Abstract
Alpha-1 antitrypsin (A1AT) is a 52 kDa serine protease inhibitor that is synthesized in and secreted from the liver. Although it is present in all tissues in the body the present consensus is that its main role is to inhibit neutrophil elastase in the lung. A1AT deficiency occurs due to mutations of the A1AT gene that reduce serum A1AT levels to <35% of normal. The most clinically significant form of A1AT deficiency is caused by the Z mutation (Glu342Lys). ZA1AT polymerizes in the endoplasmic reticulum of liver cells and the resulting accumulation of the mutant protein can lead to liver disease, while the reduction in circulating A1AT can result in lung disease including early onset emphysema. There is currently no available treatment for the liver disease other than transplantation and therapies for the lung manifestations of the disease remain limited. Gene therapy is an evolving field which may be of use as a treatment for A1AT deficiency. As the liver disease associated with A1AT deficiency may represent a gain of function possible gene therapies for this condition include the use of ribozymes, peptide nucleic acids (PNAs) and RNA interference (RNAi), which by decreasing the amount of aberrant protein in cells may impact on the pathogenesis of the condition.
Introduction
Alpha-1 antitrypsin (A1AT) is an acute phase 52kDa 418 amino acid glycoprotein that is primarily synthesized and secreted by the hepatocytes in the liverCitation1 though it is also actively transcribed and secreted in smaller amounts by cells including neutrophils, mononuclear phagocytes, enterocytes,Citation2 and human respiratory epithelial cells.Citation3
It is present in all tissues of the body with its primary role being to inhibit the enzyme neutrophil elastase (NE).Citation4 It also inhibits to a much lesser degree trypsin, chymotrypsin, cathepsin G, plasmin, thrombin, tissue kallikrein, factor Xa, plasminogen, and proteinase 3 ().Citation5
Table 1 The A1AT alleles and their effects
A1AT has other pathobiologically relevant functions in addition to elastase inhibition. There is a direct pro-survival effect of A1AT through inhibition of lung alveolar endothelial cell apoptosis. Primary pulmonary endothelial cells internalize human A1AT which co-localizes with and inhibits staurosporine-induced caspase-3 activation. A1AT is among several serum proteins that can rescue serum withdrawal-induced apoptosis.Citation6 Inhibition of structural alveolar cell apoptosis by A1AT represents a novel protective mechanism of this serpin against emphysema.Citation6
A1AT has also been reported to play an immunoregulatory role, to inhibit neutrophil superoxide production, to induce the release of macrophage-derived interleukin-1 (IL-1) receptor agonist, and to increase hepatocyte growth factor production in human lung fibroblasts in vitro. In vivo A1AT has been shown to protect against tumor necrosis factor-α (TNFα) or endotoxin-induced lethality and in a mouse model of lung inflammation and connective tissue breakdown. It was shown that A1AT binds to the secreted enteropathogenic Escherichia coli proteins EspB and EspD thereby reducing their hemolysis of red blood cells. Thus A1AT may not only afford protection against proteolytic injury, but may also have the potential to neutralize microbial activities and to exert effects on the regulation of innate immunity. In this regard there is growing evidence that A1AT is also able to inhibit lipopolysaccharides (LPS)-induced inflammatory responses in vitro and in vivo.Citation7
A1AT is a member of the serine proteinase inhibitor (Serpin) family of proteins. This family also includes α-1 antichymotrypsin, α-2 antiplasmin, plasminogen activator I, thyroxine-binding globulin and angiotensin, among others. Serpins are single chain proteins containing a conserved domain structure of 370–390 residues, usually flanked by amino or carboxyl terminal extensions. The serpins exist in a metastable state and their energy is released by cleavage of their reactive centre. A target protease binds to and cleaves the reactive centre of the serpin which then transposes the protease to the other end of the serpin molecule. In the process the protease is crushed against the serpin, with a loss of structural integrity that ensures its destruction.Citation8
A crucial factor in the destruction is the strength of the bond between serpin and protease.Citation8 Inhibitory serpins interact with their target proteinase at the reactive site which is located within a loop structure 30–40 amino acids from the carboxyl terminus.Citation9
The specificity of a serpin for its protease is determined by the side chain of the amino acid in the reactive centre of the serpin. The mature A1AT protein consists of a single chain of amino acids with three N-asparaginyl-linked complex carbohydrate side chains.Citation5 As already stated the target protein of A1AT is NE. A1AT has a methionine at its reactive centre which enables it to inhibit NE as NE cleaves at methionyl residues.Citation8 The predominant feature of cleaved, inactive serpins is the presence of an anti-parallel six-stranded β-sheet (the A sheet) in which the central strand contains several residues upstream from the cleavage site.Citation9
With 80 million neutrophils from the pulmonary arterial circulation travelling through human lungs every second, the pulmonary parenchymal stroma is exposed to an array of proteolytic enzymes including proteinase-3, cathespin G and NE. Of these NE is thought to be the most important in the development of pulmonary emphysema. It is a 29 kDa, 220 amino acid, single chain polypeptide glycoprotein synthesized in promyelocytes and packaged in neutrophil azurophilic granules. NE is released in large quantities in response to neutrophil activationCitation10 and is one of the few human enzymes capable of cleaving insoluble, cross-linked elastin which modulates the elastic recoil of tissues, including the alveolar walls of the lower respiratory tract.Citation5 After an inflammatory event, in normal circumstances, the NE is inhibited and controlled by A1AT.
