Abstract
Mucopolysaccharidosis type II (MPS II, Hunter syndrome) is a heterogeneous, progressive X-linked recessively inherited lysosomal storage disease that is caused by a deficiency of the enzyme iduronate-2-sulfatase, resulting in abnormal tissue accumulation of the glycosaminoglycans, dermatan sulfate and heparan sulfate. The disorder results from mutations in IDS, which is located at Xq28. Over 300 pathogenic mutations have been identified to date. The management of MPS II requires multidisciplinary care because of the many affected organ systems. Replacement of functional enzyme to involved tissues has been a focus of various therapies for several decades. The transplantation of hematopoietic stem cells provides enzymatic reconstitution in many target tissues, but the clinical response has been disappointing. Recently, enzyme replacement therapy with recombinant human iduronate-2-sulfatase (idursulfase, Elaprase®; Shire HGT Pharmaceuticals, Cambridge MA, USA), was approved by the in the US and Europe as a safe and effective treatment for individuals with MPS II. This review presents a comprehensive overview of MPS II and summarizes the recent literature on therapy for the disease.
Introduction
The mucopolysaccharidoses are a group of lysosomal storage diseases characterized by a deficiency of enzymes responsible for the stepwise degradation of glycosaminoglycans. With the exception of mucopolysaccharidosis type II (MPS II; Hunter syndrome), an X-linked recessively inherited disorder, all are inherited in an autosomal recessive manner. MPS II is a heterogeneous, progressive lysosomal storage disease that results from deficiency of the enzyme, iduronate-2-sulfatase, which cleaves an O-linked sulfate from dermatan sulfate and heparan sulfate (CitationNeufeld and Muenzer 2001). Consequently, dermatan sulfate and heparan sulfate progressively accumulate in the lysosomes, leading to the phenotypic features observed in the disease. The disease occurs with an incidence of approximately 1.3 cases per 100,000 male live births in Germany and the Netherlands (CitationPoorthuis et al 1999; CitationBaehner et al 2005). Approximately two-thirds of cases exhibit central nervous system (CNS) involvement, representing more severe phenotypes of the disease (CitationWraith et al 2007, Citation2008). A few affected females have also been reported, often in association with extremely skewed X-inactivation (CitationTuschl et al 2005). The disorder results from mutations in IDS, which is located at Xq28. Over 300 mutations have been identified to date, many of which are private and associated with a significant new mutation rate in the male genome (CitationFroissart et al 2007).
Hematopoietic stem cell transplantation (HSCT) has been performed in numerous individuals with varying degrees of severity of MPS II, but limited success and significant risk. Enzyme replacement therapy (ERT) involves the intravenous administration of a purified, carbohydrate-modified, recombinant human enzyme into individuals in whom the enzyme is missing or defective. The recombinant enzyme undergoes receptor mediated cell uptake and subsequent intracellular trafficking into the lysosomes, where it carries out its specific function. ERT has been used successfully for the primary treatment of Gaucher disease, Fabry disease, and mucopolysaccharidosis types I and VI. Recombinant iduronate-2-sulfatase (idursulfase, Elaprase®; Shire HGT Pharmaceuticals, Cambridge, MA, USA) was approved in July, 2006 in the US and January, 2007 in Europe as a safe and effective treatment for individuals with MPS II.
This review presents a comprehensive overview of MPS II and summarizes the recent literature on therapy, specifically ERT, for the disease.
Clinical features
Two clinical forms of MPS II have been historically recognized, attenuated and severe, which were primarily distinguished by the presence of CNS involvement. However, MPS II is more appropriately considered a heterogeneous disorder with a broad spectrum of clinical features. Even individuals with attenuated phenotypes of the disease may experience significant morbidity and limitation of life expectancy.
Individuals with attenuated phenotypes of MPS II do not exhibit CNS impairment. A significant body of work prepared by Young and Harper in the 1980s reported a mean age of recognition of symptoms of 4.3 years, the most common presenting feature being coarse facial appearance (CitationYoung and Harper 1982; CitationYoung et al 1982). More recently, Schwartz and colleagues reported a median age of recognition of symptoms of 3.25 years in their patients with attenuated disease (CitationSchwartz et al 2007). Despite reported mean ages of death from 17 to 21.7 years in this population, survival well into adulthood is not unexpected (CitationYoung and Harper 1982; CitationYoung et al 1982; CitationSchwartz et al 2007). Conversely, individuals with severe MPS II demonstrate clinically significant and progressive CNS involvement. Symptoms associated with severe MPS II are usually recognized earlier than those the attenuated phenotype. In the earlier reviews, Young and Harper reported a mean age of recognition of symptoms of approximately 2.47 years, the most common presenting feature being developmental delay. The median age of recognition of symptoms reported by Schwartz and colleagues was only slightly earlier at 2 years (CitationYoung et al 1982; CitationYoung and Harper 1983; CitationSchwartz et al 2007). Lifespan in these patients is considerably shortened, commonly as a result of pulmonary and/or cardiac disease (CitationYoung et al 1982; CitationYoung and Harper 1983). The mean ages of death from 11.8 to 14.6 years have not changed much in 20 years (CitationYoung and Harper 1983; CitationSchwartz et al 2007).
