 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Simple sequence repeats (SSRs) play important roles in gene regulation and genome evolution. Although there exist several online resources for SSR mining, most of them only extract general SSR patterns without providing functional information. Here, an online search tool, CG-SSR (Comparative Genomics SSR discovery), has been developed for discovering potential functional SSRs from vertebrate genomes through cross-species comparison. In addition to revealing SSR candidates in conserved regions among various species, it also combines accurate coordinate and functional genomics information. CG-SSR is the first comprehensive and efficient online tool for conserved SSR discovery.
Introduction
SSRs, also called simple tandem repeats (STRs) or microsatellites, are DNA segments composed of tandem repetitions of relatively short motifs. They are commonly and easily identified DNA sequences which consist of repeated units one to six base pairs in length.Citation1–Citation4 For decades, SSRs were mainly considered to be genetic markers in DNA fingerprinting and diversity studies due to their high rate of polymorphism. Nevertheless, recent studies pointed out that SSR expansions and/or contractions in protein-coding regions bring about a gain or loss of gene function through frameshift mutations.Citation2–Citation4 SSR variations in 5’UTRs could affect gene transcription and translation, whereas SSR expansions in 3’UTRs could cause transcription slippage and result in disrupting splicing and possibly disturbing cellular functions. For example, a CGG repeat pattern within the 5’UTR of the FMR1 gene is expanded in families with fragile X syndrome. When the length of SSR exceeds 200 CGGs, mental retardation occurs due to the absence of the encoded FMR protein.Citation5 Another example is a CTG expansion located in the 3’UTR of a kinase gene involved in myotonic dystrophy type 1 (DM1), a multisystemic dominantly inherited disorder. DM1 disorder affects skeletal and smooth muscle as well as the eye, heart, endocrine system, and central nervous system.Citation6 Furthermore, SSRs in introns affect transcription, mRNA splicing, or export to the cytoplasm, which have been shown that SSRs indeed possess a functional role as cis-regulatory elements. For example, a CA simple sequence repeat within the first 2000 bases in intron 1 enhances egfr transcription and involves in breast carcinogenesis.Citation7 These functional SSRs, SSRs with biological functions, play an important role in gene regulation.Citation2 Consequently, the discovery of potential functional SSRs to decipher gene regulatory networks intrigues biologists.
There are many in silico SSR mining tools and databases such as MMDBJ,Citation8 Satellog,Citation9 MRD,Citation10 SSRD,Citation11 and EuMicroSatdb. Citation12 All of these mining tools are briefly described by Aishwarya and colleagues,Citation12 but none of them allows users to retrieve potential functional SSRs using comparative genomics. These available tools either emphasize a collection of SSRs from specific organisms or provide only limited functions for various genome comparisons. Accordingly, it is still tedious for biologists to find functional SSRs from millions of SSR candidates.
Due to functional constraints, DNA regions involved in gene regulation or genome evolution are expected to be conserved among related species. It has been shown that cross-species comparison of DNA sequences can facilitate identification of candidate regulatory elements.Citation13 If SSRs possess significant biological functions, they are likely to be located in conserved regions. In this report, the proposed comparative genomics SSR discovery (CG-SSR) web service comprises eleven representative vertebrate species (human, chimpanzee, orangutan, mouse, rat, opossum, rhesus, cow, dog, zebrafish, and medaka) for constructing the fundamental SSR database. It also includes another thirteen species (cat, horse, marmoset, guinea pig, platypus, chicken, lizard, Xenopus tropicalis, tetraodon, fugu, stickleback, lamprey, lancelet) for verification of conserved regions of the retrieved SSRs. Users can evaluate the biological significance of SSRs through cross-species conservation because such comparisons are available for the various species selected as the representative model organisms in CG-SSR. CG-SSR also combines other relevant functional genomics information such as GO (Gene Ontology),Citation14 InterPro,Citation15 and Pfam.Citation16 GO provides biological annotation of genes in light of their associated biological processes, cellular components, and molecular functions. The InterPro database contains identifiable features – protein families, domains, repeats and sites – found in known proteins that can be applied to novel proteins. Through hidden Markov models and multiple sequence alignments, the Pfam database collects domain information for a large quantity of protein families. Taken together, these functional genomics resources permit biologists to decipher candidate roles for SSRs in gene regulatory networks.
