Abstract
Familial hypercholesterolemia (FH) is an autosomal dominant condition with a population prevalence of one in 300–500 (heterozygous) that is characterized by high levels of low-density lipoprotein (LDL) cholesterol, tendon xanthomata, and premature atherosclerosis and coronary heart disease (CHD). FH is caused mainly by mutations in the LDLR gene. However, mutations in other genes including APOB and PCSK9, can give rise to a similar phenotype. Homozygous FH with an estimated prevalence of one in a million is associated with severe hypercholesterolemia with accelerated atherosclerotic CHD in childhood and without treatment, death usually occurs before the age of 30 years. Current approaches for the treatment of homozygous FH include statin-based lipid-lowering therapies and LDL apheresis. Mipomersen is a second-generation antisense oligonucleotide (ASO) targeted to human apolipoprotein B (apoB)-100. This review provides an overview of the pathophysiology and current treatment options for familial hypercholesterolemia and describes novel therapeutic strategies focusing on mipomersen, an antisense apoB synthesis inhibitor. Mipomersen is distributed mainly to the liver where it silences apoB mRNA, thereby reducing hepatic apoB-100 and giving rise to reductions in plasma total cholesterol, LDL-cholesterol, and apoB concentrations in a dose-and time-dependent manner. Mipomersen has been shown to decrease apoB, LDL-cholesterol and lipoprotein(a) in patients with heterozygous and homozygous FH on maximally tolerated lipid-lowering therapy. The short-term efficacy and safety of mipomersen has been established, however, injection site reactions are common and concern exists regarding the long-term potential for hepatic steatosis with this ASO. In summary, mipomersen given alone or in combination with standard lipid-lowering medications shows promise as an adjunct therapy in patients with homozygous or refractory heterozygous FH at high risk of atherosclerotic CHD, who are not at target or are intolerant of statins.
Introduction
Familial hypercholesterolemia (FH) is the most common and severe form of monogenic hypercholesterolemia.Citation1 FH was the first genetic disease of lipid metabolism to be clinically and molecularly characterized. The main biochemical abnormality observed in FH is high levels of low-density lipoprotein (LDL)-cholesterol in plasma, due to reduced function of the LDL-receptor pathway, which removes LDL particles from the circulation into the liver. If untreated, patients develop premature coronary heart disease (CHD). The mean age of CHD is between 40 and 45 years in male and a decade later in female FH patients. Most people with FH are undiagnosed or only diagnosed after their first coronary event.
In the late 1930s, Müller was first to recognize that FH was an inherited disorder with related individuals exhibiting xanthomata, hypercholesterolemia, and premature CHD.Citation2 In the 1960s, Khachadurian showed a gene–dosage effect, with homozygotes having a more severe phenotype than heterozygotes. Citation3 A breakthrough occurred in the mid-1970s, when Brown et al elucidated the LDL-receptor pathway;Citation4 Goldstein and Brown showed that defects in the LDL-receptor cause FH,Citation5 a discovery that earned the authors the Nobel Prize in Physiology or Medicine in 1985.Citation6
Metabolic and molecular basis of FH