Genetics of A1AT deficiency
The A1AT gene is located on the q arm of chromosome 14 at position 14q32.1.Citation11,Citation12 It is a 12.2-kilobase pair gene composed of seven exons and six introns. The protein is encoded by the protease inhibitor (Pi) locus. The Pi locus is highly polymorphic, resulting in different A1AT isotypes that can be detected by electrophoresis.Citation13
There are many normal A1AT alleles, and at least four common ‘M’ alleles. The many different alleles can be either normal, null (express no A1AT), dysfunctional (nonfunctional A1AT is expressed in the serum) or deficient (serum A1AT levels <35% of normal). More than 100 different allelic variations of A1AT have been described. Over 95% of the population has one of the four M alleles.Citation14
The numerous A1AT phenotypes are named according to their migration characteristics in a pH4 to pH5 isoelectric focusing (IEF) polyacrylamide gel. Variants displaying a higher isoelectric point are conferred with letters from the beginning of the alphabet whereas those with lower isoelectric points are named from the end of the alphabet.Citation10 When two alleles have an identical IEF pattern and the sequence difference is known, the relevant residue is specifically indicated. For example; the two most common A1AT alleles, M1 (Val213) and M1 (Ala213), have IEF patterns of M1, but differ at residue 213 by the neutral amino acids Val and Ala. Some rare alleles are labeled by a letter indicating the IEF position together with the birth site of the allele (Mprocida). The null alleles are labeled “null” together with the place of origin (Nullbellingham).Citation5
One in 10 people of European descent are carriers of one of the two most common base-substitution mutations of A1AT that result in its deficiency. The more common is the S mutation (Glu264Val). In homozygotes this results in a 40% decrease in plasma A1AT concentrations. The most clinically significant mutation is the Z allele (Glu342Lys). Approximately 4% of Northern Europeans and 3% of people in the United Sates are heterozygous for the Z allele (PiMZ), with approximately 1 in 2000 being homozygotes (PiZZ). Homozygosity for the Z allele reduces plasma levels of A1AT to 10%–15% of normal. In PiZZ homozygotes, or those that co-inherit the Z allele and the S mutation plasma concentrations of A1AT will be insufficient to ensure lifetime protection of the lungs from proteolytic damage, particularly in smokers.Citation8
More than 90% of clinical cases of A1AT deficiency are caused by the PiZZ mutation. This occurs at an allelic frequency of 0.01 to 0.02 in North American white persons and 0.02 to 0.03 in Northern Europeans. The allelic frequency is lower in Southern Europeans (0.009) and African Americans (0.004). This demographic distribution also refers to the rarer clinically significant alleles. The null allele occurs with an allelic frequency of 0.00017. The reason for the predominance of the Z allele is unknown, but does raise the possibility that heterozygosity confers a survival advantage on the host.Citation10
Gene-mapping studies show that the PiZ allele probably arose in northern Europe and age estimates of the A1AT variants suggest that it first appeared 107 to 135 generations ago and could have been spread in Neolithic times. The PiS deficiency allele is older. It is estimated to have arisen 279–470 generations ago and from its high incidence on the Iberia peninsula it has been suggested that PiS could have arisen in this region.Citation15
A study of genetic epidemiologic studies from peer-reviewed literature was used to establish the worldwide racial and ethnic distribution of A1AT deficiency. The database estimated that the deficiencies would be present in a variety of populations and races.Citation16 A study of 96 Filipino infants with a history of jaundice since two weeks old, however, found an extremely low incidence of A1AT deficiency with only one patient positive for the PiS allele and negative for the periodic acid-Schiff (PAS) positive hepatic globules characteristic of the deficiency.Citation17 A similar study of 58 children with either chronic liver disease or neonatal cholestasis syndrome was carried out in India. None of the children had A1AT-deficient phenotypes.Citation18
Low plasma concentrations are also associated with two other rare variants of A1AT, Siiyama (Ser53Phe) and Mmalton (ΔPhe52), the commonest causes of A1AT deficiency in Japan and Sardinia, respectively.Citation8 Siiyama is also associated with PAS-positive, diastase-resistant inclusion bodies in the liver. The serine at position 53 is thought to contribute to the organization of the internal core element of the A1AT molecule. The deficient state in Siiyama may therefore be due to the change from an uncharged polar to a nonpolar amino acid imposed on the conserved serpin backbone which may exert severe effects on the integrity of the molecule and hence alter the intracellular processing of A1AT.Citation19
ZA1AT is the mutation which causes the most severe and most clinically relevant form of A1AT deficiency. The Z mutation (Glu342Lys) in A1AT occurs at the head of the fifth strand of the A-sheet and affects the mobility of the reactive centre loop and therefore the physical properties of A1AT. The mutation results in a unique molecular interaction between the reactive centre loop of one molecule and the gap in the A-sheet of another. This process can continue until large polymers form. This loop-sheet polymerisation of ZA1AT occurs spontaneously at 37 °C.Citation20
The loop-sheet polymerization has provided an excellent explanation for the aggregation of the Z-variant of A1AT in the liver endoplasmic reticulum (ER). The polymers of ZA1AT form aggregates. These aggregates are present as diastase-resistant PAS positive inclusions of A1AT in the ER of periportal cells. These inclusions result in a deficiency of circulating plasma A1AT, reducing plasma levels of the protein to 10%–15% of normal.Citation21
Electron microscopy of the hepatocytic inclusions in PiZZ homozygotes reveals bead-like necklaces of A1AT polymers. These polymers are resistant to the usual degradative processes of the ER because the individual molecules retain their ordered structure and hence fail to elicit the chaperone binding needed to activate degradation.Citation8 The oligosaccharide structure of the accumulated material from the PAS-positive diastase-resistant inclusions is compatible with a blockage of processing prior to entry to the Golgi. The protein accumulation occurs after core glycosylation in the endoplasmic reticulum but before the high mannose pruning which takes place in the Golgi. On extraction from the inclusions the A1AT is functionally active although this ZA1AT had only 15% of the activity of MA1AT.Citation22 The Z mutation reduces the association rate between A1AT and NE by approximately fivefold.Citation20 Therefore not only is there less A1AT in PiZZ individuals, but the population of ZA1AT molecules is less competent as an inhibitor of NE than MA1AT, and this suggests that PiZZ individuals have far less functional antielastase protection than the reduced concentration of A1AT alone would imply.Citation23
Polymerization of A1AT and consequent globular deposits are enhanced by high temperatures. Storage is speeded up also by the increase in synthesis of A1AT because polymerization is a concentration-dependent phenomenon. Consequently hepatocytic storage accelerates during febrile illness and infectious diseases (the acute-phase response) that involve either the liver or other organs. In fact in these conditions A1AT synthesis is incremental as the body temperature rises.Citation24 A1AT is an acute-phase reactant as its plasma levels increase during the host response to inflammation/tissue injury.Citation25 It should be noted however that differing results have been reported regarding the effects of temperature on the secretion/polymerization of ZA1AT and it is unlikely that there is a simple relationship between febrile episodes and phenotypic expression of liver disease in A1AT-deficient patients.
The process of intra-hepatic polymerization also underlies the mild plasma deficiency of the common S (Glu264Val) and rare I (Arg39Cys) alleles of antitrypsin and the severe plasma deficiency of the Siiyama and Mmalton alleles.Citation26
There is a strong genotype-phenotype correlation in these diseases that can be explained by the molecular instability caused by the mutation and, in particular, the rate at which the mutant forms polymers. Those mutants that cause the most rapid polymerisation cause the most retention of A1AT within the liver. This correlates with the greatest risk of liver damage and cirrhosis, and the most severe plasma deficiency.Citation27
There is prolonged hepatic retention of the A1AT Saar allele even though this allele does not have polymerogenic properties. The retention of this allele in the ER may be even more prolonged than that of ZA1AT even though A1AT Saar does not form insoluble polymers. This indicates that there could be mechanisms other than polymerization that determine whether mutant A1AT molecules are retained in the ER. It also raises the question of whether the polymerization of ZA1AT is the cause or the result of ER retention of the protein.Citation25,Citation28
Clinical manifestations of A1AT deficiency in the lung
A1AT deficiency was first described by Laurell and Eriksson in 1963,Citation29 and it is associated with a substantially increased risk for the development of pulmonary emphysema by the 4th or 5th decade of life.Citation20 It is thought that the emphysema is caused by the lack of fully active A1AT in the pulmonary interstitium which results in the unopposed action of proteases, the gradual destruction of pulmonary connective tissues and the loss of alveolar units.Citation10 It appears that patients with lower rates of secretion and low plasma A1AT levels risk rapid development of emphysema whereas those with somewhat higher rates of synthesis have better protection of alveolar tissue.