Macrocephaly is a common finding, often becoming more apparent with age (CitationYoung et al 1982). Facial features include progressive coarsening, with frontal bossing, prominent supraorbital ridge, large nose and flat nasal bridge, widely spaced teeth, thickened gingival mucosa, and macroglossia, The body habitus is typically broadly built with a short neck, broad chest, and protuberant abdomen (CitationYoung and Harper 1982, Citation1983; CitationYoung et al 1982). The skin is thick and hirsutism is common. Individuals may exhibit numerous Mongolian spots. Skin-colored papules and nodules (sometimes termed peau d’orange) may be observed, particularly on the back, upper arms, shoulders, and thighs, and among the MPS disorders, this finding is considered characteristic for Hunter syndrome.
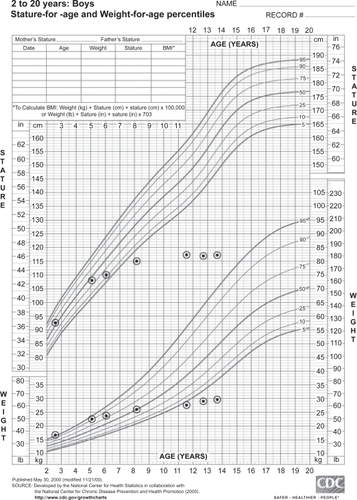
Early linear growth is typically normal to advanced, which may limit recognition of the disease. The widely recognized short stature with contractures develops relatively late when compared to features such as hepatosplenomegaly, airway disease, and developmental delays. Additionally, patients may initially be overweight for age and height ().
Figure 1 Growth chart demonstrating the typical growth pattern in patients with Hunter syndrome.
Corneal clouding, which is observed in other forms mucopolysaccharidosis, is characteristically absent in MPS II. Refraction errors are frequently identified on ophthalmologic examination (CitationSchwartz et al 2007). Progressive retinal degeneration and optic nerve disease may be observed, particularly in severely affected individuals. Papilledema may be noted these patients, even in the absence of elevated intracranial pressure, possibly a result of glycosaminoglycan accumulation within the sclera resulting in compression of the optic nerve at the intersceral level (CitationYoung and Harper 1982; CitationBeck and Cole 1984; CitationNeufeld and Muenzer 2001). Progressive, mixed conductive and sensorineural hearing loss begins in early childhood. The conductive component results from eustachian tube dysfunction, thick middle ear effusion, and effects of chronic otitis media (CitationYoung and Harper 1982; CitationYoung et al 1982; CitationYoung and Harper 1983; CitationNeufeld and Muenzer 2001).
Chronic nasal discharge and frequent upper respiratory tract infections are common findings in these patients. Progressive deposition of glycosaminoglycans in the larynx results in a characteristically hoarse, low pitched voice. Progressive accumulation of glycosaminoglycans in supraglottic tissue, tongue, tonsils, and adenoids, produces muffling of the voice, and places individuals at a significant risk of airway obstruction, particularly during sleep and perioperatively. Obstructive sleep apnea is a considerable problem for many individuals, many of whom ultimately require some form of therapeutic intervention, including tonsillectomy and/or adenoidectomy, positive pressure ventilation, or tracheostomy. Lower respiratory illnesses are more common in severely affected individuals and become progressively more severe and life threatening as the individual’s disease worsens and comorbidities arise. Additionally, rib cage rigidity and abdominal distension diminish chest excursion, further contributing to pulmonary insufficiency (CitationYoung and Harper 1982, Citation1983; CitationYoung et al 1982).
Cardiac disease presents a common source of morbidity and mortality in these patients. Accumulation of glycosaminoglycans within the cardiac valves (specifically mitral and aortic valves) results in thickening and abnormal function of the valves and impaired cardiac function. Cardiac valve replacement has been performed in these patients with success (CitationDangel 1998; CitationBhattacharya et al 2005). Additionally, progressive left ventricular hypertrophy/interventricular septal thickening may further lead to impaired cardiac function (CitationDangel 1998; CitationMohan et al 2002).
Hepatosplenomegaly is a consistent feature in MPS II. Umbilical and inguinal hernias are frequent, and may recur after surgery. Chronic diarrhea is commonly reported in severely affected individuals, and may be one of the most distressing features of the disease (CitationYoung and Harper 1983). Its etiology is unclear; however, this symptom may be the result of impaired autonomic innervation of the intestines (CitationElsner 1970).
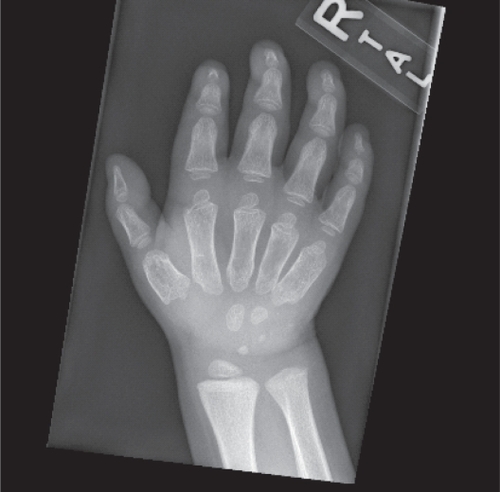
Skeletal radiographs may demonstrate dysostosis multiplex, which is characterized by macrocephaly, thickened calvarium, and J-shaped sella. The chest demonstrates wide, oar shaped ribs, and short, thickened clavicles. The spine shows anterior beaking of the lumbar vertebrae. The pelvis demonstrates wide, flared ilia, dysplastic acetabulae, small, flattened femoral heads, and coxa valga. The tubular bones exhibit thickened cortices, irregular metaphyses, and poorly developed epiphyseal centers. The phalanges are also shortened and trapezoidal in shape () (CitationSpranger et al 2002). Skeletal abnormalities also affect the jaw, leading to limited range of motion.