In the late 1970s, in vivo metabolic studies using radiolabeling showed in agreement with the underlying LDL receptor defect that patients with FH are characterized by a decreased clearance of LDL from the circulation and an increase in LDL synthesis, with changes in homozygotes being more marked than in heterozygotes, consistent with a gene dosage effect.Citation7 These findings were confirmed by subsequent stable isotope studies demonstrating that the fractional catabolic rate of LDL–apoB (and, to a lesser extent, very low-density lipoprotein [VLDL]– apoB and intermediate density lipoprotein [IDL]–apoB) is decreased in FH, with the majority of studies also showing increased production of VLDL–, IDL–, and LDL–apoB,Citation8–Citation10 with the increased VLDL–apoB production rate being more pronounced in FH homozygotes than in heterozygotes.Citation9
FH is an autosomal codominant inherited disorder of lipoprotein metabolism caused mainly by mutations in the LDL-receptor (LDLR) gene.Citation11,Citation12 More than 1000 mutations causing FH have been reported; these span the entire LDLR gene, which encodes the LDL-receptor (860 amino acids), and range from single nucleotide substitutions through to large structural rearrangements.Citation13 While the majority of FH is caused by mutations in the LDLR, amino acid changes in apoB-100 also lead to a form of FH called familial ligand-defective apoB-100.Citation14 As well as having an important structural role in LDL, apoB-100 acts as a ligand for the LDL-receptor to facilitate clearance of LDL particles from plasma. Rare heterozygous gain-of-function mutations in PCSK9 cause a severe form of FH by causing accelerated degradation of the LDL-receptor, while mutations in the adaptor protein LDLRAP1 can lead to a very rare form of autosomal recessive hypercholesterolemia.Citation12
Prevalence of FH
Heterozygous FH is estimated to affect 1 in 300–500 individuals worldwide, but is more prevalent in certain populations, where founder effects have led to one in 100 Afrikaners,Citation15 one in 170 Christian Lebanese,Citation16 and one in 270 QuébécoisCitation17 carrying an FH-causing mutation. Homozygous or compound heterozygous FH has an estimated population prevalence of one in a million, with one in 30,000 Afrikaners, one in 100,000 Christian Lebanese, and one in 275,000 Québécois.Citation15–Citation17 The epidemiology, Neolithic origins, and modern distribution of FH has been the subject of a recent review.Citation1
Biochemical and clinical features of FH
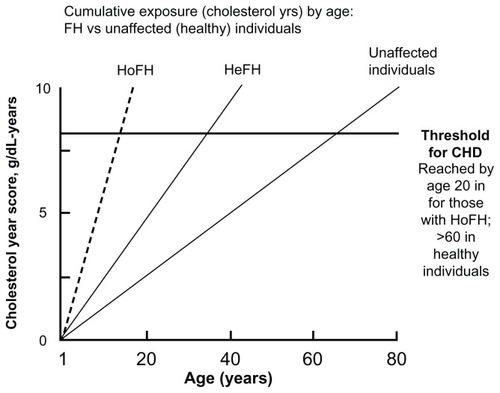
Untreated heterozygous FH patients typically have plasma LDL-cholesterol concentrations ranging from 5–12 mmol/L. The hallmark physical finding in adult patients with FH is the presence of tendon xanthomas, characteristically seen in the extensor tendons of the hands and the Achilles tendons (). Less common are xanthomas in the olecranon process and the tibial tuberosity. Corneal arcus and palpebral xanthomas may also be seen, but are less specific features of FH. About half of men and one third of women with FH experience a coronary event by the age of 60 years.Citation18,Citation19 Early atherosclerosis (observed as endothelial dysfunction and increased carotid intima-media thickness) can be seen in untreated FH children.Citation20,Citation21 The cumulative exposure in cholesterol–life years and the corresponding risk of developing CHD in FH is shown in .
Figure 1 Discrete clinical manifestations of familial hypercholesterolemia. (A) Corneal arcus and xanthelasma; (B) extensor tendon xanthomas; (C and D) Achilles tendon xanthomas.
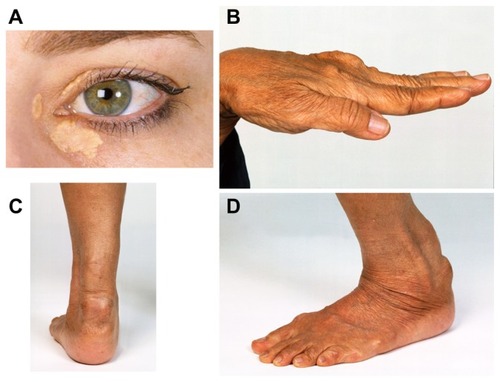
Figure 2 Cumulative LDL exposure (expressed as grams of cholesterol per year) over a lifetime in familial hypercholesterolemia patients (HeFH, HoFH) and normal individuals.
Abbreviations: CHD, coronary heart disease; HeFH, heterozygous familial hypercholesterolemia; HoFH, homozygous familial hypercholesterolemia; LDL, low-density lipoprotein.