Cigarette smoking markedly accelerates the development of emphysema in individuals with A1AT deficiency. This can be explained in part because the key Met358 in the active site of A1AT can be easily oxidized. When oxidation occurs the association rate constant of A1AT for NE is reduced 2000-fold. Cigarette smoke and inflammatory cells in the lower respiratory tract can oxidize the Met efficiently.Citation5 Studies from this laboratory have demonstrated that in addition to Met358, Met351 is also susceptible to oxidation and site-directed mutants of A1AT with alanines substituted for these key methionines are resistant to oxidative inactivation.Citation30
Major differences in pathology exist between emphysema in PiMM and PiZZ individuals. This is due to the proteolytic attack and tissue destruction present in A1AT deficiency, and the fact that in its native form ZA1AT is fivefold less efficient at inhibiting neutrophil elastase than normal MA1AT. A1AT is also locally produced on the epithelial surface of the lung. This ZA1AT polymerizes at body temperature and is both an ineffective antiprotease inhibitor and a neutrophil chemoattractant.Citation31 There is already an excess of neutrophils in the airways of individuals suffering from ZA1AT-related emphysema and the polymers cause an influx of neutrophils. The excess of neutrophils is likely due to elevated pro-inflammatory cytokines such as IL-8 and leukotriene B4. The clearance of the A1AT polymers is impaired as they are bound to the alveolar wall. This enables them to act as a chronic stimulus for neutrophil influx into ZA1AT lungs. Thus the single amino acid substitution converts an anti-inflammatory molecule into a pro-inflammatory stimulus and this contributes in part to the excessive inflammation that underlies the progressive emphysema associated with ZA1AT deficiency.Citation32
Bronchial secretions of chronic obstructive pulmonary disease (COPD) patients with A1AT deficiency were observed to have increased levels of IL-8 and, in particular leukotriene B4 (LTB4). LTB4 and IL-8 are both potent chemotactic agents capable of promoting neutrophil transendothelial migration. The absolute contribution of LTB4 and IL-8 was significantly higher in the sputum from patients with A1AT deficiency than in patients without A1AT deficiency, although the remaining sputum chemotactic activity (not accounted for by LTB4 or IL-8) did not differ significantly between the two groups.Citation33 A possible explanation for the presence of these two chemoattractants in the sputum is the presence of free elastase as elastase has been shown to stimulate LTB4 release from alveolar macrophages as the macrophages bind uninhibited NE due to the presence of surface receptors for NE.Citation34
Sputum inflammation and chemotactic activity can be influenced by variations in patient characteristics such as the degree of lung function impairment, the presence of acute exacerbations, cigarette smoking, corticosteroid treatment and high bacterial load.Citation33
Clinical manifestations of A1AT deficiency in the liver
A1AT deficiency is also associated with liver disease. The PiZ and PiMmalton mutations are those most frequently associated with liver disease. Sharp and colleagues (1969) first described cirrhosis in A1AT deficiency in 10 children from 6 families and later reported the characteristic intrahepatocyte PAS diastase-resistant inclusions.Citation35
Of PiZ adults who have been systematically followed, it appears that ~50% die at a mean age of 52 yr of severe lung disease. Those subjects have mild or no signs of liver disease. Mortality of the remaining 50% occurs ~10 yr later with signs of chronic progressive liver disease at death and milder lung disease. Among PiMZ heterozygotes, lung disease is not significantly increased but chronic progressive liver disease has been found occasionally with progression to cirrhosis and, rarely, hepatoma. A German study looking at liver carcinoma and tumor-bearing tissue found that there is an increased risk of primary liver carcinoma among PiZ heterozygotes and that PiZ-associated carcinoma may develop in noncirrhotic tissue.Citation36 However a more recent study in Florida found that there was no association between the PiMZ state and chronic liver disease in general, but among patients with liver disease there was a significantly higher number of PiMZ patients suffering from severe liver disease compared to those suffering with less severe liver disease. It was also discovered that there was a significant association between the PiMZ heterozygous state and the increased severity of liver disease and the need for transplantation in patients with hepatitis C virus (HCV). These represent interim results of a long-term study into the role of heterozygous A1AT deficiency in chronic liver disease Citation37
While homozygous inheritance of the Z deficiency allele is associated with an increased risk for both emphysema and liver disease, the homozygous inheritance of a null allele is associated with a higher risk of emphysema but no risk of liver disease.Citation5 Patients with a higher rate of synthesis of ZA1AT tend to have a greater prevalence of liver disease than patients who have a low rate of expression. These second group tend towards emphysema. A1AT deficiency is the most common genetic cause of liver disease in children and predisposes adults to chronic liver disease and hepatocellular carcinoma.Citation25
Large scale screening for A1AT deficiency in Sweden between 1972 and 1974 showed that of 200,000 neonates there were 120 PiZZ individuals, only 14 of whom had prolonged jaundice with 9 developing severe liver disease. In addition 2 PiZ-, 54 PiSZ and 1 PiS- children were identified.Citation38
The consistent occurrence of overt liver disease in newborn infants, in contrast to its only occasional occurrence in young adults, may be explained by the fact that the liver cells in the infants are less capable of degrading the polymerized protein.Citation8
While the intrahepatocytic globules of A1AT are nontoxic in themselves, being present in all PiZ individuals including those who never develop liver disease, the accumulation does hamper the natural defenses of hepatocytes through an engorgement of the cellular synthetic pathway. This leads to a toxic gain-of-function. The accumulated ZA1AT is subjected to degradation by the ER-associated degradation pathway (ERAD).Citation39 The proteasome also plays an important role in the ER degradation of ZA1AT in many cell types.Citation40 Autophagy represents a nonproteasomal mechanism by which the protein is degraded.Citation41 Where the protein is not degraded at a high enough rate the ER stress response is activated. The three main functional components of the ER stress response are the ER overload response (EOR), the unfolded protein response (UPR) and apoptosis.Citation14 Endoplasmic reticulum stress responses have evolved to be protective, however when they are ineffective, toxic damage occurs demonstrating how these responses can be described as a double edged sword.Citation42 Interestingly human hereditary chromatosis, a genetic liver disease associated with iron overload is also characterized by ER stress.Citation43
This derangement of cellular functions renders hepatocytes more prone to damage by external insults which can have particularly damaging effects on the neonatal liver due to its relatively immature structure and function.Citation24 In early childhood the most common presentation of A1AT deficiency’s effect on the liver is prolonged jaundice. Abnormal liver function is largely self-limiting, but it can sometimes persist into adolescence. It is estimated that 5%–10% of A1AT deficient patients over the age of 50 will develop cirrhosis. A study examining adult patients with A1AT deficiency and chronic liver disease revealed a late onset of symptomatic hepatic abnormalities. 68% of the patients studied were 60 years or older when the liver disease was discovered. There seemed to be a later onset of the liver disease in the heterozygotes. At the time of diagnosis, the hepatic condition usually was advanced.Citation44
The liver damage occurs through a gain-of-function mechanism, unlike the lung disease, which is due to loss-of-function. The retention of the mutant ZA1AT molecules in the ER triggers a series of events that are eventually hepatotoxic.Citation45 This gain-of-function mechanism is illustrated in ZA1AT transgenic mice. The liver disease is apparent in the mice even though normal levels of antielastases expressed by endogenous genes are still present, therefore the liver injury can not be attributed to loss of function.Citation46
The variability in the severity of liver disease and the age of onset among patients with A1AT deficiency may be explained in part by individual variations in episodes of inflammation and hence to increased synthesis of A1AT.Citation8 A study that involved the transfection of human skin fibroblasts with ZA1AT from patients with and without liver disease found that there was a marked delay in the degradation of the accumulated protein in the cells from those subjects with liver disease, compared to those without liver disease. Human skin fibroblasts do not express the endogenous A1AT gene, but presumably express genes involved in the processing of secretory proteins. This result would suggest that there are other genetic traits that affect the fate of the ZA1AT molecule within cells.Citation45 Virus-induced liver disease in deficient subjects frequently assumes a chronic course because of the deranged immunologic system of these subjects. Studies have demonstrated that PiZ subjects with liver disease have anomalies of immunomodulation and anomalous activation of the complement cascade.Citation24