Methods
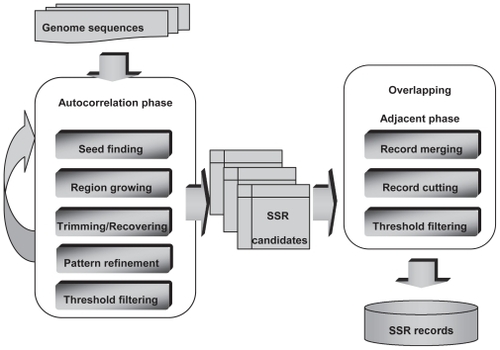
To construct the database for CG-SSR, vertebrate genome sequences and gene coordinates were obtained from Ensembl Genome Browser.Citation17 Whole genomes were scanned based on an efficient correlation method for SSR mining that is composed of two major phases including autocorrelation and overlapping adjacent phases (see Supplementary materials). In the first autocorrelation phase, the CG-SSR performs seed finding, region growing, trimming, recovering, pattern refinement, and noise filtering processes to discover all possible SSR repeats as initial candidates. For the second phase, the system verifies the overlapping records and confirms no redundant patterns by employing merging, cutting and threshold filtering processes. illustrates the flowchart of CG-SSR searching algorithm and a detailed description of the developed methods and examples can be found in the Supplementary materials.
Figure 1 The flowchart of CG-SSR searching algorithm.
The core algorithm for discovering SSR patterns from genome sequences employs autocorrelation methodology. The basic concept assumes that the target sequence contains repeat substrings with basic patterns of length N within a range from 1 to 6. If we shift the target sequence with N nucleotides to its right and compare to the original sequence, all repeat patterns can be discovered since they overlapped with their shifted sequence at least within N nucleotides continuously. Based on the observation of such overlapping, we can easily detect the repeating locations by shifting and matching the whole target sequence without knowing the nucleotide contents of repeated patterns. If the continuously matched nucleotides are longer than the shifting length N, at least one repeat with the basic pattern length N is proved to exist in the target sequence. Hence, exact matched strings can be identified and considered as the seeds of perfect SSR candidates. These seeds were then extended by region growing techniques to formulate imperfect SSR sequences. To identify imperfect SSRs with various tolerant conditions, a module applied forward region growing method to perform neighboring comparison. As long as the coordinates and contents of the perfect repeat patterns were obtained, the forward region growing processes were performed by examining the right-hand-side neighboring nucleotides and skipping noise-like nucleotides which did not belong to the perfect repeat patterns. As no extra basic unit pattern could be found by continuously extending the verification on right-hand-side neighboring nucleotides, the searching processes terminated. After the region growing processes, the boundary detection through trimming/recovering operations was applied to delimit the appropriate range of each SSR sequence. In this module, verifying pattern boundaries on both sides was achieved by left-hand-side trimming and right-hand-side recovering processes. The right-hand-side trimming processes were achieved by backward scanning from the rightmost nucleotide of the primitively searched SSR to its last noninterrupted basic pattern. Once the last perfect basic pattern was found, the partial and noninterrupted patterns on its right-hand-side were recalled for its longest representation. For the left-hand-side of SSR string verification, this module examined only on the length of an assigned basic pattern which could not be recovered by insertion, deletion, or substitution examinations employed in the previous autocorrelation phase. In the module of pattern refinement, if a basic unit pattern is a repeated sequence itself, trivially, the pattern will be identified as SSRs within different unit lengths during autocorrelation processes. For example, a tri-nucleotide SSR string of length 60 could be identified as well as a hexa-nucleotide SSR string of length 30 from the first phase computation. Hence, for eliminating the redundant cases of this type, all self-repeating basic patterns were double checked and reduced to its smallest unit size.