Homozygous FH, a more severe form of the disorder, is associated with severe hypercholesterolemia (typically plasma LDL-cholesterol ranging from 15–24 mmol/L) with widespread accelerated atherosclerotic CHD as early as childhood and without treatment, death usually occurs before age 30.Citation22 Patients with homozygous FH also exhibit aortic stenosis and atherosclerotic plaques involving the aortic root and supravalvular regions.Citation23
Diagnostic criteria for FH
The clinical diagnosis of FH is based on a personal and family history, physical examination findings, and plasma cholesterol concentrations. However, there are no internationally agreed criteria for the phenotypic diagnosis of FH. Three diagnostic criteria currently in use are the Dutch Lipid Clinic Network criteria, the Simon Broome Register Group criteria, and the Make Early Diagnosis – Prevent Early Death (MED-PED) criteria.Citation24–Citation26 These criteria differ in their need for DNA testing and in their diagnostic effectiveness. There are also no internationally agreed phenotypic diagnostic criteria for homozygous FH, although these have been recently reviewed by Raal and Santos, who describe that homozygous FH is generally based on the presence of xanthomata before the age of 10 years and an untreated LDL-cholesterol of >13.0 mmol/L.Citation27
Screening for FH
FH meets the World Health Organization criteria for systematic screening.Citation28 A variety of strategies have been proposed to screen for FH.Citation29 These include: (1) universal population screening; (2) opportunistic screening of patients consulting for unrelated reasons in primary care; (3) opportunistic screening of patients admitted to hospital with premature CHD; or (4) systematic screening of first-degree relatives of people diagnosed with FH. We have recently shown that the community laboratory has the potential to assist with opportunistic FH screening.Citation30
Current approaches for the treatment of homozygous FH
The cornerstone of treatment for FH is diet and lifestyle modifications and pharmacotherapy. 3-hydroxy-3-methylglutaryl–coenzyme A reductase inhibitors (statins) are by far the most common and effective drugs with which to treat FH. However, statins require some residual LDL receptor function, thus they are not effective in receptor-negative homozygous FH. Statins decrease atherosclerotic CHD and have been shown to be cost-effective in the treatment of FH. Higher risk patients who require greater LDL-cholesterol and apoB reductions will require combination therapy to achieve therapeutic targets; especially ezetimibe, but also niacin, fibrates, and bile acid-binding resins. Therapeutic efficacy, safety, medication adherence, and compliance should be monitored closely. Effective lipid-lowering therapy has been demonstrated to reduce both mortality and major adverse cardiovascular events in individuals with homozygous FH.Citation31 However, trials confirming additional mortality and morbidity benefits over and above that of LDL-cholesterol lowering are required. Ezetimibe has been shown to effectively lower LDL-cholesterol when added to a statin, however, this did not lead to an improvement in carotid intima-media thickness over a 2-year follow-up period.Citation32
LDL apheresis is a radical form of treatment for FH that entails the extracorporeal removal of apoB-containing lipoproteins from the circulation. LDL apheresis is indicated for patients with homozygous or compound heterozygous FH, as well as for patients with heterozygous FH with documented CHD who are refractory to pharmacotherapy.Citation33,Citation34 Importantly, LDL apheresis improves CHD outcomes, progression of atherosclerosis and aortic fibrosis, endothelial function, and coagulation and is supported by international guidelines.Citation35 Homozygous patients who are not suitable for LDL apheresis should be considered for liver transplantationCitation36 or enrolment in clinical trials of novel cholesterol-lowering treatments. It is difficult, if not impossible, to treat a patient with FH to the currently recommended LDL-cholesterol targets.Citation37 Therefore, it is imperative that new therapeutic strategies be developed to lower LDL-cholesterol.
Novel strategies for the treatment of homozygous FH
Several new therapeutic strategies to lower LDL-cholesterol have been developed for patients with homozygous FH. These include inhibitors of PCSK9, microsomal triglyceride transfer protein (MTTP), and cholesteryl ester transfer protein (CETP), as well as mipomersen, the focus of this review.
PCSK9 inhibitors
PCSK9, a serine protease that binds to the LDL-receptor promoting its degradation, is an important regulator of LDL metabolism. PCSK9 gain-of-function mutations are rare and cause FH,Citation38 whereas more common loss-of-function mutations cause low LDL-cholesterol and atheroprotection.Citation39,Citation40 A compound heterozygote and a homozygote for PCSK9 nonsense mutations each had a very low LDL-cholesterol at ~0.4 mmol/L.Citation41,Citation42
Statins are known to upregulate PCSK9 potentially limiting their efficacy in reducing LDL-cholesterol levels. PCSK9 inhibitors, including antibodies, short-interfering RNA and antisense oligonucleotides are in development as a strategy for lowering plasma LDL-cholesterol and enhancing the LDL-cholesterol-lowering ability of statins.Citation43–Citation45 However, PCSK9 inhibitors require some residual LDL-receptor function, thus this approach is limited to heterozygous FH and those homozygotes with reduced, but not absent LDL-receptor function.
MTTP inhibitors
MTTP is a chaperone that facilitates apoB-containing lipoprotein assembly and secretion.Citation46 MTTP plays a critical role in incorporating hepatic triglyceride with apoB leading to the formation of VLDL, which is subsequently the vehicle of lipid export from the liver.
Abetalipoproteinemia is an extreme form of MTTP inhibition characterized by marked hypocholesterolemia, absence of LDL-cholesterol and apoB, and low triglyceride concentrations. In addition, increased transaminases due to hepatic steatosis, acanthocytosis and fat-soluble vitamin deficiency are found. MTTP is a molecular target for therapies to decrease LDL-cholesterol, and apoB and MTTP inhibitors have potential as lipid-regulating and antiatherosclerotic agents.Citation47,Citation48
Lomitapide is a small-molecule MTTP inhibitor designed as an oral, once-daily treatment for homozygous FH. Lomitapide has been granted orphan drug status by the US Food and Drug Administration (FDA), and, in March 2012, Aegerion Pharmaceuticals (Cambridge, MA) filed a New Drug Application (NDA) with the FDA and a Marketing Authorization Application with the European Medicines Agency (EMA) for lomitapide. A long-term safety and efficacy study for lomitapide in homozygous FH showed that the 23 patients who completed the 78-week trial achieved a 38% reduction in LDL-cholesterol, and about half of apheresis patients either reduced or eliminated apheresis therapy.Citation49 However, therapies for MTTP inhibition have also increased plasma transaminases and caused hepatic steatosis,Citation50 the long-term significance of which is unknown.