Cirrhosis is favored over restitution as the proteases released by leukocytes during the inflammatory response to the hepatocytic necrosis cannot be properly counteracted and collagen and reticulum fibers are improperly rearranged during the healing process. Increased numbers of collagen bundles have been observed in the space of Disse and around hepatocytes in A1AT-deficient patients. An insufficient defense against reactive oxygen metabolites has been hypothesized to perpetuate the liver damage.Citation24
The accumulation of ZA1AT can cause liver damage in transgenic PiZZ mice where the degree of liver damage correlates with the amount of ZA1AT accumulated in the liver of the different pedigrees of mice. Although 40% of the PiZZ mice tested were seropositive for mouse hepatitis virus (MHV), comparison of the PiZZ mice with and without MHV showed no difference in liver damage. The degree of liver damage was therefore not influenced by the MHV seropositivity; rather, it was related only to the presence of accumulated ZA1AT protein.Citation46
Male gender and obesity have been found to predispose A1AT deficient individuals to liver disease.Citation47 The association of the hepatitis virus with A1AT deficiency is less clear. An Austrian study of A1AT deficient patients with chronic liver disease found that 62% of those with cirrhosis were HCV positive and 33% showed evidence of hepatitis B virus (HBV) infection.Citation48 Another study found that the odds of having a heterozygous Z phenotype increased in patients with HCV compared to a control population, where patients with hepatitis C or B virus were 3.6 times more likely to have a heterozygous Z phenotype than a normal phenotype.Citation49
Some studies, however, have found no association between hepatitis C infection and A1AT deficiency.Citation50 A study into the PiMZ population found that the prevalence of A1AT PiMZ was no greater in hepatitis C patients than in the general population.Citation51
The nonsteroidal anti-inflammatory drug (NSAID) indomethacin causes an increase of liver damage in transgenic ZA1AT expressing mice. It is estimated that NSAIDs are used by 15–20 million people in the United States on a long-term basis. The administration of indomethacin increases hepatic injury as indicated by the increase in hepatocellular proliferation and increased activation of caspase-9. Indomethacin-induced injury is associated with activation of IL-6-STAT3 signaling, increased expression of A1AT mRNA and greater accumulation of polymerized ZA1AT in the liver. This indicates that environmental factors such as exogenous medication administration can significantly potentiate the liver injury associated with ZA1AT hepatic accumulation; NSAIDs may be especially injurious to patients with A1AT deficiency, possibly by increasing the expression and accumulation of the hepatotoxic mutant protein.Citation52 As the Z mutation may lead to worse liver injury in the setting of other liver diseases interplay of cholestasis and the PiZZ phenotype was investigated in transgenic mice. A bile duct ligation (BDL) was carried out on mice possessing a transgenic alpha1-ATZ mutation and littermate controls. PiZ transgenic mice undergoing BDL developed more liver fibrosis than wild-type mice after BDL. Increased apoptosis was also noted in PiZ BDL mice.Citation53 This shows that PiZ transgenic mice are more susceptible to liver fibrosis induced by cholestasis from BDL. The Z mutation of A1AT may act as a modifier gene in patients with concurrent liver diseases such as cystic fibrosis and cholestasis. Similarly the co-existence of the Z mutation and C282Y HFE (the protein involved in human hereditary hemochromatosis) act as potential disease modifiers with respect to each other in terms of expression on ER stress responses.Citation43
PiZZ subjects with liver disease are prone to develop a hepatocellular type of carcinoma. Mild or severe hepatocytic dysplasia is not an infrequent sign found in biopsy specimens. Usually liver cancer develops in a patient with cirrhosis or other chronic liver disease. Occasionally however, PiZZ patients with hepatocellular carcinoma do not have cirrhosis. Usually globules are situated in the periportal hepatocytes, but in the cirrhotic nodules they are spread throughout.Citation24
A1AT deficiency may also affect other human proteins. A recent case report involved a patient presenting with polycythemia (a known paraneopaetic manifestation of hepatoma which requires the presence of alpha-fetopro [AFP]) but with normal serum levels of AFP. This patient was found to have A1AT deficiency. Infants with neonatal hepatitis and A1AT deficiency do not have raised AFP. It was postulated that this is because A1AT is a rate limiting factor in the production of AFP. The authors raised the possibility of A1AT deficiency resulting in normal AFP in adults with hepatocellular carcinoma.Citation54
Overall 10% of PiZZ neonates develop hepatitis and cholestasis. Cholestasis usually occurs in the first two months, though it may persist for up to eight months.Citation55 In early childhood patients may present with jaundice in which the stools generally contain no yellow or green pigment, indicating cholestasis and mimicking biliary atresia. All patients have hepatomegaly and about 50% also have splenomegaly. Approximately 5% of the patients present with an increased bleeding tendency. This is due to vitamin K deficiency caused by the cholestasis-induced malabsorption. Less commonly children present later in childhood with hepatosplenomegaly or with cirrhosis.Citation13
Current therapeutics
The only corrective therapy for A1AT deficient patients with severe liver disease currently is liver transplantation. The most recent study shows that the 1-, 3-, and 5-year patient survival is 89%, 85%, and 83%, respectively, for adults after transplantation versus 92%, 90%, and 90% for pediatric patients after transplantation.Citation56 In cases where transplantation is not possible there are palliative benefits of portocaval shunting. The beneficial effects of both breast feeding and vitamin E supplementation in cholestatic infants have been suggested.Citation10
In 2003 the American Thoracic Society published their first new set of guidelines for the management of obstructive lung disease associated with A1AT deficiency since 1989. They recommended standard treatments as per nonhereditary emphysema.
Augmentation therapy has been given for the treatment of lung disease associated with A1AT deficiency since 1987. The levels of A1AT in serum, bronchoalveolar lavage fluid and the pulmonary interstitium, required to provide protection against proteolytic lung destruction, have been estimated from those observed in PiSZ persons who have lower than normal levels of A1AT but no demonstrable increase in premature lung disease. These levels can be reached and maintained in PiZZ and null individuals by intermittent infusions of plasma-purified A1AT.Citation10
Gene therapeutics
As emphasized by the ATS in 2003 there is no therapy available other than liver transplantation for individuals with advanced A1AT-deficiency liver disease. To address this challenge there have been a number of investigations into alternative methods to treat the liver disease. As the adverse effects of A1AT deficiency are due to the expression of a single gene, the specific knockdown of this gene has high potential for the treatment of the deficiency. Gene therapy may be a possibility for treatment of the liver disease. Gene therapy was originally designed as a means to provide therapeutic merit by introducing genetic material (DNA or RNA) encoding a protein that is missing or defective into a patient’s cells or tissues. Newer gene therapy approaches aimed at inhibiting the expression of a target mutant gene include RNA interference (RNAi) mediated through small interfering RNA (siRNA), peptide nucleic acids, and ribozymes. These approaches can be used to stop the production of the mutant Z protein, hence stopping the accumulation of the protein in the liver and providing protection against liver disease.
A recent study investigated the effects of small DNA fragments (SDFs) on A1AT. This is the DNA technology of small fragment homologous replacement (SFHR) which is versatile in that sequences can be directly altered, inserted or deleted. Treatment involves direct conversion of the mutant sequence to a wild-type genotype, thereby restoring the normal phenotype. Small DNA fragments are introduced into the cell and recombine with the genomic DNA at a targeted site, thereby producing a specific change in the sequence. SDFs including normal M and ZA1AT sequences were generated and transfected into peripheral blood monocytes from PiM subjects and PiZ subjects. The defective gene could be corrected in ZA1AT monocytes in vitro with SDFs, and treatment was associated with an increase in A1AT secretion. The development of this methodology to repair the gene defect in hepatocytes could have beneficial effects on secretion, thereby protecting both the lung and liver.Citation57
RNAi and siRNA
Post-transcriptional gene silencing (PTGS) was first observed in petunias at the end of the 1980s. Attempts to enhance the hue of petals via the introduction of a chimeric gene unexpectedly resulted in the production of completely white petals with corresponding knockdown in mRNA levels. The developmental timing of mRNA expression of the endogenous gene was not altered by introduction of an exogenous gene, but the level of the mRNA produced by the endogenous gene was seen to decrease 50-fold from wild-type levels. Somatic reversion of plants with white flowers to phenotypically parental violet flowers was associated with a coordinate rise in the steady state levels of the mRNAs produced by both the endogenous and introduced genes.Citation58 This led to the discovery of RNA interference (RNAi).
