Abstract
Paroxysmal nocturnal hemoglobinuria (PNH) is a clonal non-malignant hematological disease characterized by the expansion of hematopoietic stem cells (HSCs) and progeny mature cells, whose surfaces lack all the proteins linked through the glycosyl-phosphatidyl inositol anchor. This defect arises from an acquired somatic mutation in the X-linked phosphatidylinositol glycan class A gene, with subsequent clonal expansion of the mutated HSCs as a result of a concomitant, likely immune-mediated, selective pressure. The disease is characterized by complement-mediated chronic intravascular hemolysis, resulting in hemolytic anemia and hemosiderinuria; capricious exacerbations lead to recurrent gross hemoglobinuria. Additional cardinal manifestations of PNH are a variable degree of bone marrow failure and an intrinsic propensity to thromboembolic events. The disease is markedly invalidating, with chronic symptoms requiring supportive therapy – usually including periodical transfusions; possible life-threatening complications may also ensue. The biology of PNH has been progressively elucidated in the past few years, but therapeutic strategies remained unsatisfactory for decades, the only exception being stem cell transplantation, which is restricted to selected patients and retains significant morbidity and mortality. Recently, a biological agent to treat PNH has been developed – the terminal complement inhibitor eculizumab – which has been tested in a number of clinical trials, with exciting results. All the data from worldwide clinical trials confirm that eculizumab radically modifies the symptoms, the biology, and the natural history of PNH, strongly improving the quality of life of PNH patients.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH), first described in the 18th century, is characterized by the typical clinical triad of hemolytic anemia, bone marrow failure, and propensity to thromboembolism (CitationRotoli and Luzzatto 1989; CitationDunn et al 2000; CitationRotoli et al 2006; CitationHill et al 2007a). It is a rare disease, with a worldwide prevalence estimated in the range of 1–5 cases per million (CitationRosse 1996) regardless of ethnicity; an increased prevalence is reported in some regions which also harbor higher incidence of aplastic anemia (eg, Thailand and some other Asian countries) (CitationPramoonjago et al 1999). While the original denomination focused on a single component of the disease – namely intravascular hemolysis and subsequent hemoglobinuria – its complexity emerged in subsequent studies, and remained a riddle for generations of investigators. In the past 3 decades most pathogenic mechanisms have been unraveled, although some biological aspects have not been elucidated yet.
PNH is a hemolytic anemia resulting from the clonal expansion of one or a few abnormal hematopoietic stem cells (HSCs) (CitationOni et al 1970). This defect was identified by genetic studies in the 1990s as a mutation in the X-linked phosphatidylinositol glycan class A (PIG-A) gene (Takeda et al 1993; CitationLuzzatto et al 1997). PNH is therefore an acquired genetic blood disorder; however, the PIG-A mutation is likely insufficient to cause the disease and additional events may be involved (CitationRotoli and Luzzatto 1989), as noted below. Indeed, the genetic abnormality does not explain the entire clinical phenotype of PNH, especially marrow failure and thrombosis. Here we will review the natural history of PNH, focusing on clinical manifestations and their specific pathogenic mechanisms, aiming to give a theoretical background for targeted therapeutic strategies. In addition, we will discuss the clinical results from recent international trials using the complement inhibitor eculizumab (Soliris®) as treatment for PNH patients.