After all frameshifts were completed, redundant segments were removed to eliminate possible overlapping patterns. The final noise filtering module was implemented to remove imperfect SSRs which contain impure variations higher than the defined threshold proportion. In the proposed system, two thresholding parameters of minimum length and maximum noise rate were decided in advance for verifying the qualification of a searched SSR record. The first thresholding value confirms the total length of a candidate SSR record which includes the repeats and the tolerant nucleotides, whereas the second thresholding parameter inspects the noise rate of the identified SSR records. The definition of noise rate is indicated as
where LPatternLength represents the length of basic unit pattern, NRepeatTime the number of repeats of complete pattern (exact the same as the basic unit pattern), NIncompletePattern the number of nucleotides of incomplete patterns (partial subset of the basic unit pattern) in the SSR, and LTotalLength the total length of the identified SSR. Hence, a perfect SSR possesses “0” noise rate and higher noise rate represents more tolerant nucleotides appeared in the identified SSR strings. The default setting of noise rate for the initial database in CG-SSR is 0.2 which represents 20% of the total nucleotides recognized as the maximum noisy base pairs for each SSR segment.
For the second phase of overlapping adjacent verification, the SSR candidates were re-inspected based on their locations and basic unit patterns. Merging and cutting operations on neighboring SSR candidates enhanced the consecutive relationship and centralized the diverse representations. On the condition of two overlapping SSR candidates, the system firstly verified the overlapped records if they possessed identical basic unit patterns. If two or more SSR records possessed an identical pattern, the recombination of such candidates were concatenated after evaluating the criterion of noise threshold requirements. On the other hand, if these overlapped SSR candidates of the same pattern length did not possess identical basic unit pattern, they usually resulted from the tolerant conditions. Hence, the system combined such two SSR candidates to make sure no entry was redundant. Finally, a threshold filter was applied again to satisfy the requirements of minimum SSR length and maximum noise rate. It is also true that two SSR candidates of different basic unit patterns and lengths might overlap. The ambiguity usually arose on the transistion positions of two consecutive basic patterns. In this system, two SSR strings of different lengths of basic unit patterns would be categorized and stored as two independent SSRs. Only two overlapping adjacent SSRs of the same basic pattern were merged for efficient and effective consideration.
When all of the SSR patterns in each genome were identified respectively, by comparing identical SSR patterns in the conserved regions among various species, the SSR patterns with high occurrence rates were considered as the candidates of important functional SSRs.
Results and discussion
In the retrieval processes, all perfect and imperfect SSRs were verified and annotated with accurate coordinate information of upstream, downstream, 5’UTR, 3’UTR, protein-coding, intron, and intergenic regions. Moreover, for identifying candidates of functional SSRs, the system integrated comparative genomics methods in which information from crossspecies comparison was obtained from the UCSC Genome BrowserCitation18 to provide coordinates for cross-species conserved regions. Currently, 24 species were included in the CG-SSR system for comparison. Through cross-species conservation, all conserved SSRs were identified and displayed in an explicit table. Furthermore, the biological function of each gene can refer to GO, InterPro, and Pfam resources for further comprehensive analyses. The developed CG-SSR web server is freely available at: http://cgssr.cs.ntou.edu.tw/. There are four major functions provided by the system: SSR Discovery, SSR on Transcripts, Comparative Genomics, and SSR Searching Tool.
SSR Discovery
It provides all loci of perfect and imperfect SSRs on a designated chromosome of a specified genome. Users can allocate precise locations of SSRs by selecting a target species, the number and range of chromosome, parameter of minimum length, and/or specific patterns of interested SSRs.
SSR on Transcripts
It collects all genes which possess perfect and imperfect SSRs found on their exon regions. Users can allocate precise locations of SSRs by selecting a target species, the number of chromosome, and the transcript IDs. (All transcript IDs were defined in Ensembl release 48, Dec. 2007).
Comparative Genomics
It provides a tool for searching all perfect and imperfect SSRs on conserved regions or orthologous genes among various species. From the query keywords or transcript IDs, the system provides precise and complete information of SSRs including coordinates, lengths, basic unit patterns, regions, flanking sequences, and corresponding primers.
SSR Searching Tool
It provides an online SSR searching tool for discovering all possible SSRs located in the uploaded multiple DNA sequences with respect to required parameter settings.
The detail guidelines of each subsystem can be obtained by clicking on the “tips” icon in each web interface. These guidelines provide helpful information to a user who is looking for a particular point of interest in SSR discovery. In conclusion, abundant resources for functional genomics, cross-species comparison, accurate coordinate annotation, flanking sequences and corresponding primers have been well integrated to provide applicable information for users. In this study, the numbers of retrieved imperfect and perfect SSRs (≥10 base pairs with repeated unit lengths of 1–6 base pairs) by CG-SSR and the number of identifiable gene characteristics and protein families from several well known databases for eleven representative vertebrate species are listed in the . All detailed results are available online at the CG-SSR website. shows the statistical distributions of various repeated unit patterns for eleven model species. As can be seen from the diagram, similarly distributed proportions of different unit lengths of SSR patterns occur for all species and the most amounts of perfect and imperfect SSR repeats is the dinucleotide patterns with the default parameter settings of minimum length of 10 base pairs and noise rate of 20%.