CETP inhibitors
CETP transfers cholesteryl esters from high-density lipoprotein (HDL) to the apoB-containing lipoproteins. Individuals with CETP deficiency due to mutations in CETP have high levels of HDL-cholesterol. Inhibition of CETP is a target to increase HDL-cholesterol and potentially reduce atherosclerosis.Citation51,Citation52 CETP inhibition could potentially correct abnormalities in HDL-functionality in homozygous FH.Citation53
Torcetrapib, the first CETP inhibitor to be studied in a Phase III trial, was discontinued due to a number of off-target side effects, namely elevations in systolic blood pressure and increases in plasma aldosterone, sodium, and bicarbonate, and reductions in potassium, leading to increased mortality.Citation54 Dalcetrapib, a structurally unrelated CETP “modulator,” did not show major side effects; however, a lack of clinically meaningful benefit has meant that further testing of the drug has been halted. Anacetrapib added to statin therapy has been demonstrated to reduce LDL-cholesterol by a further ~40% and increase HDL-cholesterol by ~140%.Citation55 The onus is now on anacetrapib to show that CETP inhibition can prevent coronary events, which is currently being investigated in the REVEAL trial.Citation56
Mipomersen
Mipomersen (Kynamro™ [mipomersen sodium]; ISIS Pharmaceuticals, Carlsbad, CA), developed under the name ISIS 301012, is a second-generation antisense oligonucleotide (ASO), which is administered by subcutaneous injection in a formulation with 0.9% sodium chloride and targets apoB-100 mRNA in the liver. Mipomersen has been granted orphan drug status by the FDA for the treatment of homozygous FH. In May 2012, an NDA for mipomersen was filed with the FDA by Genzyme Corporation (Cambridge, MA), who had previously filed mipomersen for marketing approval as a treatment for homozygous and severe heterozygous FH with the EMA in July 2011.
Mode of action
Mipomersen consists of a 20-mer 2′-O-methoxyethyl modified nucleotide complementary and specific to human apoB-100 mRNA. It distributes mainly to the liver, where it forms a duplex with the target mRNA, causing the mRNA to be cleaved by RNase H and therefore unable to be translated to apoB-100. Hepatic apoB mRNA silencing gives rise to reductions in hepatic apoB and plasma total cholesterol, LDL-cholesterol, and apoB concentrations in a dose- and time-dependent manner.Citation57 The estimated median effective concentration (EC50) in human liver is 81 ± 122 μg/g.
Pharmacokinetics
Pharmacokinetic studies have indicated that mipomersen has near complete systemic absorption and is rapidly and extensively distributed to tissues (volume of distribution in humans 48.3 L/kg).Citation58 Animal studies show that the highest levels of the oligonucleotide are found in liver and kidney.Citation58 In plasma, greater than 85% of mipomersen is bound to plasma proteins. Animal studies suggest that plasma and liver concentrations of mipomersen are in equilibrium with a ratio of 1:6000.Citation57
The half-life of mipomersen in humans has been calculated as approximately 30 days.Citation57 Urinary excretion of the drug is minimal in the first 24 hours. Oligonucleotide metabolites, consisting of between seven and 14 nucleotides of the parent compound, can be detected in urine along with mipomersen. No potential pharmacokinetic interactions of mipomersen occur with coadministration of either simvastatin or ezetimibe, and nor does mipomersen inhibit cytochrome P450 enzyme activities.Citation59
Clinical efficacy
Phase I trials showed that, in healthy individuals with mild dyslipidemia, mipomersen produces rapid and dose- dependent prolonged reductions in serum apoB, total cholesterol and LDL-cholesterol, with the apoB and LDL-cholesterol concentrations remaining below baseline for up to 3 months after the last dose.Citation60
Subsequent Phase II and III trials have demonstrated that mipomersen is an effective lipid-lowering therapy in FH (). A Phase II trial in 44 heterozygous FH patients already taking conventional lipid-lowering therapy demonstrated that 300 mg doses of mipomersen could safely, further reduce plasma apoB and LDL-cholesterol concentrations by one-third.Citation61 Patients were randomized to receive subcutaneous mipomersen (50, 100, 200 or 300 mg) or placebo on days 1, 4, 8 and 11, followed by once-weekly injections to a total of 6 weeks. Significant reductions in apoB (23% and 33%) and LDL-cholesterol (21% and 34%) were observed in the 200 mg and 300 mg dose groups. While there was a trend towards reductions in lipoprotein(a) (Lp(a)), an important risk factor for coronary artery disease in FH that is highly heritable,Citation62–Citation64 these changes were not significant. Triglycerides and HDL-cholesterol levels were not significantly affected by mipomersen treatment. Four patients in the 300 mg group continued out to 13 weeks in an extension study, showing reduced apoB and LDL-cholesterol, each by 37% from baseline levels. These parameters remained below baseline for more than 3 months after the last dose.