Pathophysiology
As mentioned above, PNH has a well-recognized genetic cause that is neither inherited nor transmitted to the progeny (Takeda et al 1993; CitationLuzzatto et al 1997). It develops through a somatic mutation in the PIG-A gene occurring in HSC(s), which originate progeny mature blood cells with the bizarre feature of a lack of several proteins from their surface. This abnormality was first described in the 1980s (CitationNicholson-Weller et al 1983; CitationSelvaraj et al 1988), and rapidly became the hallmark of PNH, although its relationship with the pathophysiology of the disease remained obscure at that time. Subsequently, it became clear that all the proteins missing from the PNH cell surface share a common mechanism for attaching to the cell membrane, which was identified in a specific glycolipid structure named glycosyl-phosphatidyl inositol (GPI) anchor (CitationMahoney et al 1992). In 1993, using complementation of GPI-anchored protein deficient cell lines and expression cloning, Kinoshita and colleagues first isolated the cDNA of the PIG-A gene (Takeda et al 1993; CitationMiyata et al 1993). PIG-A is a housekeeping gene located on the short arm of the X chromosome (Xp22.1); the organization of the genomic gene was described in 1994 (CitationBessler et al 1994). The PIG-A gene encodes an enzyme essential, in combination with at least two other proteins, to transfer N-acetyl glucosamine to phosphatidyl inositol; this is the very first step of the glycosyl-phosphatidyl inositol (GPI)-anchor biosynthesis (CitationHirose et al 1992; CitationTakahashi et al 1993). All mutations identified in PNH patients affect the PIG-A gene; no abnormalities were found in any of the other numerous genes involved in the subsequent metabolic steps of the GPI-anchor synthesis. In fact, PIG-A is the only X-linked gene, and a single mutational event is sufficient to impair GPI-anchor biosynthesis (also in females, as a result of X-chromosome functional inactivation). As an exception, an autosomal recessive inherited GPI deficiency resulting from a mutation of the PIG-M gene has been recently described in two kindreds; however, the phenotype was clearly different from that of PNH, with a partial GPI deficiency resulting in a clinical syndrome characterized by propensity to thrombosis and seizures, in the absence of significant hemolysis (CitationAlmeida et al 2006; CitationAlmeida et al 2007).
The role of the GPI-anchor in cell biology seems to be very important, as supported by its high conservation in eukaryotic cells, including yeast and trypanosome; however, the functional implications of the GPI-anchoring of proteins, and of the GPI-anchor itself, are not completely understood. It is known that GPI-anchored proteins (GPI-APs) are not randomly distributed on the cell surface, but rather constitute membrane subdomains enriched in specific lipids, such as cholesterol and sphyngolipids; these structures, called rafts, seem to be involved in membrane trafficking and remodeling, including capping and endo-, exo-, and potocytosis, as well as in signal transduction (eg, immunological synapse) (CitationGoebel et al 2002).
As a result of the GPI-anchor deficiency, PNH cells lack all GPI-APs on their surface (); so far, more than 25 different proteins sharing the GPI-anchoring system have been described on blood cells () (CitationRotoli et al 1993), their specific functions being only partially known. However, some GPI-APs are functionally well-described proteins, and have been essential to fully unravel the pathogenic mechanisms of some clinical manifestations of PNH (). In fact, among the GPI-APs there are two complement-regulatory proteins, namely CD59/MIRL (Membrane Inhibitor of Reactive Lysis) (CitationZalman et al 1987; CitationOkada et al 1990) and CD55/DAF (Decay Accelerating Factor) (CitationNicholson-Weller et al 1983; CitationNicholson-Weller et al 1985). They act at different levels of the complement cascade (): CD55 controls early complement activation, inhibiting C3 and C5 convertases (CitationBurge et al 1981; CitationMedof et al 1984), while CD59 interferes with the terminal effector complement, blocking the incorporation of C9 onto the C5b-C8 complex, forming the membrane attack complex (MAC) (CitationShin et al 1986; CitationZalman et al 1986). The deficiency of such proteins leads to the increased susceptibility of PNH red blood cells (RBCs) to complement mediated lysis, resulting in the typical intravascular hemolysis (CitationHolguin et al 1989). In addition, hypersensitivity to complement activation in vitro leads to red cell destruction upon serum acidification, a feature utilized as a diagnostic assay for PNH for several decades (the HAM test, recently replaced by flow cytometry). A low level of complement activation is a constant physiological phenomenon greatly amplified during inflammatory or infectious diseases; thus, PNH erythrocytes chronically undergo intravascular hemolysis in PNH patients due to CD55 and CD59 deficiency, with a lifespan reduced to 10% compared with that of normal RBCs (CitationWiedmer et al 1993). GPI-AP deficiency on RBCs may be complete (PNH type III cells) or partial (PNH type II cells), resulting in different sensitivity to complement mediated lysis, as initially described by Rosse (CitationRosse and Dacie 1966; CitationRosse 1990). However, particular aspects of hemolysis in PNH are still debated; for instance, it is still unclear why exacerbations occur mainly during the night. In a functional hierarchy, CD59 was demonstrated as playing a major role (CitationWilcox et al 1991). This is also supported by the observation of rare patients harboring inherited deficiency of a single molecule (CitationYamashina et al 1990; CitationShichishima et al 1999); in fact, a unique CD59-deficient patient had a PNH-like picture, whereas subjects deficient in CD55 on red cell membrane (ie, those carrying the rare Inab blood group phenotype) showed no clinically significant hemolysis.