RNA interference (RNAi) is the evolutionary-conserved process of sequence-specific, post-transcriptional gene silencing in animals and plants, initiated by double-stranded RNA (dsRNA) that is homologous in sequence to the silenced gene.Citation59 RNAi is exclusively found in eukaryotes. This is because in pro-karyotes RNase III, which is a very potent and fast ds-specific RNase, degrades dsRNA substrates as short as 12 bp.Citation60
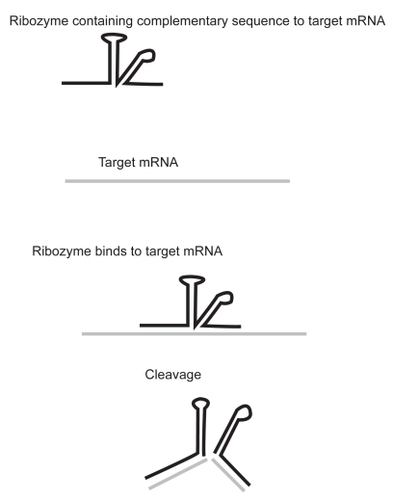
Although the pathway of RNAi is under constant investigation, many of the underlying mechanisms have been clarified. The process is divided into distinct steps (). The initiation of post transcriptional gene silencing occurs on processing of a dsRNA by an enzyme known as Dicer into ~22nt guide sequences. These guide sequences are known as small interfering RNA (siRNA). The siRNA then forms part of a multi-component nuclease, the RNA-induced silencing complex (RISC), and instructs it to destroy specific mRNAs.Citation61 Fractionation indicates that RISC and the process generating siRNAs from dsRNA are separate, though the two enzymes responsible may interact at some point during the silencing process. RNA III family members are among the few nucleases that show specificity for dsRNA.Citation62 Dicer is evolutionary conserved in worms, flies, plants, fungi, and mammals.Citation63
Figure 1 Mechanism of RNA interference. dsRNA are processed by Dicer to produce siRNA of 21–23 nucleotides in length. The dsRNA unwinds, allowing one strand to bind to the RNA-induced silencing complex (RISC). Binding of the antisense RNA strand activates the RISC to cleave mRNAs containing a homologous sequence.
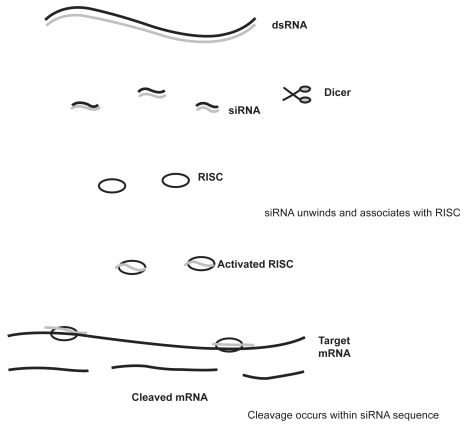
Figure 2 PNA-induced gene inhibition. PNA can bind with DNA leading to anti-gene inhibition, or it can bind with RNA leading to antisence inhition.
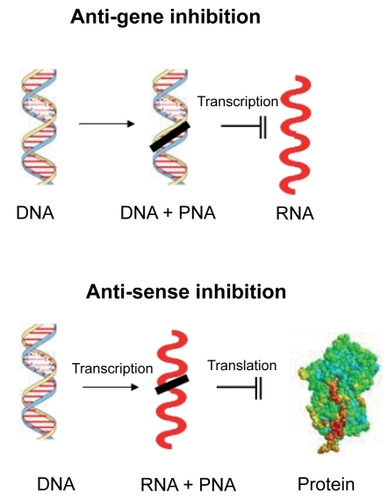
Figure 3 Mechanism of action of ribozymes. Ribozymes with a section of sequence complementary to the target mRNA will bind the mRNA and cleave it.
In actively proliferating cells siRNA activity lasts for up to 1 week, whereas in terminally differentiated cells such as neurons, the silencing can last for up to 3 weeks.Citation64 This transiency is due to the fact that mammalian cells lack the RNA-dependent RNA polymerases that amplify siRNAs in nonmammalian cells such as Caenorhabditis elegans. As a result the siRNA duplexes become progressively diluted as cells divide.Citation65 This could be an ideal therapeutic window for many chronic diseases, for although it would necessitate repeat administrations of siRNA every few weeks this would be a short enough term to lessen concerns regarding the long-term toxicity of permanently silencing a gene, while still a long enough treatment time to be a worthwhile therapy.Citation66 This would be particularly useful for the liver disease associated with A1AT deficiency.
The delivery in vivo of siRNAs by liposomes has proven successful. The complexation of a siRNA targeting the Bcl-2 oncogene with liposomes resulted in the successful inhibition of tumor formation in mice.Citation67 Liposomes were also used to demonstrate that RNAi can be used successfully in vivo to significantly reduce functional expression of the V2R in the mouse kidney.Citation68 Liposomes have been used in vivo in a number of studies in mouse models and may represent a potential method to deliver siRNA successfully.
The delivery in vivo of siRNAs by cationic lipids has been described. Other strategies for local or systemic siRNA administration have been explored including chemical modifications of siRNA molecules, electropulsation, polyamine or other basic complexes, atelocollagen, virosomes and certain protein preparations.
In addition it is also possible to target siRNAs to specific organs including the liver. This can prevent unwanted effects on normal host tissues. Targeted delivery can be achieved by local administration or by injection via catheterization of the vascular supply to particular organs. Chemically modified siRNA could possibly also be covalently linked to ligands for cell-surface receptors, enabling delivery only to cells bearing these receptors.Citation66 The delivery of siRNA into model mammalian organisms has been achieved by intravenous injection or by electroporation of synthetic siRNAs directly into target tissues and organs.Citation69 In rodents the rapid infusion of siRNA by hydrodynamic injection has seen successful transgene delivery in the liver with expression found in up to 40% of hepatocytes.Citation70
Cruz and colleagues reported successful knockdown of A1AT in cell lines using A1AT siRNAs.Citation71 They also successfully knocked down the expression of ZA1AT in ZA1AT overexpressing transgenic mice.
Pulmonary disease in A1AT deficiency is due to low serum levels of A1AT. Knockdown of ZA1AT expression would be of no benefit here, as it would further reduce the serum levels of circulating A1AT. As a gene therapy alternative to augmentations therapy the normal A1AT gene has been successfully introduced into the striated muscle cells of animals using an AAV vector both in vitro and in vivo.Citation72,Citation73 A1AT gene replacement therapy may be of benefit to the pulmonary disease as the A1AT administered is in the form of MA1AT and trials are ongoing. ZA1AT knockdown and MA1AT replacement together could potentially be a very effective way of treating A1AT deficiency in PiZZ individuals. The major challenges to this therapy will include targeted delivery and chronicity.