Table 1 Effects of mipomersen on plasma apoB, LDL-cholesterol, triglyceride, HDL-cholesterol, and Lp(a) in FH
The efficacy and safety of 200 mg/week mipomersen has also been assessed in homozygous FH.Citation65 Patients (mean age: 31 years) were all on stable maximum-tolerated lipid- lowering therapy, and over 80% had genetically confirmed homozygous or compound heterozygous FH. Of the 51 patients enrolled, all 17 assigned to the placebo group completed the 26-week treatment period, compared to 28 of the 34 assigned to mipomersen. The reasons given for withdrawal included injection site reactions (n = 2), rash, alanine aminotransferase (ALT) increase, noncompliance, and consent withdrawn. In the mipomersen group, the mean LDL-cholesterol reduced from 11.4 mmol/L at baseline to 8.4 mmol/L (−25%), with reductions near maximum at week 17. However, there was variability in changes to LDL-cholesterol, which ranged between 2% to −82%, and was independent of baseline LDL-cholesterol, age, race, or sex. Significant reductions in Lp(a) (−31%) and triglyceride levels (−17%), and an increase in HDL-cholesterol (15%), were also observed.
Visser et al examined the effect of mipomersen in 33 statin-intolerant patients at high risk for cardiovascular disease.Citation66 Over half of the patients were FH heterozygotes. Treatment with 200 mg/week mipomersen for 26 weeks resulted in a 47% decrease in LDL-cholesterol, ranging from −19% to −77%. This was predominantly the result of a reduction in small LDL particles (−56%; P = 0.001 vs placebo) rather than large LDL particles (−4%; P < 0.017 vs placebo). While triglycerides and Lp(a) levels were significantly reduced by mipomersen treatment, HDL-cholesterol and apoA-I concentrations did not change.
Safety
The majority of patients treated with mipomersen experience mild, transient injection site reactions, and about one-third experience flu-like symptoms. However, the focus of mipomersen’s safety concerns has been on hepatic steatosis, as this was the major issue with suppression of VLDL production using MTTP inhibitors. Elevated plasma liver transaminases are common in patients on mipomersen therapy, and may be associated with the development of steatosis.
The impact of 200 mg/week mipomersen on intrahepatic triglyceride content, assessed by proton magnetic resonance spectroscopy (1H-MRS), was studied in 21 patients with heterozygous FH.Citation67 Patients with a baseline liver fat greater than 5% were excluded from the trial. Liver fat content was assessed at baseline, where the mean intrahepatic triglyceride content was 1.2%, and again at weeks 4 and 15. The most common adverse events were injection site reactions, which affected all patients in the mipomersen group (19% of injections) compared to 73% of the placebo (9% of injections). Flu-like symptoms were observed in 70% of the mipomersen group and 18% of placebo. While no clinically significant increases in ALT were observed, the mean intrahepatic triglyceride content increased 0.8 percentage points in the mipomersen group at week 15, compared to a decrease of 0.1 percentage points in the placebo group (P = 0.051). In one patient, liver fat increased from 0.6% at baseline to 5.7% at week 15, but this appeared to be reversible, decreasing to 2.5% at week 35.
In a Phase III trial of homozygous FH patients, 1H-MRS quantification of liver fat content was performed at baseline and only remeasured in patients with increases in ALT greater than three times the upper reference limit.Citation65 This corresponded to four patients (12%) in the mipomersen group, and none in the placebo group. While two of these patients did not show an increase in hepatic steatosis, a third showed an increase in hepatic fat content from 9.6% to 24.8%. Mipomersen treatment gave a strong lipid-lowering response in this patient, with a 75% reduction in LDL-cholesterol to <1.8 mmol/L achieved by week 13. The fourth patient had increased liver transaminases at baseline and again at week 17 where the protocol-defined stopping rule was met and dosing stopped.
In a trial of 33 statin-intolerant patients, the majority with FH, 81% of mipomersen-treated patients had increases in ALT above the upper limit of normal, compared to 25% of the placebo group.Citation66 One-third of mipomersen-treated patients showed persistent elevations in liver transaminases greater than three times the upper reference limit. One patient in the mipomersen group met a stopping rule with liver transaminases elevated more than 10 times the upper reference limit in week 8. However, the patient’s intrahepatic triglyceride content was only 0.8% when measured in week 9, and transaminases returned to normal within four weeks. Hepatic 1H-MRS was performed in all patients with elevated liver transaminases (>2 times upper reference limit); hepatic steatosis was detected in 12 of 14 patients in the mipomersen group compared to 1 of 1 placebo-treated patients. The median intrahepatic triglyceride content was 24.4% in the mipomersen group, ranging from 0.8% to 47.3%.