Figure 1 Paroxysmal nocturnal hemoglobinuria (PNH) pathophysiology. A. PIG-A mutations: mutation occurring in the PIG-A gene may affect the synthesis or the function of the encoded protein, resulting in the impairment of the first step of GPI-anchor biosynthesis. As a result, all proteins linked to the cellular membrane through this mechanism are lacking from the surface. B. The dual pathophysiology theory: PIG-A gene mutation may occur without provoking the PNH disease. To induce the clonal expansion necessary for developing clinical PNH, additional factors should occur, which create conditions permissive to expansion of mutated cells. Such extrinsic factors are postulated similar to those inducing stem cell exhaustion in aplastic anemia, which include immune mediated attack of hematopoiesis.
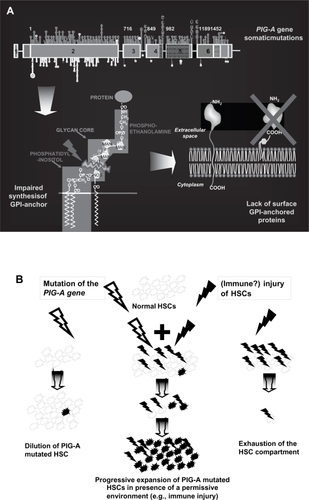
Figure 2 Effect of glycosyl-phosphatidyl inositol anchored proteins (GPI-AP) deficiency on blood cell populations. A. The lack of the complement regulators CD59 and CD55 on red blood cells (RBCs) accounts for their susceptibility to complement mediated lysis resulting in the clinical hallmark of PNH, intravascular hemolysis. B. Deficiency of complement regulators on platelets may account for their putative propensity to activation and aggregation, possibly leading to increased clot formation and thromboembolism. Thrombophilia in PNH may also be related to NO consumption occurring in the presence of increased free Hb resulting from hemolysis, as well as to the impairment of the fibrinolytic system due to the lack of membrane uPAR (see text). C. The lack of anchored proteins GPI-AP has been postulated as putative cause of the expansion of the PNH clone over normal hematopoiesis; the presence of a GPI-linked immune target or a more generic reduced sensitivity to immune effector mechanisms has been hypothesized to explain the immune escape of the PNH clone, but definitive evidence is still lacking.
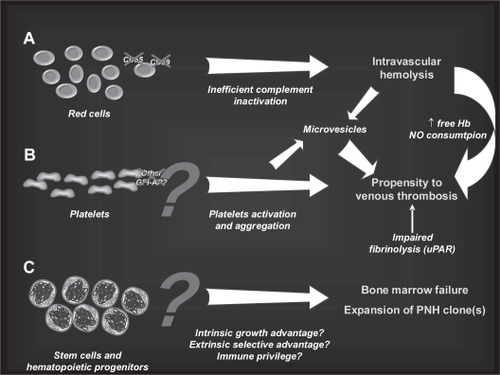
Figure 3 Mechanism of hemolysis in paroxysmal nocturnal hemoglobinuria red blood cells (PNH RBCs). The activation of both the alternative and the classical pathways of the complement cascade converge on the activation of a C5 convertase; PNH RBCs lack CD59, which normally inhibits the assembly of membrane attack complex (MAC), and undergo hemolysis. In addition, the early alternative pathway is dysregulated on PNH as a result of the absence of the C3 convertase inhibitor CD55; thus, PNH RBCs experience uncontrolled complement activation through the alternative pathway. This may explain C3 accumulation on PNH RBCs when hemolysis is blocked in presence of the terminal complement inhibitor eculizumab.