Figure 2 X-ray of the hand demonstrating the typical radiographic findings in patients with Hunter syndrome. The small density over the proximal hypothenar eminence in this image represents a foreign body.
A progressive, destructive arthropathy may affect all the joints, particularly the hips, eventually leading to decreased range of motion and significant disability. Physical therapy may provide some benefit in preserving joint function (CitationWraith et al 2008); however, surgical intervention may eventually be required to stabilize and/or repair the damaged joints. Progressive joint contractures may be quite debilitating. Contractures of the hands, eventually give rise to a claw-like hand deformity. Carpal tunnel syndrome is a common, albeit frequently overlooked manifestation that may become quite severe, ultimately requiring surgical intervention (CitationHaddad et al 1997). Progressive motor impairment into early adulthood has been documented using Functional Independence Measures (Kato et al 2007).
Progressive neurologic deterioration characterizes the severe phenotypes of MPS II with slow global acquisition of developmental milestones during the preschool years. Behavior problems, particularly aggressiveness and hyper-activity, may be significant and quite taxing on care givers (CitationYoung and Harper 1981). A plateau occurs around age 6, followed by progressive neurological deterioration. In the late stages of the disease, individuals are severely impaired and dependent upon caregivers for all their needs (CitationYoung and Harper 1983). Pyramidal tract disease, ie increased muscle tone, exaggerated deep tendon reflexes, and ankle clonus has been observed (CitationYoung and Harper, 1982, Citation1983; CitationYoung et al 1982). Seizure activity, reported in 18% of individuals in the Hunter Outcome Survey (HOS) registry (CitationWraith et al 2007) appears in the first decade of life, and is usually amenable to antiepileptic therapy. Communicating hydrocephalus may develop during the end of first decade of life, further contributing to the neurologic deterioration (CitationNeufeld and Muenzer 2001).
In individuals with attenuated disease, cognitive deterioration does not occur and the prevalence of neurologic manifestations is decreased compared to severely affected individuals. Nonetheless, abnormal MRI findings, including volume loss and white matter lesions, are seen in individuals with the attenuated form (CitationParsons et al 1996). Individuals are expected to have normal intelligence to mild delays. However, certain medical comorbities, ie, deafness and decreased joint mobility, may contribute to problems in school. The behavioral problems observed in severely affected individuals do not occur; however, these individuals may have psychological problems related to their medical comorbidities and adjustment to their disease (CitationYoung and Harper 1981).
Spinal cord compression is one of the most serious complications of MPS II, and can lead to permanent neurologic damage or death, if not detected and treated early. This may result from foramen magnum and vertebral canal stenosis, meningeal thickening from glycosaminoglycan accumulation, and less commonly from atlanto-axial subluxation (CitationParsons et al 1996; CitationO’Brien et al 1997).
The work of Young and Harper (CitationYoung and Harper 1981, Citation1982, Citation1983; CitationYoung et al 1982) and others has contributed significantly to our understanding of the clinical features of MPS II. However, these studies were conducted prior to the availability of modern supportive and definitive medical care. Comprehensive studies addressing the effectiveness of newer medical techniques in treating patients with MPS II are only beginning to appear in the literature. It is anticipated that rare disease registries such as the Hunter Outcome Survey, sponsored by Shire HGT Pharmaceuticals, will provide an opportunity to assess both the natural history and effect of interventions on patients currently cared for in participating centers. Preliminary findings from the HOS as well as a similar review of the clinical manifestations and treatment of MPS II were recently reported by Wraith and colleagues (CitationWraith et al 2008).
Molecular
Iduronate-2-sulfatase, a member of the sulfatase family, catalyzes the removal of the sulfate group from the 2- position of L-iduronic acid in dermatan sulfate and heparan sulfate in lysosomes (CitationNeufeld and Muenzer 2001). IDS, which is located at Xq28, spans approximately 24 kb, consists of 9 exons and 8 introns, and has a coding sequence of 2.3 kb. A pseudogene, IDS2, located approximately 20 kb distal to the gene, contains sequences homologous to exon II, exon III, intron 2, and a chimeric intron3-intron7 (CitationFroissart et al 2002). Its exact function is unclear; however, pathogenic structural rearrangements between IDS2 and IDS have been identified in individuals with MPS II. Sequence analysis of human IDS cDNA predicts the translation of a 550 amino acid precursor peptide, including a 25 amino acid amino-terminal signal sequence and eight additional amino acids that are removed from the proprotein (CitationWilson et al 1990).
IDS has a strong sequence homology to other human sulfatases, including arylsulfatases A, B, and C, and glucosamine-6-sulfatase (CitationWilson et al 1990). All members of the sulfatase family undergo post-translational modification in the endoplasmic reticulum during which a conserved cysteine residue located within the active site is converted into Cα-formylglycine by formylglycine-generating enzyme (FGE). Cα-formylglycine is essential for catalytic activity of the sulfatases. Mutations in SUMF1, which codes for FGE, results in severely reduced activity of the sulfatases, leading to multiple sulfatase deficiency, an autosomal recessive disorder, the phenotype of which combines features of disorders resulting from individual sulfatase deficiencies (CitationDierks et al 2003).
