multiple sclerosisArch Phys Med Rehabil19846531351386703889
- SmithMMArnettPAFactors related to employment status changes in individuals with multiple sclerosisMult Scler200511560260916193900
- HeesenCNawrathLReichCFatigue in multiple sclerosis: an example of cytokine mediated sickness behaviour?J Neurol Neurosurg Psychiatry2006771343916361589
- ZiemssenTKümpfelTKlinkertWEFNeuhausOHohlfeldRGlatiramer acetate-specific T-helper 1- and 2-type cell lines produce BDNF: Implications for multiple sclerosis therapyBrain2002125112381239112390966
- PattenSBBeckCAWilliamsJVABarbuiCMetzLMMajor depression in multiple sclerosis: A population-based perspectiveNeurology200361111524152714663036
- SadovnickADRemickRAAllenJDepression and multiple sclerosisNeurology19964636286328618657
- FeinsteinAAn examination of suicidal intent in patients with multiple sclerosisNeurology200259567467812221156
- TsaiSGlatiramer acetate could be a potential antidepressant through its neuroprotective and anti-inflammatory effectsMed Hypotheses200769114514817196755
- ZiemssenTSchrempfWGlatiramer acetate: mechanisms of action in multiple sclerosisInt Rev Neurobiol20077953757017531858
- ZiemssenTReichmannHSchneiderHPresence of glatiramer acetate-specific TH2 cells in the cerebrospinal fluid of patients with multiple sclerosis 12 months after the start of therapy with glatiramer acetateJ Neurodegen Regen200811922
- VarkonyHWeinsteinVKlingerEThe glatiramoid class of immunomodulator drugsExpert Opin Pharmacother200910465766819245345
- CohenJARovarisMGoodmanADLadkaniDWynnDFilippiMRandomized, double-blind, dose-comparison study of glatiramer acetate in relapsing-remitting MSNeurology2007681293994417372130
- ComiGCohenJAFilippiMResults from a phase III, one-year, randomized, double-blind, parallel-group, dose-comparison study with glatiramer acetate in relapsing-remitting multiple sclerosisSession Presentation: World Congress on Treatment and Research in Multiple SclerosisMontreal, Canada [P79]. 2008 September 17–20
- PolmanCHO’ConnorPWHavrdovaEA randomized, placebocontrolled trial of natalizumab for relapsing multiple sclerosisN Engl J Med2006354989991016510744
- NgCYMadsenJCRosengardBRAllanJSImmunosuppression for lung transplantationFront Biosci2009141627164119273152
- CookSVermerschPComiGSafety and tolerability of cladribine tablets in relapsing-remitting multiple sclerosis during the 96-week, phase III, double-blind, placebo-controlled CLARITY studySession Presentation: 25th Congress of the European Committee for Treatment and Research in Multiple SclerosisDusseldorf, Germany [88]. 2009 September 9–12
- KapposLCohenJPelletierJon behalf of the TRANSFORMS (TRial Assessing injectable interferoN vs FTY720 Oral in RRMS) Study GroupSafety findings from a 12-month phase III study (TRANSFORMS) comparing oral fingolimod (FTY720) and intramuscular interferon beta-1a for relapsing-remitting multiple sclerosisPoster Presentation: 25th Congress of the European Committee for Treatment and Research in Multiple SclerosisDusseldorf, Germany [P807]. 2009 September 9–12
- FishmanJAInfection in solid-organ transplant recipientsN Engl J Med2007357252601261418094380
- KawasakiHCarreraCJPiroLDSavenAKippsTJCarsonDARelationship of deoxycytidine kinase and cytoplasmic 5’-nucleotidase to the chemotherapeutic efficacy of 2-chlorodeoxyadenosineBlood19938135976018094016
- RobakTBlonskiJZGora-TyborJCladribine alone and in combination with cyclophosphamide or cyclophosphamide plus mitoxantrone in the treatment of progressive chronic lymphocytic leukemia: report of a prospective, multicenter, randomized trial of the Polish Adult Leukemia Group (PALG CLL2)Blood2006108247347916551966
- RobakTBłońskiJZKasznickiMCladribine with or without prednisone in the treatment of previously treated and untreated B-cell chronic lymphocytic leukaemia – Updated results of the multicentre study of 378 patientsBr J Haematol2000108235736810691866
- MartinMGWelchJSAugustinKHladnikLDiPersioJFAbboudCNCladribine in the treatment of acute myeloid leukemia: a single-institution experienceClin Lymphoma Myeloma20099429830119717379
- HirotaYYoshiokaATanakaSImbalance of deoxyribonucleoside triphosphates, DNA double-strand breaks, and cell death caused by 2-chlorodeoxyadenosine in mouse FM3A cellsCancer Res19894949159192563234
- CarsonDAWassonDBTaetleRYuASpecific toxicity of 2-chlorodeoxyadenosine toward resting and proliferating human lymphocytesBlood19836247377436136305
- BeutlerESipeJCRomineJSKoziolJAMcmillanRZyroffJThe treatment of chronic progressive multiple sclerosis with cladribineProc Natl Acad Sci U S A1996934171617208643695
- ChesonBDInfectious and immunosuppressive complications of purine analog therapyJ Clin Oncol1995139243124487666104