Peptide nucleic acids
Although peptide nucleic acids were originally conceived as agents for double-stranded DNA binding, they were first exploited for gene therapy drug design. PNAs were first described by Nielson et al in 1991.Citation74 They are synthetic DNA analogs in which the phosphodiester backbone has been replaced by repetitive units of the pseudo-peptide polymer N-(2-aminoethyl) glycine to which the purine and pyrimidine bases are attached by a methyl carbonyl linker.Citation75 PNAs hybridize to complementary DNA or RNA in a sequence-dependent manner, according to the Watson–Crick hydrogen bonding scheme. In contrast to DNA, PNA can bind in either a parallel or antiparallel manner.Citation76 The inhibition of gene expression by PNA is known as anti-gene inhibition when it occurs as a result of PNA binding to DNA, and as antisense inhibition when it occurs as a result of PNA binding to RNA. The synthetic uncharged peptide backbone provides PNA with unique hybridization characteristics. There is no electrostatic repulsion when PNAs hybridize to their target nucleic acid sequence and this gives a higher stability to the PNA–DNA or PNA–RNA duplexes than the natural homo- or heteroduplexes.Citation77 PNAs are also not degraded by nucleases or proteases and therefore, cannot be directly used as primers, or be copied.Citation75
PNAs can form strand invasion complexes or result in strand displacement in DNA and thus lead to the inhibition of gene transcription. Several in vitro studies have shown that the binding of PNA or bis-PNA to complementary DNA can efficiently block transcriptional elongation and inhibit the binding of transcriptional factors.
The interaction of PNAs with mRNA is independent of the RNA secondary structure. The antisense effects of PNAs occur through the steric interference of RNA processing or transport into the cytoplasm or through the interference of translation. This interference is caused by the binding of PNA to the mRNA. The antisense inhibition caused by siRNAs is due to RNase mediated degradation, however, PNAs are not substrates for RNase.Citation78
PNAs could constitute highly efficient compounds for gene therapy as the oligomers have good stability and a strong binding affinity for DNA and RNA. They are also nontoxic, even at relatively high concentrations. However, as with siRNA, progress in the use of PNAs as tools for regulating gene expression slows at the in vivo stage. This is due to the slow uptake of naked PNA by living cells. Modifications of PNAs have led to significant improvements in the uptake of PNA in eukaryotic cells. PNAs can be coupled to DNA oligomers, to receptor ligands or more efficiently to peptides such as cell penetrating peptides which are rapidly internalized by mammalian cells.Citation79 Such coupling markedly improves the uptake of PNAs by cells. As with siRNA liposomes can be of use to improve the uptake of PNAs when PNAs are incorporated into the liposomes.Citation80
Ribozymes
Another method of inhibiting the expression of a gene is through the use of ribozymes. Ribozymes are naturally occurring catalytic RNA molecules that cleave RNA with high specificity. They behave like enzymes though there is no protein involved. The catalytic properties of RNA were discovered in the 1980s by Thomas Cech and Sidney Altman and the term ‘ribozyme’ was coined in 1982.Citation81
Many ribozymes have either a hammerhead or hairpin shaped active center. Synthetically designed and specifically targeted hammerhead and hairpin ribozymes have been shown to be promising gene-targeting reagents. These ribozymes contain both a catalytic RNA domain that cleaves the target mRNA and a substrate-binding domain with a sequence antisense to the target mRNA sequence. Therefore, these gene-targeting ribozymes bind to the mRNA sequence through Watson–Crick interactions between the target sequence and the antisense sequence in the substrate-binding domain of the ribozyme.Citation82
Ribozymes bind to their targets with a much higher specificity than siRNAs, which can potentially produce off-target effects. There is no risk of an interferon response with ribozymes, unlike with siRNA. As with siRNA and PNAs there are problems using ribozymes to knockdown gene expression in vivo. The activity in vivo is not usually high enough to achieve the desired effects.
Human hepatoma cells were transduced with ribozymes to inhibit the expression of the mutant A1AT gene and express a modified A1AT gene. This ribozyme-mediated gene therapy did show inhibition of the mutant gene and expression of the modified A1AT gene; however the efficiency of this method is low. Citation83
Perspective
Gene therapy has been under investigation since the emergence of restriction enzymes. Retroviral vectors were investigated and the first human trials began in the late 1980s.Citation84 Gene therapy with recombinant adenovirus (rAd) has since developed and recombinant adenovirus (rAAV) trials were carried out in cystic fibrosis (CF) patients in 1993.Citation85 rAAV trials were initiated in CF patients in 1995.Citation86 Lentivirus and recombinant herpesvirus vectors have also been used. rAd, rAAV, lentivirus and recombinant herpesvirus vectors are capable of gene transfer and expression in vivo in nondividing cells.Citation87 Nonviral gene transfer methods include DNA conjugates, and naked DNA injection and these came into use in gene therapy in the 1990s.
There are drawbacks to all of these methods of gene therapy. Wild-type Ad triggers innate and acquired immune responses. Recombinant Ad also triggers these responses in a dose-dependent manner. Gamma-retrovirus vectors have caused leukemia-like syndromes when used on bone-marrow stem cells. Recombinant AAV vectors can generate adaptive immune responses to the retained viral capsid proteins. Aerosol administration of nonviral vectors has been associated with an influenza-like syndrome and the efficacy of these systems in vivo is low and transient.
The only indications of clinically significant efficacy with gene therapy have been with onco-retrovirus vectors and a first-generation rAd vector, but these systems are associated with important limiting toxicities.
Gene therapy targeted to the liver has also been attempted. A study into the effectiveness of adenoviral delivery of interferon alpha to rats that were induced to develop human hepatocellular carcinoma and liver cirrhosis found that targeting of IFN-alpha expression to the liver significantly reduced liver tumor volume and ameliorated liver cirrhosis.Citation88 In addition pseudotyped AAV vectors for the liver-specific expression of human blood coagulation factor IX (hFIX) have been investigated and expression in immunocompetent mice ranged from 36% to more than 2000% of normal. Expression was dose- and time-dependent.Citation89 With respect to gene-targeted therapeutics for liver disease in general the efficacy of novel strategies still needs to be further validated in animal studies and confirmed in clinical trials. Particularly important are their efficient and safe delivery to the target tissue and the limitation of unintended harmful nonspecific effects such as transient or sustained inflammation.
siRNA, PNA and Ribozymes represent potential gene knockdown methods and with the ongoing investigation into their administration in vivo remains only a matter of time until a valid gene therapy emerges. For A1AT-deficient individuals this development would be most welcome. The liver disease caused by the polymerization of ZA1AT and the inflammatory effects due to the chemotactic characteristics of polymerized ZA1AT in the lung could both potentially be ameliorated by the knockdown of ZA1AT gene and protein expression in these organs. With the continuing investigations into A1AT augmentation therapy the adverse effects of A1AT deficiency may soon be completely overcome.
Disclosure
The authors report no conflicts of interest in this work.