The Human Gene Mutation Database (HGMD) presently documents 345 disease causing mutations in IDS (CitationKrawczak and Cooper 1997; CitationStenson et al 2003), many of which are private. They include missense, nonsense, splice site mutations, small insertions and deletions, partial and whole gene deletions, and large rearrangements (CitationStenson et al 2003). Approximately 12% of cases arise de novo from maternal meiosis (CitationRathmann et al 1996; CitationVafiadaki et al 1998). Approximately 80% of all cases of MPS II result from small deletions, insertions, or single base substitutions, while the remaining 20% of cases result from large scale deletions in IDS or structural rearrangements (CitationHopwood et al 1993; CitationFroissart et al 2007). Point mutations account for approximately 50% of all mutations, and tend occur more frequently in exons III, VIII, and IX, particularly at CpG sites (CitationRathmann et al 1996).
The identification of genotype-phenotype correlations in MPS II has been challenging for several reasons. Measured IDS activity in plasma or circulating cells does not differentiate between severe or attenuated forms, suggesting that the differences in disease threshold fall below the range of measurable enzyme activity. There is currently no standardized severity scoring system, although these tools are being developed as therapeutic options are brought into clinical application. There is broad degree of genotypic heterogeneity, and a significant number of private mutations exist in this disease (CitationFroissart et al 2002, Citation2007). Despite these limitations, certain genotype/phenotype correlations have been described. Large gene alterations (deletions and rearrangements) are typically associated with severe disease phenotypes because of a complete absence of functional enzyme. Other genes, including FMR-1 may also be deleted, further contributing to the severe phenotypes, particularly development of mental retardation (CitationFroissart et al 2007).
Splice site mutations affecting consensus sequences, small deletions or insertions causing frameshifts, and nonsense mutations are generally associated with intermediate and severe phenotypes (CitationHopwood et al 1993; CitationFroissart et al 1998, Citation2002; CitationVafiadaki et al 1998). However, there are exceptions: The nonsense mutation Q513X is associated with an attenuated phenotype because the encoded amino acid product lacks 19 C-terminal amino acids normally removed during protein maturation (CitationFroissart et al 2002). The effects of splice site mutations are variable, depending on the location and nature of the mutation and presence of normal transcripts. Affected individuals may produce the normal gene transcript in sufficient, albeit low, levels to avoid developing a severe phenotype.
The effects of missense mutations and small in-frame deletions and insertions are often difficult to predict, and expression studies are sometimes needed to differentiate a polymorphism from a disease-causing mutation (CitationFroissart et al 2002). Missense mutations that do not significantly impact protein folding and/or enzyme activity are predicted to result in attenuated phenotypes. Conversely, missense mutations affecting sequences highly conserved among the sulfatases generally result in a severe disease phenotype (CitationFroissart et al 1998).
Animal studies
A knockout mouse model of MPS II (ids y/-) has been developed by replacing exon 4 and a portion of exon 5 of IDS with the neomycin resistance gene (CitationMuenzer et al 2002; CitationGarcia et al 2007b). Affected mice exhibit many of the features observed in humans, including elevated urine glycosaminoglycan excretion, accumulation of glycosaminoglycans in tissues and organs, and deficient iduronate-2-sulfatase enzyme activity. Clinical features include hepatosplenomegaly, progressive skeletal abnormalities, and premature death (CitationGarcia et al 2007b).
Garcia and colleagues studied the effects of idursulfase on the IdS-KO (ids y/-) mouse model (CitationGarcia et al 2007a). Idursulfase infused at various doses (0.1, 0.25, 0.5, and 1.0 mg/kg) and dosing frequencies (once, weekly, every other week, and monthly), resulted in a reduction in urine and tissue glycosaminoglycan levels. Additionally, liver weight was significantly reduced after 24 weeks of therapy in mice treated with 1 mg idursulfase weekly and every other week. This study provided further proof of the principle that some histological and biochemical features of MPS II can be effectively reversed with enzyme replacement therapy.
A gene therapy technique using the AAV2/8 vector to selectively deliver human IDS cDNA to the liver in the ids y/- mouse model resulted in a restoration of plasma and tissue IDS activity in the treated mice (CitationCardone et al 2006). Consequently, normalization of urine glycosaminoglycan levels and clearance of glycosaminoglycan accumulation within the visceral tissues was observed. Corrections of the skeletal and locomotor defects were also observed in treated mice. Interestingly, partial clearance of glycosaminoglycans and improved IDS activity in the CNS were also reported, including uptake of human IDS into the lysosomes of the cortex and cerebellum. These results suggest that significant overexpression of IDS in peripheral (liver) tissue may allow penetration of IDS at low levels past the blood brain barrier.
Friso and colleagues (CitationFriso et al 2005) evaluated an alternate delivery system by intraperitoneally implanting alginate microcapsules containing C2C12 murine myoblasts overexpressing IDS into ids y/- mice. Detectable levels of IDS in plasma and tissues of implanted ids y/- mice were found at 3 days and 2 weeks, respectively and persisting at 8 weeks of therapy. Urine glycosaminoglycan excretion was significantly decreased in implanted knockout mice between weeks 4 and 6 of therapy. Additionally, a significant decrease in tissue glycosaminoglycan accumulation was noted biochemically in the kidney and spleen after 8 weeks of therapy. Decreased tissue glycosaminoglycan accumulation was also apparent histologically in the liver, spleen, and kidney after 8 weeks of therapy.