- LeistTPVermerschPThe potential role for cladribine in the treatment of multiple sclerosis: clinical experience and development of an oral tablet formulationCurr Med Res Opin200723112667267617880754
- RiceGPAFilippiMComiGCladribine and progressive MS: clinical and MRI outcomes of a multicenter controlled trialNeurology20005451145115510720289
- SipeJCCladribine for multiple sclerosis: review and current statusExp Rev Neurother200556721727
- BrousilJARobertsRJSchleinALCladribine: an investigational immunomodulatory agent for multiple sclerosisAnn Pharmacother200640101814182116985095
- CostelloKSipeJCCladribine tablets’ potential in multiple sclerosis treatmentJ Neurosci Nurs200840527528018856248
- GiovannoniGComiGCookSDResults from the CLARITY study: a phase III randomized, double-blind study to evaluate the safety and efficacy of oral cladribine in relapsing-remitting multiple sclerosis (RRMS)Session Presentation: 61st Annual Meeting of the American Academy of NeurologySeattle, WA [Abstract LBS.001]. 2009 April 25–May 2
- GiovannoniGComiGCookSfor the CLARITY Study GroupA placebo-controlled trial of oral cladribine for relapsing multiple sclerosisN Engl J Med Published at NEJM.org 1202010 (DOI: 10.1056/NEJMoa0909494).
- Merck Serono submits application for cladribine tablets as a potential oral short-course multiple sclerosis therapy in the United States [press release]. Merck Serono; 2009930
- RomineJSSipeJCKoziolJAZyroffJBeutlerEA double-blind, placebo-controlled, randomized trial of cladribine in relapsing-remitting multiple sclerosisProc Assoc Am Physicians1999111135449893155
- The Multiple Sclerosis Resource CentreEU asked to approve oral multiple sclerosis treatment, cladribine200993 http://msrc.co.uk/index.cfm?fuseaction=show&pageid=1629 Accessed October 1, 2009.
- ColesAJCoxALe PageEThe window of therapeutic opportunity in multiple sclerosis: evidence from monoclonal antibody therapyJ Neurol200625319810816044212
- MuraroPABielekovaBEmerging therapies for multiple sclerosisNeurotherapeutics20074467669217920549
- MoreauTThorpeJMillerDPreliminary evidence from magnetic resonance imaging for reduction in disease activity after lymphocyte depletion in multiple sclerosisLancet199434489182983017914262
- CAMMS223 Trial InvestigatorsColesAJCompstonDAAlemtuzumab vs interferon beta-1a in early multiple sclerosisN Engl J Med2008359171786180118946064
- ColesAAlemtuzumab treatment benefit is durable: primary efficacy outcomes of CAMMS223 at 4 yearsPoster Presentation: 25th Congress of the European Committee for Treatment and Research in Multiple Sclerosis [P890]. Dusseldorf, Germany2009 September 9–12
- Rituxan [package insert]South San Francisco, CA USA 94080, 992009
- CrossAHStarkJLLauberJRamsbottomMJLyonsJRituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patientsJ Neuroimmunol20061801–2637016904756
- WaubantESpotlight on Anti-CD20Int MS J200815192518713565
- HauserSLWaubantEArnoldDLB-cell depletion with rituximab in relapsing-remitting multiple sclerosisN Engl J Med2008358767668818272891
- CarsonKREvensAMRicheyEAProgressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports projectBlood2009113204834484019264918
- BuddeKSchu̇tzMGlanderPFTY720 (fingolimod) in renal transplantationClin Transplant20062017 Suppl172417100697
- MandalaSHajduRBergstromJAlteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonistsScience2002296556634634911923495
- KahanBDKarlixJLFergusonRMPharmacodynamics, pharmacokinetics, and safety of multiple doses of FTY720 in stable renal transplant patients: a multicenter, randomized, placebo-controlled, phase I studyTransplantation20037671079108414557756
- ChoiJWHerrDLeeCWTeoSKennedyGChunJS1P1 receptor signalling on cells of astrocytic lineages in experimental autoimmune encephalomyelitis: a role in disease progression and the efficacy of fingolimod (FTY720)Session Presentation: World Congress on Treatment and Research in Multiple SclerosisMontreal, Canada [P29]. 2008 September 17–20
- ForrestMSunSHajduRImmune cell regulation and cardiovascular effects of sphingosine 1-phosphate receptor agonists in rodents are mediated via distinct receptor subtypesJ Pharmacol Exp Ther2004309275876814747617
- KapposLAntelJComiGOral fingolimod (FTY720) for relapsing multiple sclerosisN Engl J Med20063551124114016971719
- O’ConnorPMontalbanXAntelJOral fingolimod (FTY720) in multiple sclerosis: two-year results of a phase II extension studyNeurol2009727379
- KapposLRadueEWO’ConnorPfor the FREEDOMS Study GroupOral fingolimod (FTY720) in patients with relapsing multiple sclerosis: 3-year results from a phase II study extensionPoster Presentation: World Congress on Treatment and Research in Multiple SclerosisMontreal, Canada [P72]. 2008 September 17–20
- CohenJBarkhofFComiGfor the TRANSFORMS Study Group Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. Published at NEJM.org January 20, 2010 (10.1056/NEJMoa0907839).