References
- RogersJKalshekerNWallisSThe isolation of a clone for human alpha 1-antitrypsin and the detection of alpha 1-antitrypsin in mRNA from liver and leukocytesBiochem Biophys Res Commun19831163753826606425
- MolmentiEPPerlmutterDHRubinDCCell-specific expression of alpha 1-antitrypsin in human intestinal epitheliumJ Clin Invest199392202220348408656
- HuCPerlmutterDHCell-specific involvement of HNF-1beta in alpha(1)-antitrypsin gene expression in human respiratory epithelial cellsAm J Physiol Lung Cell Mol Physiol2002282L757L76511880302
- TravisJOwenMGeorgePIsolation and properties of recombinant DNA produced variants of human alpha1-proteinase inhibitorJ Biol Chem1985260438443893872299
- CrystalRGBrantlyMLHubbardRCCurielDTStatesDJHolmesMDThe alpha1-antitrypsin gene and its mutationsChest1989951962082642408
- PetracheIFijalkowskaIZhenLA novel antiapoptotic role for alpha1-antitrypsin in the prevention of pulmonary emphysemaAm J Respir Crit Care Med20061731222122816514110
- NitaIMSerapinasDJanciauskieneSMAlpha1-antitrypsin regulates CD14 expression and soluble CD14 levels in human monocytes in vitroInt J Biochem Cell Biol2007391165117617448722
- CarrellRWLomasDAAlpha1-antitrypsin deficiency – A model for conformational diseasesN Engl J Med2002364455311778003
- PotempaJKorzusETravisJThe serpin superfamily of proteinase inhibitors: structure, function, and regulationJ Biol Chem199426915957159608206889
- CoakleyRJTaggartCO’NeillSMcElvaneyNGAlpha1-antitrypsin deficiency: biological answers to clinical questionsAm J Med Sci2001321334111202478
- DarlingtonGJAstrinKHMuirheadSPDesnickRJSmithMAssignment of human alpha-1 antitrypsin to chromosome 14 by somatic cell hybrid analysisProc Natl Acad Sci U S A1982798708736801664
- SchroederWTMillerMFWooSLCSaundersGFChromosomal localization of the human alpha1-antitrypsin gene (PI) to 14q31–32Am J Hum Genet1985378688723876766
- KokKWahabPHouwenRHJHeterozygous alpha-1 antitrypsin deficiency as a co-factor in the development of chronic liver disease: a reviewNeth J Med20076516016617519511
- GreeneCMMillerSDWCarrollTAlpha-1 antitrypsin deficiency: a conformational disease associated with lung and liver manifestationsJ Inherit Metab Dis200831213418193338
- SeixasSGarciaOTrovoadaMJSantosMTAmorimARochaJPatterns of haplotype diversity within the serpin gene cluster at 14q32.1: insights into the natural history of the alpha1-antitrypsin polymorphismHum Genet2001108203011214903
- de SerresFJWorldwide racial and ethnic distribution of alpha1-antitrypsin deficiencyChest20021221818182912426287
- TanJJCutiongco-dela PazEMAvilaJMCGregorioGVLow Incidence of alpha1-antitrypsin deficiency among Filipinos with neonatal cholestatisJ Paediatr Child Health20064269469717044896
- KhannaRAlamSSherwaniRAroraSAroraNKMalikAAlpha-1 antitrypsin deficiency among Indian children with liver disordersIndian J Gastroenterol20072519119316974034
- SeyamaKNukiwaTTakabeKTakahashiHMiyakeKKiraSSiiyama (Serine 53 (TCC) to phenylalanine 53 (TTC)) a new alpha1-antitrypsin-deficient variant with mutation on a predicted conserved residue of the serpin backboneJ Biol Chem199126612627126321905728
- LomasDAEvansDLStoneSRChangWSCarrellRWEffect of the Z mutation on the physical and inhibitory properties of alpha1-antitrypsinBiochem1993325005088422359
- LomasDAEvansDLFinchJTCarrellRWThe mechanism of Z alpha1-antitrypsin accumulation in the liverNature19923576056091608473
- BathurstITravisJGeorgePMCarrellRWStructural and functional characterization of the abnormal Z alpha1-antitrypsin isolated from human liverFEBS Lett19841771791836333994
- BurrowsJAWillisLKPerlmutterDHChemical chaperones mediate increased secretion of mutant alpha 1-antitrypsin (alpha 1-AT) Z: A potential pharmacological strategy for prevention of liver injury and emphysema in alpha 1-AT deficiencyProc Natl Acad Sci U S A2000971796180110677536
- MassiGPathogensis and pathology of liver disease associated with alpha1-antitrypsin deficiencyChest1996110251S255S8989160
- PerlmutterDHLiver injury in alpha1-antitrypsin deficiency: an aggregated protein induces mitochondrial injuryJ Clin Invest20021101579158312464659
- CarrellRWAlpha 1-Antitrypsin: molecular pathology, leukocytes, and tissue damageJ Clin Invest198678142714313537008
- LomasDAAntitrypsin deficiency, the serpinopathies, and chronic obstructive pulmonary diseaseProc Am Thorac Soc2006349950216921127
- LinLSchmidtBTeckmanJHPerlmutterDHA naturally occurring nonpolymeric mutant of alpha1-antitrypsin characterised by prolonged retention in the endoplasmic reticulumJ Biol Chem2001276338933389811427540
- LaurellCBErikssonSHypo-alpha-1-antitrypsinemiaVerh Dtsch Ges Inn Med19647053753914294270
- TaggartCCervantes-LaureanDKimGOxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activityJ Biol Chem2000275272582726510867014
- MulgrewATTaggartCCLawlessMWZ α1-antitrypsin polymerizes in the lung and acts as a neutrophil chemoattractantChest200412551952195715136414
- MahadevaRAtkinsonCLiZPolymers of Z alpha1-antitrypsin co-localize with neutrophils in emphysematous alveoli and are chemotactic in vivoAm J Pathobiol2005166377387
- WoolhouseIBayleyDStockleyRASputum chemotactic activity in chronic obstructive pulmonary disease: effect of alpha1-antitrypsin deficiency and the role of leukotriene B4 and interleukin 8Thorax20025770971412149532
- HubbardRCFellsGGadekJPacholokSHumesJCrystalRGNeutrophil accumulation in the lung in alpha 1-antitrypsin deficiency. Spontaneous release of leukotriene B4 by alveolar macrophagesJ Clin Invest1991888918971653278
- SharpHLBridgesRAKrivitWFreierEFCirrhosis associated with alpha-1-antitrypsin deficiency: a previously unrecognized inherited disorderJ Lab Clin Med1969739349394182334
- ZhouHOrtiz-PallardóMEKoYFischerEPIs heterozygous alpha-1-antitrypsin deficiency type PIZ a risk factor for primary liver carcinoma?Cancer2000882668267610870048
- RegevAGuaquetaCMolinaEGDoes the heterozygous state of alpha-1 antitrypsin deficiency have a role in chronic liver diseases? Interim results of a large case-control studyJ Pediatr Gastroenterol Nutr200643S30S3516819398
- SvegerTLiver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infantsN Engl J Med1976294131613211083485
- McCrackenAAKarpichevIVErnagaJEWernerEDDillinAGCourchesneWEYeast mutants deficient in ER-associated degradation of the Z variant of alpha-1-protease inhibitorGenetics1996144135513628978025
- TeckmanJHBurrowsJHidvegiTSchmidtBHalePPerlmutterDHThe proteasome participates in degradation of mutant alpha-1 antitrypsin Z in the endoplasmic reticulum of hepatoma-derived hepatocytesJ Biol Chem2001276448654487211577074
- TeckmanJHPerlmutterDHRetention of mutant alpha-1 antitrypsin Z in the endoplasmic reticulum is associated with an autophagic responseAm J Physiol Gastrointest Liver Physiol2000279G961G97411052993
- LawlessMWMankanAKGraySGNorrisSEndoplasmic reticulum stress – a double edged sword for Z alpha-1 antitrypsin deficiency hepatoxicityInt J Biochem Cell Biol2008401403141418353704
- LawlessMWMankanAKWhiteMO’DwyerMJNorrisSExpression of hereditary hemochromatosis C282Y HFE protein in HEK293 cells activates specific endoplasmic reticulum stress responsesBMC Cell Biol200783017650303