Therapy
The management of MPS II requires lifelong attention to the multisystemic involvement by a team of specialists experienced in the management of this disease since none of the therapeutic options currently available result in complete resolution of morbidity.
HSCT has been successful in improving certain disease manifestations in patients with mucopolysaccharidosis type I (MPS I, Hurler syndrome), including visceral manifestations and attenuation of neurologic disease progression when performed early (ie, less than 2 years of age), and is considered a mainstay in the treatment of severe MPS I. HSCT has been performed in numerous individuals with MPS II. In some cases, individuals demonstrate improvement of the visceral and skeletal manifestations of the disease, ie, decreased urine glycosaminoglycan excretion, decreased liver and spleen volumes, diminished facial coarsening, improved respiratory function, and increased joint mobility (CitationCoppa et al 1995; CitationMcKinnis et al 1996; CitationMullen et al 2000; CitationFroissart et al 2002). However, HSCT does not appear to mitigate the progressive neurodegenerative disease often present in MPS II (CitationCoppa et al 1995; CitationShapiro et al 1995; CitationMcKinnis et al 1996; CitationVellodi et al 1999). Additionally, this technique is associated with a significant risk of morbidity and mortality. For these reasons, and with the advent of enzyme replacement therapy (discussed below), HSCT has limited, if any, utility in the treatment of MPS II.
Elaprase® (idursulfase; Shire HGT Pharmaceuticals, Cambridge, MA) was approved in the US in July, 2006 and Europe in January, 2007 for the treatment of individuals with MPS II at a recommended dose of 0.5 mg/kg administered once weekly as an intravenous infusion. Idursulfase is a purified form of iduronate-2-sulfatase produced in a human cell line via recombinant DNA technology. The 525 amino acid glycoprotein contains 8 N-linked glycosylation sites that are occupied by 2 bis mannose-6-phosphate containing oligosaccharide chains (CitationMuenzer et al 2006). These mannose-6-phosphate (M6P) chains bind to M6P receptors on the cell surface, allowing for subsequent internalization and trafficking to the lysosomes, where the enzyme is catalytically active. The enzyme activity of idursulfase is dependent upon the post-translational modification of cysteine to Cα-formylglycine within the enzyme’s active site. This modification was demonstrated in 50% of the molecules of idursulfase (CitationMuenzer et al 2006). Idursulfase also contains complex, highly sialylated glycans that prolong the circulating half-life of the drug (CitationMuenzer et al 2006).
The safety and efficacy of idursulfase was evaluated in a randomized, double-blinded, placebo controlled phase I/II clinical trial (CitationMuenzer et al 2007). In this study, 12 participants, between the ages of 6 and 20, who met clinical and biochemical criteria for MPS II and were able to cooperate with all study procedures (which excluded severely affected MPS II patients), received idursulfase at doses of 0.15, 0.5, or 1.5 mg/kg or placebo infused once per week over a period of 24 weeks, followed by a 6-month open-label extension study in which all participants received idursulfase. Clinical endpoints were evaluated after 48 weeks of therapy with active agent and compared with the three patients observed over 24 weeks of placebo infusions.
Urine glycosaminoglycan excretion was reduced within 2 weeks of initiating idursulfase, and continued to be significantly reduced after 48 weeks of therapy (p < 0.0001). A majority of patients experienced reductions of urine GAGs to near normal levels, while two individuals achieved urine GAGs within the normal range after 6 months. Liver and spleen volumes were significantly decreased in the pooled study population after 24 and 48 weeks of therapy (p < 0.01 and p < 0.001, respectively). Sixty-six percent of individuals with baseline hepatomegaly and all individuals with baseline splenomegaly experienced a normalization of organ volumes. Finally, after 48 weeks of therapy, average walking distance of the combined groups increased by an average of 48 m (p = 0.013).
In a follow-up phase II/III clinical study, the safety and efficacy of idursulfase was evaluated in 96 patients, ages 5–31, with MPS II (CitationMuenzer et al 2006). In this study design, patients were randomized to receive placebo or idursulfase 0.5 mg/kg weekly or every other week, for 53 weeks, followed by an open-label extension. Participants met clinical and biochemical criteria for MPS II, were able to cooperate with all study procedures (excluding severely affected MPS II patients), and had a baseline forced vital capacity (FVC) less than 80% of predicted. The primary endpoint of the study was a 2-component composite score of %FVC and the 6-minute walk test.
After 53 weeks of therapy, patients in the weekly and every other week idursulfase groups exhibited significant improvement in the 2-component composite endpoint compared to placebo (p = 0.0049 and p = 0.0416, respectively). However, there was no difference between the two idursulfase treatment groups. The weekly idursulfase treatment group experienced a 37 m increase in distance walked in the 6-minute walk test (P = 0.013) compared to placebo. Additionally, the weekly idursulfase treatment group experienced a 2.7% increase in predicted %FVC (p = 0.065) and a 160 mL increase in absolute FVC (p = 0.001) compared to placebo at 53 weeks.