- Novartis Pharmaceutical Corporation First phase III results for FTY720, a novel oral therapy for MS, show superior efficacy compared to interferon beta-1a [press release]. Novartis; 2008 December 12.
- KapposLRadueEWO’ConnorPfor the FREEDOMS Study GroupA placebo-controlled trial of oral fingolimod in relapsing multiple sclerosisN Engl J Med Published at NEJM.org January 20, 2010 (10.1056/NEJMoa0909494).
- TallantyreEEvangelouNConstantinescuCSSpotlight on teriflunomideInt MS J2008152626818782502
- O’ConnorPWLiDFreedmanMSA phase II study of the safety and efficacy of teriflunomide in multiple sclerosis with relapsesNeurology200666689490016567708
- FreedmanMWolinskyJSByrnesWJOral teriflunomide or placebo added to interferon beta for 6 months in patients with relapsing multiple sclerosis: safety and efficacy resultsPoster Presentation: 25th Congress of the European Committee of Treatment and Research in Multiple Sclerosis [P878]. Dusseldorf, Germany2009 September 9–12
- Arava [package insert]. sanofi-aventis US LLC, Bridgewater, NJ 08807 USA, 2009
- NoseworthyJHWolinskyJSLublinFDLinomide in relapsing and secondary progressive MS: part I: trial design and clinical resultsNeurology20005491726173310802775
- TanILLycklamaÀNijeholtGJPolmanCHAdèrHJBarkhofFLinomide in the treatment of multiple sclerosis: MRI results from prematurely terminated phase-III trialsMult Scler2000629910410773855
- WolinskyJSNarayanaPANoseworthyJHLinomide in relapsing and secondary progressive MS: Part II: MRI resultsNeurol20005417341741
- BrunmarkCRunströmAOhlssonLThe new orally active immunoregulator laquinimod (ABR-215062) effectively inhibits development and relapses of experimental autoimmune encephalomyelitisJ Neuroimmunol20021301–216317212225898
- JönssonSAnderssonGFexTSynthesis and biological evaluation of new 1,2-dihydro-4-hydroxy-2-oxo-3-quinolinecarboxamides for treatment of autoimmune disorders: structure-activity relationshipJ Med Chem20044782075208815056005
- ZouLPAbbasNVolkmannISuppression of experimental autoimmune neuritis by ABR-215062 is associated with altered Th1/Th2 balance and inhibited migration of inflammatory cells into the peripheral nerve tissueNeuropharmacology200242573173911985832
- PolmanCBarkhofFSandberg-WollheimMLindeANordleONedermanTTreatment with laquinimod reduces development of active MRI lesions in relapsing MSNeurology200564698799115781813
- ComiGPulizziARovarisMEffect of laquinimod on MRI-monitored disease activity in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebocontrolled phase IIb studyLancet200837196302085209218572078
- ComiGAbramskyOArbizuTOral laquinimod in patients with relapsing-remitting multiple sclerosis: 9-month double-blind active extension of the multi-center, randomized, double-blind, parallel-group placebo-controlled studyMult Scler200814S37
- ComiGAbramskyOArbizuTfor the LAQ/5062 Clinical Advisory Board and Study GroupLong-term open extension of oral laquinimod in patients with relapsing multiple sclerosis shows favourable safety and sustained low relapse rate and MRI activityPoster Presentation: 25th Congress of the European Committee for Treatment and Research in Multiple SclerosisDusseldorf, Germany [P443]. 2009 September 9–12
- RollAReichKBöerAUse of fumaric acid esters in psoriasisIndian J Dermatol Venereol Leprol200773213313717456929
- LinkerRALeeDHStangelMGoldRFumarates for the treatment of multiple sclerosis: potential mechanisms of action and clinical studiesExp Rev Neurother200881116831690
- HöxtermannSNüchelCAltmeyerPFumaric acid esters suppress peripheral CD4- and CD8-positive lymphocytes in psoriasisDermatology199819622232309568412
- TreumerFZhuKGläserRMrowietzUDimethylfumarate is a potent inducer of apoptosis in human T cellsJ Invest Dermatol200312161383138814675187
- SchimrigkSBruneNHellwigKOral fumaric acid esters for the treatment of active multiple sclerosis: an open-label, baselinecontrolled pilot studyEur J Neurol200613660461016796584
- GoldRKapposLMillerDHSafety profile of BG00012, an oral formulation of dimethyl fumarate for patients with relapsing MSPoster Presentation: World Congress on Treatment and Research in Multiple SclerosisMontreal, Canada [P50]. 2008 September 17–20
- KapposLGoldRMillerDHEfficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb studyLancet200837296481463147218970976