- RakelaJGoldschmiedtMLudwigJLate manifestation of chronic liver disease in adults with alpha-1-antitrypsin deficiencyDig Dis Sci198732135813623500836
- WuYWhitmanIMomentumEMooreKHippenmeyerPPerlmutterDHA lag in intracellular degradation of mutant alpha-1 antitrypsin correlates with the liver disease phenotype in homozygous PiZZ alpha-1 antitrypsin deficiencyProc Natl Acad Sci U S A199491901490188090762
- CarlsonJABarton RogersBSifersRNAccumulation of PiZ alpha1-antitrypsin causes liver damage in transgenic miceJ Clin Invest198983118311902784798
- BowlusCLWillnerIZernMAFactors associated with advanced liver disease in adults with alpha1-antitrypsin deficiencyClin Gastroenterol Hepatol2005339039615822045
- PropstTPropstADietzeOJudmaierGBraunsteinerHVogelWHigh prevalence of viral infection in adults with homozygous and heterozygous alpha 1-antitrypsin deficiency and chronic liver diseaseAnn Intern Med19921176416451530195
- EigenbrodtMLMcCashlandTMDyRMClarkJGalatiJHeterozygous alpha 1-antitrypsin phenotypes in patients with end stage liver diseaseAm J Gastroenterol1997926026079128307
- ElzoukiANVerbaanHLindgrenSWidellACarlsonJAErikssonSSerine protease inhibitors in patients with chronic viral hepatitisJ Hepatol19972742489252072
- ScottBBEgnerWDoes alpha1-antitrypsin phenotype PiMZ increase the risk of fibrosis in liver disease due to hepatitis C virus infectionEur J Gastroenterol Hepatol20061852152316607148
- RudnickDAShikspwashyaOBlomenkampKTeckmanJHIndomethacin increases liver damage in a murine model of liver injury from alpha1-antitrypsin deficiencyHepatology20064497698217006946
- MencinASekiEOsawaYAlpha-1 antitrypsin Z protein (PiZ) increases hepatic fibrosis in a murine model of cholestasisHepatology2007461443145217668872
- OwenDRSivakumarRSuhE-SSeevaratnamMAlpha-1-antitrypin deficiency resulting in a hitherto unseen presentation of hepatocellular carcinoma: polycythemia but with normal alpha fetoproteinWorld J Gastroenterol2006124906490716937479
- SokolRJFat-soluble vitamins and their importance in patients with cholestatic liver diseasesGastroenterol Clin North Am1994236737057698827
- KemmerNKaiserTZachariasVNeffGWAlpha-1 antitrypsin deficiency: outcomes after liver transplantationTransplant Proc2008401492149418589136
- McNabGLAhmadAMistryDStockleyRAModification of gene expression and increase in alpha1-antitrypsin secretion after homologous recombination in alpha1-antitrypsin deficient monocytesHum Gene Ther2007181171117717937578
- NapoliCLemieuxCJorgensenRIntroduction of a chimeric chal-cone synthase gene into petunia results in reversible co-suppression of homologous genes in transPlant Cell1990227928912354959
- ElbashirSMMartinezJPatkaniowskaALendeckelWTuschlTFunctional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysateEMBO J2001206877688811726523
- BrantlSAntisense-RNA regulation and RNA interferenceBiochim Biophys Acta20021575152512020814
- HammondSMBernsteinEBeachDHannonGJAn RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cellsNature200040429329610749213
- RobertsonHDWebsterREZinderNDPurification and properties of ribonuclease III from Escherichia coliJ Biol Chem196824382914865702
- BernsteinECaudyAAHammondSMRole for a bidentate ribonuclease in the initiation step of RNA interferenceNature200140936336611201747
- OmiKTokunagaKHohjohHLong-lasting RNAi activity in mammalian neuronsFEBS Lett2004558899514759522
- LeungRKWhittakerPARNA interference: from gene silencing to gene-specific therapeuticsPharmacol Ther200510722223915908010
- LiebermanJSongELeeS-KShankarPInterfering with disease: opportunities and roadblocks to harnessing RNA interferenceTrends Mol Med2003939740413129706
- YanoJHirabayashiKNakagawaSAntitumor activity of small interfering RNA/cationic liposome complex in mouse models of cancerClin Cancer Res2004107721772615570006
- HassanATianYZhengWJiHSandbergKVerbalisJGSmall interfering RNA-mediated functional silencing of vasopressin V2 receptors in the mouse kidneyPhysiol Genomics20052138238815784697
- DillonCPSandyPNencioniAKisslerSRubinsonDAVan ParijsLRNAi as an experimental and therapeutic tool to study and regulate physiological and disease processesAnnu Rev Physiol20056714717315709955
- LiuFSongYLiuDHydrodynamics-based transfection in animals by systemic administration of plasmid DNAGene Ther199961258126610455434
- CruzPEMuellerCCossetteTLIn vivo post-transcriptional gene silencing of alpha-1 antitrypsin by adeno-associated virus vectors expressing siRNALab Invest20078789390217592477
- SongSMorganMEllisTSustained secretion of human alpha-1-antitrypsin from murine muscle transduced with adeno-associated virus vectorsProc Natl Acad Sci U S A19989514384143889826709
- FlotteTRRecombinant adeno-associated virus gene therapy for cystic fibrosis and alpha-1 antitrypsin deficiencyChest200212198121
- NielsenPEEgholmMBergRHBuchardtOSequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamideScience1991254149715001962210
- PellestorFPaulasovaPThe peptide nucleic acids (PNAs), powerful tools for molecular genetics and cytogeneticsEur J Hum Genet20041269470015213706
- RasmussenHKastrupJSNielsenJNNielsenJMNielsenPECrystal structure of a peptide nucleic acid (PNA) duplex at 1.7A resolutionNat Struct Biol19974981019033585
- JensenKKOrumHNielsenPENordenBHybridization kinetics of peptide nucleic acids (PNA) with DNA and RNA studied with BIAcore techniqueBiochemistry199736507250779125529
- KnudsenHNielsenPEAntisense properties of duplex- and triplex-forming PNAsNucleic Acids Res1996244945008602363
- CutronaGCarpanetoEMUliviMEffects in live cells of a c-myc anti-gene PNA linked to a nuclear localization signalNat Biotechnol20001830030310700145
- NastruzziCCortesiREspositoELiposomes as carriers for DNA–PNA hybridsJ Control Release20006823724910925132
- KrugerKGrabowskiPJZaugAJSandsJGottschlingDECechTRSelf-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of TetrahymenaCell1982311471576297745
- TrangPKimKLiuFDeveloping RNase P ribozymes for gene-targeting and antiviral therapyCell Microbiol2004649950815104592
- OzakiIZernMALiuSWeiDLPomerantzRJDuanLRibozyme-mediated specific gene replacement of the alpha1-antitrypsin gene in human hepatoma cellsJ Hepatol199931536010424283
- RosenbergSAAebersoldPCornettaKGene transfer into humans-immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transductionN Engl J Med19903235705782381442
- CrystalRGMcElvaneyNGRosenfeldMAAdministration of an adenovirus containing the human CFTR cDNA to the respiratory tract of individuals with cystic fibrosisNat Genet1994842517527271
- FlotteTRCarterBConradCA phase I study of an adeno-associated virus-CFTR gene vector in adult CF patients with mild lung diseaseHum Gene Ther19967114511598773517
- FlotteTRGene therapy: the first two decades and the current state-of-the-artJ Cell Physiol200721330130517577203
- HuangKWHuangYCTaiKFChenBHLeePHHwangLHDual therapeutic effects of interferon-alpha gene therapy in a rat hepatocellular carcinoma model with liver cirrhosisMol Ther2008161681168718665156
- GrimmDZhouSNakaiHPreclinical in vivo evaluation of pseudotyped adeno-associated virus vectors for liver gene therapyBlood20031022412241912791653