Figure 2 Distributions of various repeated unit patterns from mononucleotide to hexanucleotide for 11 vertebrate model species. The number of SSR records was identified based on the parameters of a minimum length of 10 base pairs and a maximum noise rate of 20%.
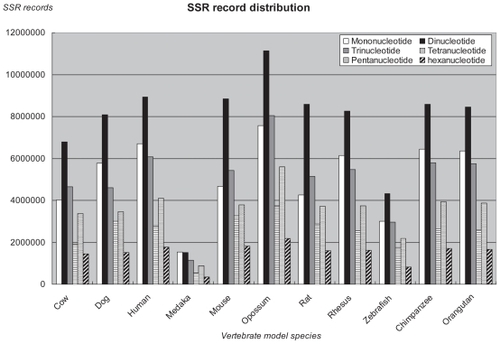
Table 1 The total number of verified SSRs in the CG-SSR database, and the number of identifiable gene characteristics and protein families from GO, InterPro, and Pfam databases for 11 representative vertebrate species
To illustrate the practical applications of CG-SSR for identifying potential functional SSRs through cross-species comparison, several well known functional SSRs were collected and shown in . According to comparative genomics rules, functional elements are likely to be located in conserved regions. Notably, several functional SSRs discovered by our system were located in coding regions, UTR regions, and even in intron regions. The identification of functional SSRs in conserved regions of several species is evidence of their common features.
Table 2 Illustrations of practical applications of CG-SSR in identifying potential functional SSRs through cross-species comparison. Taking human species as an example, several well known functional SSRs could be retrieved and annotatedCitation4
Taking ATM gene (ataxia-telengiectasia mutated gene) as an example (Ensembl transcript ID: ENST00000278616), 118 SSRs longer than 20 nucleotides were found. The comparative genomics mechanism provides an efficient way to select potential functional SSRs from among the 118 candidates. For instance, a T repeat exhibited a high degree of conservation among 17 species although it is located in an intron which is generally considered as a nonfunctional region. Interestingly, such a specific intronic mutation of T repeat has been reported to cause aberrant splicing and abnormal transcription in colon tumors.Citation19 Similarly, some CAG repeats in coding regions can be found and proved as functional SSRsin the HD (Ensembl transcript ID: ENST00000355072), DRPLA (ATN1, Ensembl transcript ID: ENST00000356654), SCA1 (ATXN1, Ensembl transcript ID: ENST00000244769), SCA2 (ATXN1, Ensembl transcript ID: ENST00000377611), SCA3 (ATXN3, Ensembl transcript ID: ENST00000340660), and SCA6 (CACNA1A, Ensembl transcript ID: ENST00000325084). To extract the highly conserved SSRs and verify as the potential functional motifs, users can input the Ensembl transcript ID directly or type the keywords in the query textbox under the “Comparative Genome” website. If an abbreviated keyword cannot be found, users can try the entire gene name to retrieve its corresponding transcript IDs from the specified gene set. Consequently, exactly and partially matched genes were listed in a table. Therefore, one can click on the retrieved transcript IDs and check/uncheck the checkbox of “intron” attribute before sending the query. According to the parameter settings, those retrieved SSRs will be listed in ascending order by chromosome number. To exploit potential functional SSRs of a gene, users are suggested to select SSRs in accordance with the number of conserved species. For example, from the first six genes listed in , the CAG repeats appeared in shifted or complementary patterns are considered as potential functional motifs because they were highly conserved in 12 to 22 species from CG-SSR. Indeed, these CAG repeat expansions in coding regions were demonstrated to bring about various neuronal diseases.Citation2 These findings suggest that cross-species comparison can be used to identify potential functional SSRs in both coding and noncoding regions.