Conclusion
Mipomersen is an antisense apoB synthesis inhibitor that is currently in Phase III development for FH as a new treatment to lower apoB and LDL-cholesterol in patients at high risk of atherosclerotic CHD. Mipomersen is distributed mainly to the liver where it silences apoB mRNA, thereby reducing hepatic apoB-100 and giving rise to reductions in plasma total cholesterol, LDL-cholesterol, and apoB concentrations in a dose- and time-dependent manner. Unlike statins, mipomersen is not dependent on the LDL receptor for its mechanism of action.
Mipomersen has been shown to decrease apoB, LDL-cholesterol and Lp(a) in patients with heterozygous and homozygous FH on maximally tolerated lipid-lowering therapy. Furthermore, mipomersen has the potential to reduce the frequency of LDL apheresis. The short-term efficacy and safety of mipomersen has been established, however, injection site reactions are a common occurrence and concern exists regarding the long-term potential for hepatic steatosis with this ASO and more detailed safety evaluations on human liver function are required.
The requirement for injection of mipomersen could present a barrier to patient uptake of this therapy. Further studies are required to develop an alternative delivery mode that would make this form of ASO available to a wider spectrum of patients who require to be treated to lower target levels of LDL-cholesterol either because current therapy is inadequate or that they cannot tolerate statins.
In summary, mipomersen given alone or in combination with standard lipid lowering medications shows promise as an adjunct therapy in patients with homozygous or refractory heterozygous FH at high risk of atherosclerotic CHD, who are not at target or are intolerant of statins. The long-term efficacy and safety and cost-effectiveness of mipomersen need to be demonstrated.
Acknowledgment
JRB is supported by a Practitioner Fellowship from the Royal Perth Hospital Medical Research Foundation***.
Disclosure
The authors report no conflicts of interest in this work.
References
- LiyanageKEBurnettJRHooperAJvan BockxmeerFMFamilial hypercholesterolemia: epidemiology, Neolithic origins and modern geographic distributionCrit Rev Clin Lab Sci201148111821657943
- MüllerCXanthomata, hypercholesterolemia, angina pectorisActa Med Scand19388975
- KhachadurianAKThe inheritance of essential familial hypercholesterolemiaAm J Med19643740240714209286
- BrownMSKovanenPTGoldsteinJLRegulation of plasma cholesterol by lipoprotein receptorsScience198121244956286356261329
- GoldsteinJLBrownMSThe LDL receptorArterioscler Thromb Vasc Biol200929443143819299327
- BrownMSGoldsteinJLA receptor-mediated pathway for cholesterol homeostasisScience1986232474634473513311
- BilheimerDWStoneNJGrundySMMetabolic studies in familial hypercholesterolemia. Evidence for a gene-dosage effect in vivoJ Clin Invest1979642524533222811
- CummingsMHWattsGFUmplebyMHennessyTRQuineyJRSonksenPHIncreased hepatic secretion of very-low-density-lipoprotein apolipoprotein B-100 in heterozygous familial hypercholesterolaemia: a stable isotope studyAtherosclerosis1995113179897755658
- MillarJSMaugeaisCIkewakiKComplete deficiency of the low-density lipoprotein receptor is associated with increased apolipoprotein B-100 productionArterioscler Thromb Vasc Biol200525356056515637307
- TremblayAJLamarcheBRuelILIncreased production of VLDL apoB-100 in subjects with familial hypercholesterolemia carrying the same null LDL receptor gene mutationJ Lipid Res200445586687214967814
- FaizFHooperAJvan BockxmeerFMMolecular pathology of familial hypercholesterolemia, related dyslipidemias and therapies beyond the statinsCrit Rev Clin Lab Sci201249111722214202
- SoutarAKNaoumovaRPMechanisms of disease: genetic causes of familial hypercholesterolemiaNat Clin Pract Cardiovasc Med20074421422517380167
- LeighSEFosterAHWhittallRAHubbartCSHumphriesSEUpdate and analysis of the University College London low density lipoprotein receptor familial hypercholesterolemia databaseAnn Hum Genet200872Pt 448549818325082
- SoriaLFLudwigEHClarkeHRVegaGLGrundySMMcCarthyBJAssociation between a specific apolipoprotein B mutation and familial defective apolipoprotein B-100Proc Natl Acad Sci U S A19898625875912563166
- SeftelHCBakerSGSandlerMPA host of hypercholesterolaemic homozygotes in South AfricaBMJ198028162416336367437743