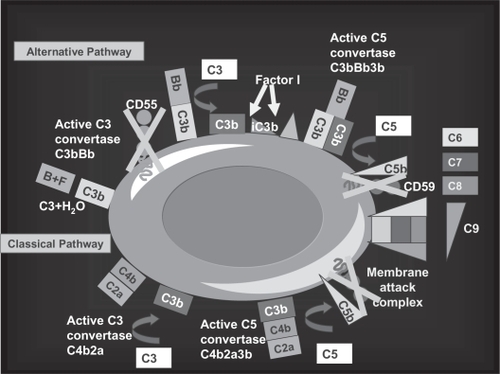
Table 1 Glycosyl-phosphatidyl inositol (GPI)-linked proteins on blood cells and their function
CD55 and CD59 are deficient on RBCs, but also on white blood cells (WBCs) and platelets (CitationNicholson-Weller et al 1985); however, as far as WBCs are concerned, there is no evidence of reduced leukocyte lifespan in PNH patients, probably because nucleated cells have an additional transmembrane (ie, non GPI-linked) complement inhibitor (CD46) (CitationChristmas et al 2006). In contrast, uncontrolled complement activation on platelets has been hypothesized as a possible explanation for another clinical hallmark of PNH, namely the propensity to thromboembolic events (). In fact, complement activation might lead to platelet activation and aggregation, enhancing clot formation; however, no demonstration of such a mechanism has been provided up to now, and other different mechanisms have been postulated. In PNH, microvesicles are known to be released upon hemolysis and complement activation from RBCs (CitationHugel et al 1999), WBCs (monocytes) and platelets (CitationWiedmer et al 1993; CitationSimak et al 2004), and even from the endothelium; their procoagulant action is commonly accepted, but their specific role in the pathophysiology of thromboembolisms in PNH still needs to be documented. An additional mechanism of thrombophilia in PNH might be the impairment of the fibrinolytic system, due to the lack of membrane-bound urokinase-type plasminogen activator receptor (uPAR), which is GPI-linked, and to the excess of its soluble form (CitationNinomiya et al 1997; CitationSloand et al 2006). Finally, thrombophilia in PNH may arise from the build-up of cell-free plasma hemoglobin due to hemolysis (CitationOlsen et al 1996; CitationSchafer et al 2004). This may occur through ability of free hemoglobin to scavenge nitric oxide from plasma, blocking its inhibitory action on platelet aggregation and ahesion to vascular endothelium (CitationRother et al 2005). Obviously, additional acquired or inherited risk factors may increase individual predisposition to thrombosis; genetically, the Factor V Leiden mutation and the 677 C>T methylenetetrahydrofolate reductase gene variant (as well as MTHFR polymorphisms leading to hyperhomocysteinemia) may have a relevant role, although this has not been proven so far (CitationNafa et al 1996).
Looking at PNH biology in depth, we have discussed the original molecular defect in HSCs and some subsequent functional abnormalities of the progeny blood cells leading to specific clinical manifestations; however, what is the missing link and, likely, the true mystery of PNH is the reason for the expansion of GPI-deficient clones within the context of a bone marrow failure syndrome. Of note, hematopoiesis in PNH is a mosaicism of phenotypically normal HSCs and PIG-A mutated HSCs; in addition, regardless of the genetic defect, hematopoiesis is impaired in almost all PNH patients, even in the presence of a broad clinical heterogeneity (CitationRotoli et al 1982; CitationMaciejewski et al 1997). As mentioned above, PNH is a true disease of the HSC, as marked by the PIG-A mutation; however, the HSC defect does not entirely explain the phenotype. In fact, a series of observations suggest that the PIG-A mutation is necessary but not sufficient per se to cause the disease:
A few (10–50 cells per million) circulating granulocytes harboring the PNH phenotype may be demonstrated also in healthy individuals by flow cytometry, and their specific PIG-A mutation may be identified by a nested PCR technique (CitationAraten et al 1999; CitationHu et al 2005).
GPI-AP deficient lymphocytes were detected in lymphoma patients shortly after treatment with alem-tuzumab (a monoclonal antibody that recognizes the GPI-AP CD52), and progressively disappeared after treatment withdrawal (Hertestein et al 2005).
The expansion of the aberrant PNH clone seen in PNH patients could not be reproduced in several murine models developed with the aim of creating the disease (CitationRosti et al 1997; CitationJasinski et al 2001; CitationKeller et al 2001), even when embryonic stem cells were employed. Thus, at least in these models, the PNH clone does not seem to have any intrinsic proliferative advantage to overcome normal hematopoiesis (CitationJasinski et al 2001; CitationKeller et al 2001); this is also confirmed by several data on in vitro growth of patient-derived PNH and wild-type hematopoietic progenitors (CitationYoung and Maciejewski 2000).