After retrieving functional SSR candidates, CG-SSR provides functional genomics resources—GO, InterPro, and Pfam—to help users ascertain the potential biological function of each SSR. Using the involvement of WISP2 in signal transduction as an example (Ensembl transcript ID: ENST00000396767), information provided by GO implicates WISP2 in cell growth. Interestingly, experimental verification suggests that this A repeat indeed has tumor suppressor function.Citation4 Users can efficiently use these functional genomics resources to obtain clues about putative roles of SSRs in gene regulatory networks.
In summary, CG-SSR comprises accurate coordinate, cross-species comparison, and functional genomics resources. It is a comprehensive, efficient and user-friendly online tool for identifying conserved SSRs as potential functional motifs in vertebrates.
Supplementary information: How does CG-SSR searching algorithm work?
Definition of SSR
Simple sequence repeats (SSRs), also called microsatellites, are nucleotide segments with basic repeat pattern of 1–6 base pairs in length. The searching algorithm of CG-SSR is designed for discovering all perfect and imperfect (with tolerant) SSRs from various genomic sequences efficiently and effectively.
Algorithm description
Figure 1 depicts the flowchart of CG-SSR searching algorithms which is composed of two major phases: (I) autocorrelation and (II) overlapping adjacent phases. The first autocorrelation phase including seed finding, region growing, trimming/ recovering, refining, and threshold filtering processes discovers all possible SSR patterns as fundamental candidates. The second overlapping adjacent phase including record merging, record cutting, and threshold filtering processes verifies overlapped segments and confirms no redundant perfect/imperfect SSR patterns. Details of each procedure of these two phases are described in the following sections:
Autocorrelation phase
Autocorrelation seed finding
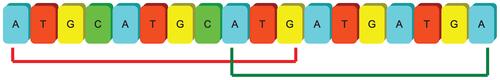
The core algorithm for discovering SSR patterns from genome sequences employs autocorrelation methodology. The basic concept assumes that the target sequence contains repeat substrings with basic patterns of length N within a range from 1 to 6. If we shift the target sequence with N nucleotides to its right and compare to its original the original sequence, all repeat patterns can be discovered since they overlapped with their shifted sequence at least within N nucleotides continuously. Based on the observation of such overlapping, we can easily detect the repeating locations by shifting and matching the whole target sequence without knowing the nucleotide contents of repeated patterns. If the continuously matched nucleotides are longer than the shifting length N, at least one repeat with the basic pattern length N is proved to exist in the target sequence. Therefore, the context of the basic pattern can be extracted directly from the target sequence where the repeats appear. The program performs frame shifting processes from length 1 to 6 base pairs to identify mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide SSRs, respectively. Figure 2 illustrates an example of autocorrelation processes for identifying seeds of repeat patterns of length N = 3. The number of continuously matched nucleotides reveals the exact length of perfect SSR repeats. However, these discontinuous perfect patterns were interrupted due to various tolerant conditions, therefore categorized as imperfect SSRs. In order to identify both perfect and imperfect SSRs, the proposed algorithms firstly considered the coordinates of perfect repeats and employed the location information as input for the following modules.
Neighboring comparison from region growing
To identify imperfect SSRs with various tolerant conditions, the proposed algorithm applied forward region growing method to enable neighboring comparison. As long as the coordinates and contents of the perfect repeat patterns were obtained, the forward region growing processes were performed by examining the right-hand-side neighboring nucleotides and skipping noise-like nucleotides which did not belong to the perfect repeat patterns. The searching processes stopped until no extra basic unit pattern could be found by continuously extending the verification on right-hand-side neighboring nucleotides.
There are three types of sequence tolerance for imperfect SSRs shown in Figure 3: insertion, deletion, and substitution. The insertion case is categorized into two different types: the inserted nucleotides located between two basic unit patterns or appeared inside a basic unit pattern. Deletions and substitutions, apparently recognized by analyzing the length and contents of insertion based on string comparison, can be considered as the insertion in different forms.
Right trimming and left recovering processes
In this module, verifications on both sides include left-hand-side trimming and right-hand-side recovering processes. Both processes are described as follows:
Right-hand-side trimming
Once an SSR sequence was allocated from seed finding and forward region growing, a trimming process was applied to examine its right-hand-side of extended SSR records for a guaranteed representation of noninterrupted pattern. This process was achieved by backward scanning from the right end of the previously searched SSR to its last noninterrupted basic pattern. Once the last perfect basic pattern was found, the following, partial, and noninterrupted patterns on its right side were recalled for its longest representation, such as the “AT” substring on the right end of the designated SSR in Figure 4a.