After 53 weeks of therapy, urine glycosaminoglycan levels were significantly decreased in both idursulfase treatment groups compared to placebo (p < 0.0001). After 53 weeks of therapy, 40.6% of individuals in both idursulfase treatment groups experienced normalization of urine GAG levels, while urine GAG levels approached the upper end of normal in a majority of the remaining patients treated with idursulfase. Liver and spleen volumes diminished significantly from baseline in both idursulfase treatment groups, compared with placebo (p < 0.0001). Approximately 80% of individuals with baseline hepatomegaly who received treatment with idursulfase (weekly and every other week) had normal liver volumes after 18 and 53 weeks of therapy.
Idursulfase was generally well tolerated in the phase I/II and phase II/III clinical trials. The most frequently reported adverse events included fever, headache, cough, pharyngitis, upper respiratory tract infection, nasal congestion, nausea, vomiting, abdominal pain, and diarrhea. Of the adverse events considered possibly related to idursulfase therapy, infusion-related events were the most common. The incidence of infusion-related events was greatest between weeks 4 and 12 and decreased subsequently. These events were generally managed by stopping the initial infusion and premedicating with antihistamines and/or corticosteroids prior to subsequent infusions. If a reaction occurred during subsequent infusions, the infusion was stopped and restarted after the reaction resolved. Two deaths occurred during the phase II/III study, neither of which was considered to be related to idursulfase therapy.
IgG antibodies to idursulfase were detected in approximately 50% of patients, the highest prevalence of which occurred after 27 weeks of therapy (44.4% of patients). However, only 31.7% of patients in the idursulfase treatment groups remained antibody positive after 53 weeks of therapy. Although the presence of IgG anti-idursulfase antibodies was associated with an approximately two thirds reduction in urine glycosaminoglycan excretion in the phase II/III clinical trial, there was reportedly no association between the presence of antibodies and adverse events or clinical assessments. IgE antibodies to idursulfase were not detected in either study.
Patients receiving ERT should be monitored carefully during the infusion and for a period afterwards for an infusion related reaction to idursulfase. Additionally, easy access to emergency equipment/medications by skilled providers should be available. Initial infusion-related reactions are usually managed by discontinuing the infusion and providing antihistamines and/or corticosteroids. Subsequent infusions may be managed by slowing the infusion rate and pre-treating with antihistamines and/or corticosteroids. If a subsequent reaction occurs, the infusion should be stopped and appropriate treatment provided. Depending upon the severity of the reaction, the infusion may be restarted upon resolution of the reaction.
Patients with compromised respiratory function or acute respiratory disease may be at an increased risk of experiencing a severe infusion-related reaction. Recently, two patients experienced late-emergent anaphylactoid reactions approximately 24 hours after having experienced initial anaphylactic reactions during enzyme infusions. Thus, prolonged observation times are indicated in certain populations (Elaprase® package insert).
Idursulfase has been studied only in patients with attenuated disease. Consequently, the effects of idursulfase on CNS disease have not been formally evaluated. However, idursulfase does not cross the blood–brain barrier; thus it is not expected to have a significant effect on the neurological features of MPS II. Whether ERT should be offered to individuals with CNS manifestations is unclear. Wraith and colleagues suggest that patients with severe CNS manifestations be offered ERT for a trial period of 12–18 months, after which time a decision is made whether to continue with the therapy (CitationWraith et al 2008).
Idursulfase has not been studied in patients younger than 5 years of age. Many patients under 5 years are currently being treated and there is no reason to think that ERT would be less effective for the visceral manifestations in these young patients. Evaluation of the long term efficacy and safety of idursulfase in these populations are needed.
While ERT is expected to provide a benefit to patients receiving the therapy, it is not without a significant financial and emotional burden. ERT using idursulfase is expensive, with an estimated average annual cost of US$491,999.04 in a 30 kg child, based upon the US average wholesale price obtained from the Red Book Database (http://micromedex.com/products/redbook/database/). However, the actual cost of ERT will vary depending upon the individual’s weight, and amount of drug prescribed. Additionally, other fees, including a markup on the drug from the hospital, private office and infusion center, and for supplies may be incurred. Insurance coverage may present a significant issue for patients. For example, premiums may become unaffordable for individuals with private insurance coverage. Additionally, the lifetime maximums in many policies will be exceeded in several years given the expense of the drug and other costs. Shire HGT Pharmaceuticals has developed a Patient Assistance Program that offers assistance to qualified patients on a case by case basis, primarily based on financial need. Private organizations, eg, the National Organization for Rare Disorders, also have programs to assist families with financial costs related to ERT and healthcare coverage (CitationBurrow et al 2007).
ERT infusions in a hospital/infusion center environment frequently require patients and their families to take time off of work/school to receive treatments, further contributing to their emotional/financial burden. In patients who have previously tolerated infusions without reactions, or mild reactions easily treated with antipyretics/antihistamines and/or slowed infusion rates, transferring patients to receiving infusions in the home setting is a plausible option (CitationWraith et al 2008). However, this transition should be performed with consideration given these patients’ complex medical histories, the “black box warning” on the label, and recent reports of late-emergent anaphylactoid reactions in patients treated with idursulfase. In patients who are transitioned to home infusions, a skilled home care nurse should be present and easy access to emergency equipment/medications in case of reactions should be available.
Alternative methods aimed at directly targeting enzyme to the CNS, ie, intrathecally, have been previously evaluated in MPS I animal models (CitationDickson et al 2007). A study evaluating the safety and efficacy of intrathecal ERT in the treatment of certain spinal cord manifestations of MPS I is currently recruiting participants. Intrathecal trials of ERT for MPS II are planned. However, the differences between the cognitive and behavioral manifestations in MPS I and II, and the difference in response to HSCT are reminders that a therapy with efficacy in one of the MPS disorders may have different outcomes in another.