- FahedACSafaRMHaddadFFHomozygous familial hypercholesterolemia in Lebanon: a genotype/phenotype correlationMol Genet Metab2011102218118821145767
- MoorjaniSRoyMGagneCHomozygous familial hypercholesterolemia among French Canadians in Quebec ProvinceArteriosclerosis1989922112162923577
- SlackJRisks of ischaemic heart-disease in familial hyperlipoproteinaemic statesLancet196927635138013824188273
- StoneNJLevyRIFredricksonDSVerterJCoronary artery disease in 116 kindred with familial type II hyperlipoproteinemiaCirculation19744934764884813182
- TonstadSJoakimsenOStensland-BuggeERisk factors related to carotid intima-media thickness and plaque in children with familial hypercholesterolemia and control subjectsArterioscler Thromb Vasc Biol19961689849918696963
- WiegmanAde GrootEHuttenBAArterial intima-media thickness in children heterozygous for familial hypercholesterolaemiaLancet2004363940636937015070569
- KhachadurianAKUthmanSMExperiences with the homozygous cases of familial hypercholesterolemia. A report of 52 patientsNutr Metab19731511321404351242
- SummersRMAndrasko-BourgeoisJFeuersteinIMEvaluation of the aortic root by MRI: insights from patients with homozygous familial hypercholesterolemiaCirculation19989865095189714107
- Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register GroupBMJ199130368078938961933004
- World Health OrganizationFamilial Hypercholesterolaemia (FH)Report of a second WHOT ConsultationGenevaSeptember 4, 1998GenevaWorld Health Organization Human Genetics Programme1999 Available from http://whqlibdoc.who.int/hq/1999/WHO_HGN_FH_CONS_99.2.pdfAccessed August 1, 2012
- WilliamsRRHuntSCSchumacherMCDiagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular geneticsAm J Cardiol19937221711768328379
- RaalFJSantosRDHomozygous familial hypercholesterolemia: Current perspectives on diagnosis and treatmentAtherosclerosis2012223226226822398274
- WilsonJMJungnerGPrinciples and Practice of Screening for GiseaseGeneva, SwitzerlandWorld Health Organization1968
- BenderRBellDAHooperAJScreening for familial hypercholesterolaemiaPathology201244212212822228254
- BellDAHooperAJBenderROpportunistic screening for familial hypercholesterolaemia via a community laboratoryAnn Clin Biochem Epub September 21, 2012
- RaalFJPilcherGJPanzVRReduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapyCirculation2011124202202220721986285
- KasteleinJJAkdimFStroesESSimvastatin with or without ezetimibe in familial hypercholesterolemiaN Engl J Med2008358141431144318376000
- ThompsenJThompsonPDA systematic review of LDL apheresis in the treatment of cardiovascular diseaseAtherosclerosis20061891313816546196
- ThompsonGRLipoprotein apheresisCurr Opin Lipidol201021648749121206339
- ThompsonGRCatapanoASahebSSevere hypercholesterolaemia: therapeutic goals and eligibility criteria for LDL apheresis in EuropeCurr Opin Lipidol201021649249820935563
- MoiniMMistryPSchilskyMLLiver transplantation for inherited metabolic disorders of the liverCurr Opin Organ Transplant201015326927620489626
- PijlmanAHHuijgenRVerhagenSNEvaluation of cholesterol lowering treatment of patients with familial hypercholesterolemia: a large cross-sectional study in The NetherlandsAtherosclerosis2010209118919419818960
- AbifadelMVarretMRabesJPMutations in PCSK9 cause autosomal dominant hypercholesterolemiaNat Genet200334215415612730697
- BennMNordestgaardBGGrandePSchnohrPTybjaerg-HansenAPCSK9 R46L, low-density lipoprotein cholesterol levels, and risk of ischemic heart disease: 3 independent studies and meta-analysesJ Am Coll Cardiol201055252833284220579540
- CohenJCBoerwinkleEMosleyTHJrHobbsHHSequence variations in PCSK9, low LDL, and protection against coronary heart diseaseN Engl J Med2006354121264127216554528
- HooperAJMaraisADTanyanyiwaDMBurnettJRThe C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African populationAtherosclerosis2007193244544816989838
- ZhaoZTuakli-WosornuYLagaceTAMolecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygoteAm J Hum Genet200679351452316909389
- McKenneyJMKorenMJKereiakesDJHanotinCFerrandACSteinEASafety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/ REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapyJ Am Coll Cardiol201259252344235322463922
- SteinEAGipeDBergeronJEffect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trialLancet2012380293622633824
- SteinEAMellisSYancopoulosGDEffect of a monoclonal antibody to PCSK9 on LDL cholesterolN Engl J Med2012366121108111822435370