Left-hand-side recovering
For the left-hand-side of SSR string verification, the system examined only the length of an assigned basic pattern which could not be recovered by insertion, deletion or substitution examinations employed in the previous autocorrelation phase. An example of left-hand-side recovering process was shown in Figure 4b. The “ATG” pattern in the leftmost position could not be recognized through the first autocorrelation module, but it could be identified after the verification of left-hand-side recovering process.
Threshold filtering
Two thresholding parameters were applied to verify if an SSR record was qualified for the requirements. One was the parameter of minimum length and the other was the maximum noise rate. The first thresholding filter checked the total length of a candidate SSR record which included the repeat contents and the tolerant nucleotides. The default argument in the system was 10 nucleotides.
The second thresholding parameter inspected the noise rate of an identified SSR record. The definition of noise rate is
where LPatternLength represents the length of basic pattern; NRepeatTime represents the number of repeats of complete pattern; NIncompletePattern denotes the number of nucleotides of incomplete patterns in the SSR: LTotalLength is the total length of an identified SSR.
A perfect repeat SSR possesses “0” noise rate, and higher noise rate represents more tolerant nucleotides appeared in an identified SSR strings. The default setting is 0.2 which represents 20% of the total nucleotides recognized as noisy base pairs for the selected SSR string. Examples of noise rate calculation are shown in Figure 5.
Pattern refinement
If a basic unit pattern is a repeat sequence itself, trivially, the pattern will be identified as SSRs with different lengths during autocorrelation processes. For example, a tri-nucleotide SSR string of length 60 could be identified as well as a hexa-nucleotide SSR string of length 30 from the first phase computation. Hence, for eliminating the redundant cases of this type, all self-repeating basic patterns were double checked and reduced to its smallest unit size. In Figure 6, an example of self-repeating SSR was shown and the basic unit pattern “TG” was considered as the fundamental SSR pattern with the smallest unit size for its final representation. Therefore, the SSR string was categorized as a di-nucleotide SSR record instead of a tetra-nucleotide or a hexa-nucleotide.
To achieve robust performance of CG-SSR system, the program skipped the mono-nucleotide SSR seed-finding module. Frameshifting of one nucleotide caused overwhelming false alarm results and wasted too much time on following evaluation processes. However, the mononucleotide SSR could be retrieved by performing from frameshifting of two to six nucleotides and verified by self-repeating analysis. The mononucleotide SSRs could be successfully and efficiently identified after this refinement processes.
It was noticed that a shifting operation applied on an identified SSR sequence formulated another new repeat sequence within a different basic unit pattern. It is shown in Figure 7 as an example, “ATG”, “TGA”, and “GAT” were different basic unit patterns for three SSR sequences. However, it should be defined that these three basic unit patterns were considered as an identical pattern as long as their contexts can be exactly matched after shifting operations.
Overlapping adjacent phase
In this phase, the SSR candidates were re-inspected based on their locations and basic unit patterns. Merging and cutting operations on neighboring SSR candidates enhance the consecutive relationship and centralize the diverse representations. The proposed filtering operations are described as follows:
Record merging and cutting
On the condition of two overlapping SSR candidates, the system firstly verified the overlapped records if they possessed identical basic unit patterns. If two or more SSR records possessed an identical pattern, the recombination of such candidates were concatenated after evaluating the criterion of noise threshold requirements. On the other hand, if these overlapped SSR candidates of the same pattern length did not possess identical basic unit pattern, they usually resulted from the tolerant conditions. Hence, the system combined such two SSR candidates to make sure no entry was redundant. Finally, a threshold filter was applied again to satisfy the requirements of minimum SSR length and maximum noise rate.
It is also true that two SSR candidates of different basic unit patterns and lengths might overlap. The ambiguity usually arose on the transistion positions of two consecutive basic patterns. In Figure 8, an example of SSR transistion from a tetra-nucleotide SSR to a tri-nucleotide SSR was shown, and the concatenation resulted from the tolerant conditions between two SSR strings. In such system, two SSR strings of different lengths of basic unit patterns would be categorized and stored as two independent SSRs. Only two overlapping adjacent SSRs of the same basic pattern were merged for efficient and effective consideration.