Newer treatment modalities, ie, pharmacological chaperone and substrate reduction therapy, are being developed for patients with other lysosomal storage diseases, including Gaucher disease (CitationCox et al 2000; CitationElstein et al 2004; CitationYu et al 2007a, Citationb). Pharmacological chaperone therapy is a therapeutic approach in which small molecules selectively bind to and stabilize the structure of target proteins in cells, allowing for improved folding and trafficking to the lysosomes and increased protein activity. Substrate reduction therapy is a therapeutic approach in which in which a pharmacologic agent decreases the production of the molecule that typically accumulates within the cells in individuals with a lysosomal storage disease so that the patient’s residual enzyme may catabolize the accumulated material. There are no current clinical trials examining these therapies in MPS II, and the prospects for chaperone therapy are low if there is no residual enzyme to protect.
Gene therapy offers significant potential for the treatment/cure of genetic diseases such as MPS II and several attempts to express iduronate sulfatase through gene therapy approaches have been investigated (CitationStroncek et al 1999; CitationHong et al 2003). Although gene therapy remains a viable potential option, safety issues and problems with robust expression in vivo remain barriers to its clinical application.
Conclusions
MPS II is a heterogeneous, progressive X-linked lysosomal storage disease, the management of which requires lifelong attention to the multisystem involvement, Recent clinical trials have demonstrated that enzyme replacement therapy with intravenous idursulfase is a safe and effective treatment modality for the somatic, but likely not the CNS manifestations of MPS II. As with other lysosomal storage disorders, enzyme replacement therapy represents a treatment rather than a cure for this disease; long term studies evaluating the efficacy of this therapy are indicated.
Approaches which are likely to ameliorate the progressive CNS manifestations in children with severe MPS II are still in preclinical stages, and will be a needed addition to the therapeutic armamentarium for this condition.
It is anticipated that rare disease registries such as the HOS will provide a mechanism through which experience beyond clinical trials can be documented and made available to centers which care for these patients.
Disclosures
Neither author has any conflicts of interest to disclose.
References
- BaehnerFSchmiedeskampCKrummenauerF2005Cumulative incidence rates of the mucopolysaccharidoses in GermanyJ Inherit Metab Dis281011716435194
- BeckMColeG1984Disc oedema in association with Hunter’s syndrome: ocular histopathological findingsBr J Ophthalmol6859046430340
- BhattacharyaKGibsonSCPathiVL2005Mitral valve replacement for mitral stenosis secondary to Hunter’s syndromeAnn Thorac Surg801911216242483
- BurrowTAHopkinRJLeslieND2007Enzyme reconstitution/replacement therapy for lysosomal storage diseasesCurr Opin Pediatr196283518025928
- CardoneMPolitoVAPepeS2006Correction of Hunter syndrome in the MPSII mouse model by AAV2/8-mediated gene deliveryHum Mol Genet1512253616505002
- CoppaGVGabrielliOZampiniL1995Bone marrow transplantation in Hunter syndromeJ Inherit Metab Dis189127623456
- CoxTLachmannRHollakC2000Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin OGT 918. to decrease substrate biosynthesisLancet3551481510801168
- DangelJH1998Cardiovascular changes in children with mucopolysaccharide storage diseases and related disorders – clinical and echocardiographic findings in 64 patientsEur J Pediatr15753489686810
- DicksonPMcEnteeMVoglerC2007Intrathecal enzyme replacement therapy: successful treatment of brain disease via the cerebrospinal fluidMol Genet Metab9161817321776
- DierksTSchmidtBBorissenkoLV2003Multiple sulfatase deficiency is caused by mutations in the gene encoding the human Calpha.-formylglycine generating enzymeCell1134354412757705
- ElsnerB1970Ultrastructure of the rectal wall in Hunter’s syndromeGastroenterology58856624246486
- ElsteinDHollakCAertsJM2004Sustained therapeutic effects of oral miglustat Zavesca, N-butyldeoxynojirimycin, OGT 918. in type I Gaucher diseaseJ Inherit Metab Dis277576615505381
- FrisoATomaninRAlbaS2005Reduction of GAG storage in MPS II mouse model following implantation of encapsulated recombinant myoblastsJ Gene Med714829115966019
- FroissartRDa SilvaIMMaireI2007Mucopolysaccharidosis type II: an update on mutation spectrumActa Paediatr Suppl9671717391447
- FroissartRMaireIMillatG1998Identification of iduronate sulfatase gene alterations in 70 unrelated Hunter patientsClin Genet5336289660053
- FroissartRMoreira Da SilvaIGuffonN2002Mucopolysaccharidosis type II – genotype/phenotype aspectsActa Paediatr Suppl9182712572848
- GarciaARDacostaJMPanJ2007aPreclinical dose ranging studies for enzyme replacement therapy with idursulfase in a knock-out mouse model of MPS IIMol Genet Metab911839017459751