- HussainMMIqbalJAnwarKRavaPDaiKMicrosomal triglyceride transfer protein: a multifunctional proteinFront Biosci20038500506
- BurnettJRWattsGFMTP inhibition as a treatment for dyslipidaemias: time to deliver or empty promises?Expert Opin Ther Targets200711218118917227233
- HussainMMBakillahANew approaches to target microsomal triglyceride transfer proteinCurr Opin Lipidol200819657257818957879
- Aegerion Pharmaceuticals: We are Bullish on the Lomitapide Story682012 Available from: http://seekingalpha.com/article/645951-aegerion-pharmaceuticals-we-are-bullish-on-the-lomitapide-storyAccessed October 12, 2012
- CuchelMBloedonLTSzaparyPOInhibition of microsomal triglyceride transfer protein in familial hypercholesterolemiaN Engl J Med2007356214815617215532
- HooperAJBurnettJRDalcetrapib, a cholesteryl ester transfer protein modulatorExpert Opin Invest Drugs201221914271432
- HooperAJBurnettJRAnacetrapib, a cholesteryl ester transfer protein inhibitorExpert Opin Invest Drugs2012211103109
- GuerinMReverse cholesterol transport in familial hypercholesterolemiaCurr Opin Lipidol201223437738522510809
- BarterPJCaulfieldMErikssonMEffects of torcetrapib in patients at high risk for coronary eventsN Engl J Med2007357212109212217984165
- CannonCPShahSDanskyHMSafety of anacetrapib in patients with or at high risk for coronary heart diseaseN Engl J Med2010363252406241521082868
- REVEAL: Randomized EValuation of the Effects of Anacetrapib Through Lipid-modification82012 Available from: http://clinicaltrials.gov/ct2/show/NCT01252953Accessed October 12, 2012
- YuRZLemonidisKMGrahamMJCross-species comparison of in vivo PK/PD relationships for second-generation antisense oligonucleotides targeting apolipoprotein B-100Biochem Pharmacol200977591091919056355
- YuRZKimTWHongAWatanabeTAGausHJGearyRSCross-species pharmacokinetic comparison from mouse to man of a second-generation antisense oligonucleotide, ISIS 301012, targeting human apolipoprotein B-100Drug Metab Dispos200735346046817172312
- YuRZGearyRSFlaimJDLack of pharmacokinetic interaction of mipomersen sodium (ISIS 301012), a 2′-O-methoxyethyl modified antisense oligonucleotide targeting apolipoprotein B-100 messenger RNA, with simvastatin and ezetimibeClin Pharmacokinet2009481395019071883
- KasteleinJJWedelMKBakerBFPotent reduction of apolipoprotein B and low-density lipoprotein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein BCirculation2006114161729173517030687
- AkdimFVisserMETribbleDLEffect of mipomersen, an apolipoprotein B synthesis inhibitor, on low-density lipoprotein cholesterol in patients with familial hypercholesterolemiaAm J Cardiol2010105101413141920451687
- ClarkeRPedenJFHopewellJCGenetic variants associated with Lp(a) lipoprotein level and coronary diseaseN Engl J Med2009361262518252820032323
- ErqouSKaptogeSPerryPLLipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortalityJAMA2009302441242319622820
- NenseterMSLindvigHWUelandTLipoprotein(a) levels in coronary heart disease-susceptible and -resistant patients with familial hypercholesterolemiaAtherosclerosis2011216242643221376325
- RaalFJSantosRDBlomDJMipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trialLancet20103759719998100620227758
- VisserMEWagenerGBakerBFMipomersen, an apolipoprotein B synthesis inhibitor, lowers low-density lipoprotein cholesterol in high-risk statin-intolerant patients: a randomized, double-blind, placebo-controlled trialEur Heart J20123391142114922507979
- VisserMEAkdimFTribbleDLEffect of apolipoprotein-B synthesis inhibition on liver triglyceride content in patients with familial hypercholesterolemiaJ Lipid Res20105151057106220008831
- BurnettJRHooperAJCommon and rare gene variants affecting plasma LDL cholesterolClin Biochem Rev2008291112618566665
- HortonJDCohenJCHobbsHHPCSK9: a convertase that coordinates LDL catabolismJ Lipid Res200950SupplS172S17719020338
- SteinEADufourRGagneCA randomized, double-blind, placebo-controlled study to assess efficacy and safety of mipomersen as add-on therapy in heterozygous familial hypercholesterolemia patients with coronary artery diseaseEur Heart J20103189820402003
- TardifJCMcGowanMCeskaRApolipoprotein B synthesis inhibition by mipomersen reduces low-density lipoprotein cholesterol when added to maximally tolerated lipid-lowering medication in patients with severe heterozygous hypercholesterolemiaJ Am Coll Cardiol20115714E492