Figure 1 The flowchart of CG-SSR searching algorithm.
Figure 2 An example of autocorrelation process.
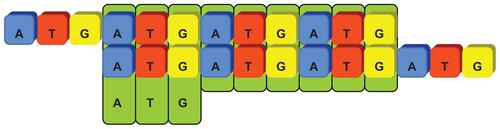
Figure 3 All tolerant cases can be considered as special cases of insertions. The substitution and deletion cases were viewed as inserting tolerant segments.
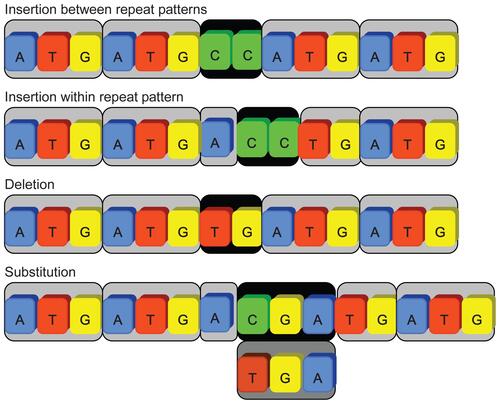
Figure 4a The right-hand-side trimming process verifies the right-hand-side of SSR to guarantee a complete, noninterrupted representation.
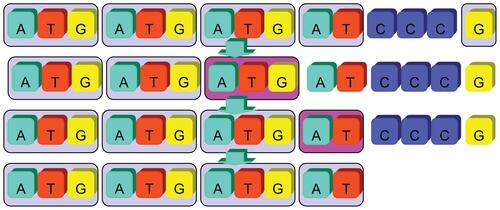
Figure 4b An example of left-hand-side recovering processes.
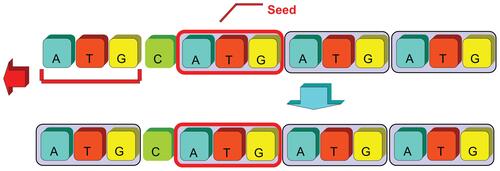
Figure 5 Examples of noise rate calculation.
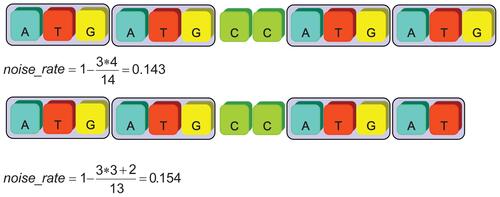
Figure 6 Self-repeating basic pattern was verified in the proposed system.
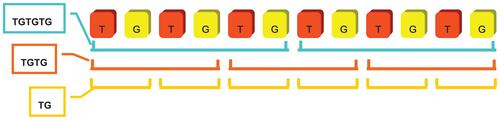
Figure 7 Different basic unit patterns due to shifting.
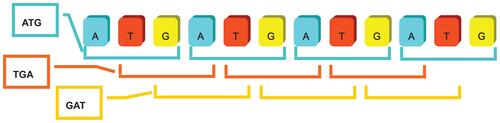
Figure 8 Ambiguity between two basic unit patterns within two different lengths.
Acknowledgments
This work was supported by the Center for Marine Bioscience and Biotechnology (CMBB) at the National Taiwan Ocean University, Keelung, Taiwan (97529002H1), and the National Science Council in Taiwan, R. O. C. (NSC96-2627- B-019-003 to T.-W. Pai).
Disclosure
This SSR is free for use and can be located at the following URL: http://cgssr.cs.ntou.edu.tw/. Supplementary information is available at the following URL: http://cgssr.cs.ntou.edu.tw/si_v2/. The authors report no conflicts of interest in this work.