- GarciaARPanJLamsaJC2007bThe characterization of a murine model of mucopolysaccharidosis II Hunter syndromeJ Inherit Metab Dis309243417876721
- HaddadFSJonesDHVellodiA1997Carpal tunnel syndrome in the mucopolysaccharidoses and mucolipidosesJ Bone Joint Surg Br79576829250742
- HongYYuSSKimJM2003Construction of a high efficiency retroviral vector for gene therapy of Hunter’s syndromeJ Gene Med5182912516048
- HopwoodJJBungeSMorrisCP1993Molecular basis of mucopolysaccharidosis type II: mutations in the iduronate-2-sulphatase geneHum Mutat2435428111411
- KrawczakMCooperDN1997The human gene mutation databaseTrends Genet1312129066272
- McKinnisEJSulzbacherSRutledgeJC1996Bone marrow transplantation in Hunter syndromeJ Pediatr12914588757575
- MohanURHayAAClearyMA2002Cardiovascular changes in children with mucopolysaccharide disordersActa Paediatr9179980412200906
- MuenzerJGucsavas-CalikogluMMcCandlessSE2007A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II Hunter syndromeMol Genet Metab903293717185020
- MuenzerJLamsaJCGarciaA2002Enzyme replacement therapy in mucopolysaccharidosis type II Hunter syndrome.: a preliminary reportActa Paediatr Suppl9198912572850
- MuenzerJWraithJEBeckM2006A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II Hunter syndromeGenet Med84657316912578
- MullenCAThompsonJNRichardLA2000Unrelated umbilical cord blood transplantation in infancy for mucopolysaccharidosis type IIB Hunter syndrome. complicated by autoimmune hemolytic anemiaBone Marrow Transplant251093710828871
- NeufeldEFMuenzerJ2001The mucopolysaccharidosesScriverCRBeaudetAlSlyWSThe metabolic and molecular basis of inherited disease8th edNew YorkMcGraw-Hill
- O’BrienDPCowieRAWraithJE1997Cervical decompression in mild mucopolysaccharidosis type II Hunter syndromeChilds Nerv Syst1387909105743
- ParsonsVJHughesDGWraithJE1996Magnetic resonance imaging of the brain, neck and cervical spine in mild Hunter’s syndrome mucopolysaccharidoses type IIClin Radiol51719238893643
- PoorthuisBJWeversRAKleijerWJ1999The frequency of lysosomal storage diseases in The NetherlandsHum Genet105151610480370
- RathmannMBungeSBeckM1996Mucopolysaccharidosis type II Hunter syndrome: mutation “hot spots” in the iduronate-2-sulfatase geneAm J Hum Genet59120298940265
- SchwartzIVRibeiroMGMotaJG2007A clinical study of 77 patients with mucopolysaccharidosis type IIActa Paediatr Suppl96637017391446
- ShapiroEGLockmanLABalthazorM1995Neuropsychological outcomes of several storage diseases with and without bone marrow transplantationJ Inherit Metab Dis18413297494400
- SprangerJWBrillPWPoznanskiA2002Bone dysplasias: An atlas of genetic disorders of skeletal developmentNew York, NYOxford University Press
- StensonPDBallEVMortM2003Human Gene Mutation Database HGMD.: 2003 updateHum Mutat215778112754702
- StroncekDFHubelAShankarRA1999Retroviral transduction and expansion of peripheral blood lymphocytes for the treatment of mucopolysaccharidosis type II, Hunter’s syndromeTransfusion393435010220258
- TuschlKGalAPaschkeE2005Mucopolysaccharidosis type II in females: case report and review of literaturePediatr Neurol32270215797184
- VafiadakiECooperAHeptinstallLE1998Mutation analysis in 57 unrelated patients with MPS II Hunter’s diseaseArch Dis Child79237419875019
- VellodiAYoungECooperA1999Long-term follow-up following bone marrow transplantation for Hunter diseaseJ Inherit Metab Dis226384810399096
- WilsonPJMorrisCPAnsonDS1990Hunter syndrome: isolation of an iduronate-2-sulfatase cDNA clone and analysis of patient DNAProc Natl Acad Sci USA87853152122463
- WraithJEBurtonBKMuenzerJ2007Clinical characteristics of patients with mucopolysaccharidosis type II: The Hunter Outcome Survey HOS. [abstract 1488]Presented at the annual meeting of The American Society of Human GeneticsOctober 25, 2007San Diego, CaliforniaURL: http://www.ashg.org/genetics/ashg06s/index.shtml
- WraithJEScarpaMBeckM2008Mucopolysaccharidosis type II Hunter syndrome.: a clinical review and recommendations for treatment in the era of enzyme replacement therapyEur J Pediatr1672677718038146
- YoungIDHarperPS1981Psychosocial problems in Hunter’s syndromeChild Care Health Dev720196793262
- YoungIDHarperPS1982Mild form of Hunter’s syndrome: clinical delineation based on 31 casesArch Dis Child57828366816147
- YoungIDHarperPS1983The natural history of the severe form of Hunter’s syndrome: a study based on 52 casesDev Med Child Neurol2548196413286
- YoungIDHarperPSNewcombeRG1982A clinical and genetic study of Hunter’s syndrome. 2. Differences between the mild and severe formsJ Med Genet19408116818348
- YuZSawkarARKellyJW2007aPharmacologic chaperoning as a strategy to treat Gaucher diseaseFebs J27449445017894779
- YuZSawkarARWhalenLJ2007bIsofagomine- and 2,5-anhydro-2,5-imino-D-glucitol-based glucocerebrosidase pharmacological chaperones for Gaucher disease interventionJ Med Chem509410017201413