References
- CharlesworthBSniegowaskiPStephanWThe evolutionary dynamics of repetitive DNA in eukaryotesNature19943712152208078581
- FondonJW3rdHammockEAHannanAJKingDGSimple sequence repeats: genetic modulators of brain function and behaviorTrends Neurosci20083132833418550185
- KashiYKingDGSimple sequence repeats as advantageous mutators in evolutionTrends Genet20062225325916567018
- LiYCKorolABFahimaTNevoEMicrosatellites within genes: structure, function, and evolutionMol Biol Evol200421991100714963101
- KennesonAZhangFHagedornCHWarrenSTReduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriersHum Mol Genet2001101449145411448936
- RanumLPWDayJWDominantly inherited, noncoding microsatellite expansion disordersCurr Opin Genet Dev20021226627112076668
- TidowNBoeckerASchmidtHDistinct amplification of an untranslated regulatory sequence in the egfr gene contributes to early steps in breast cancer developmentCancer Res2003631172117812649171
- SakaiTMiuraIYamada-IshibashiSUpdate of mouse microsatellite database of Japan (MMDBJ)Exp Anim20045315115415153678
- MissirlisPIMeadCLButlandSLSatellog: a database for the identification and prioritization of satellite repeats in disease association studiesBMC Bioinformatics 2005614515949044
- SubramanianSMadgulaVMGeorgeRMRD: a microsatellite repeats database for prokaryotic and eukaryotic genomesGenome Biol2002312 PREPRINT0011
- SubramanianSMadgulaVMGeorgeRKumarSPanditMWSinghLSSRD: simple sequence repeats database of the human genomeComp Funct Genomics2003434234518629286
- AishwaryaVGroverASharmaPCEuMicroSatdb: a database for microsatellites in the sequenced genomes of eukaryotesBMC Genomics 2007822517623061
- MarguliesEHBirneyEApproaches to comparative sequence analysis: towards a functional view of vertebrate genomesNat Rev Genet2008930331318347593
- AshburnerMBallCABlakeJAGene ontology: tool for the unification of biology. The Gene Ontology ConsortiumNat Genet200025252910802651
- MulderNJApweilerRAttwoodTKNew developments in the InterPro databaseNucleic Acids Res200735D22422817202162
- FinnRDMistryJSchuster-BöcklerBPfam: clans, web tools and servicesNucleic Acids Res200634D24725116381856
- FlicekPAkenBLBealKEnsembl 2008Nucleic Acids Res200836D70771418000006
- KarolchikDKuhnRMBaertschRThe UCSC Genome Browser Database: 2008 updateNucleic Acids Res200836D77377918086701
- EjimaYYangLSasakiMSAberrant splicing of the ATM gene associated with shortening of the intronic mononucleotide tract in human colon tumor cell lines: a novel mutation target of microsatellite instabilityInt J Cancer20008626226810738255
- ZoghbiHYOrrHTGlutamine repeats and neurodegenerationAnnu Rev Neurosci20002321723710845064
- NakamuraKJeongSYUchiharaTSCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding proteinHum Mol Genet2001101441144811448935
- MantoMUThe wide spectrum of spinocerebellar ataxias (SCAs)Cerebellum200542615895552
- YuMWYangYCYangSYHormonal markers and hepatitis B virus–related hepatocellular carcinoma risk: a nested casecontrol study among menJ Natl Cancer Inst2001931644165111698569
- YuMWYangYCYangSYAndrogen receptor exon 1 CAG repeat length and risk of hepatocellular carcinoma in womenHepatology20023615616312085360
- BraisBBouchardJPXieYGShort GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophyNat Genet1998181641679462747
- MarkowitzSWangJMyeroffLInactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instabilityScience1995268133613387761852
- ToutenhoofdSLGarciaFZachariasDAWilsonRAStrehlerEEMinimum CAG repeat in the human calmodulin-1 gene 5′ untranslated region is required for full expressionBiochim Biophys Acta199813983153209655925
- FrankePLeboyerMGänsickeMGenotype-phenotype relationship in female carriers of the premutation and full mutation of FMR-1Psychiatry Res1998801131279754690
- CummingsCJZoghbiHYTrinucleotide repeats: mechanisms and pathophysiologyAnnu Rev Genomics Hum Genet2000128132811701632
- MatsuuraTYamagataTBurgessDLLarge expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10Nat Genet20002619119411017075
- OhshimaKMonterminiLWellsRDPandolfoMInhibitory effects of expanded GAA.TTC triplet repeats from intron I of the Friedreich ataxia gene on transcription and replication in vivoJ Biol Chem199827314588145959603975
- SakamotoNOhshimaKMonterminiLPandolfoMWellsRDSticky DNA, a self-associated complex formed at long GAA*TTC repeats in intron 1 of the frataxin gene, inhibits transcriptionJ Biol Chem2001276271712717711340071