PNH hematopoiesis is certainly clonal, but not necessarily monoclonal. In fact, PNH hematopoiesis may in some cases be oligo- rather than monoclonal, as initially supported by differential susceptibility to complement lysis (CitationRosse et al 1966), subsequently by flow cytometry (Citationvan der Schoot et al 1990), and finally confirmed by PIG-A sequencing (CitationEndo et al 1996); this observation raised the question of whether the expansion of more clones carrying the same functional defect, but molecularly heterogeneous, is compatible with a random process, or rather may result from not stochastic processes, such as selection. The latter hypothesis may be supported by the observation that relapse of PNH may be sustained by clones harboring PIG-A mutations which are different from those identified at diagnosis (CitationNafa et al 1998).
According to this background, the hypothesis of a dual pathophysiology for PNH has been developed, also known as the “relative advantage” () (CitationRotoli and Luzzatto 1989; CitationLuzzatto and Bessler 1996) or the “escape” theory (CitationDunn et al 2000). According to this theory, a mutation in the PIG-A gene might be a fairly common phenomenon, with no major biological consequences, because the mutated cell has no chance of expanding in the presence of a vast majority of normal cells. In fact, no intrinsic proliferative advantage has been demonstrated in PNH hematopoietic progenitors (CitationAraten et al 2002). However, external factors may alter this equilibrium, creating an environment permissive for the expansion of PNH clone(s), and possibly leading to the occurrence of a single PIG-A mutated stem cell sustaining hematopoiesis even for the rest of the patient’s life (CitationNishimura et al 2002). The nature of such extrinsic trigger may be inferred from the observation that most PNH patients also show evidence of bone marrow failure, and that several aplastic anemia (AA) patients may harbor distinct PNH clones. Thus, it is reasonable to hypothesize that pathogenic mechanisms involved in AA may also play a pivotal role in PNH, representing the additional factor required for developing clinical PNH once a PIG-A mutation has occurred (without any specific chronologic order).
Indeed, in acquired marrow failure syndromes HSCs are known to be intrinsically normal, but subject to an extrinsic damage affecting their hematopoietic function. In idiopathic AA immunological mechanisms play a pivotal role in damaging the hematopoietic compartment, leading to HSC pool consumption or functional impairment and subsequent pancytopenia. A wealth of experimental and clinic evidence supports the presence of anti-hematopoiesis immune attack, although the target antigens are still undefined, as are the mechanisms leading to the breach in immune tolerance. Unknown triggers of autoimmunity induce a cellular immune response resulting in a preferential expansion of specific T cell clones that may damage the hematopoietic progenitors, directly or through indirect mechanisms, such as the production of inhibitory cytokines. Stem cell damage can be mediated by cytokine-transduced inhibition (mostly type I cytokines, such as interferon-γ and tumor necrosis factor-α) (CitationSato et al 1997; CitationDufour et al 2001; CitationSloand et al 2002), or by direct cell-mediated killing due to cytotoxic lymphocytes (CTLs). Such mechanisms, which may attack innocent bystander cells in addition to the primary target cells, ultimately result in apoptosis, the main key mechanism of HSC damage. Circulating and marrow CTLs have been demonstrated in vivo in AA patients, and their inhibitory effect on hematopoiesis has been documented in vitro (Zoumbos et al 1995; CitationNakao et al 1997; CitationZeng et al 2001; CitationKook et al 2002; CitationRisitano et al 2002; CitationPlasilova et al 2003); recently, we have dissected this cellular immune response at the molecular level through the identification of in vivo dominant T cell clonotypes, which are evidence of a pathogenic antigen-driven immune response (CitationRisitano et al 2004).
In PNH, an antigen-driven immune response targeting the marrow tissue may be postulated, which possibly correlates with the selective expansion of the PIG-A mutated clone; in fact, if the target on HSC membrane is a GPI-linked molecule, PNH HSC may escape this injury while normal HSC are killed. Evidence of immune derangement in PNH has been produced; as for AA, oligoclonality of the T cell pool has been reported (CitationKaradimitris et al 2000), and immunodominant pathogenic CTL clones may be detected in most PNH patients (CitationRisitano et al 2002, Citation2004; CitationPlasilova et al 2004; CitationGargiulo et al 2007), sometimes phenotypically resembling a subclinical LGL proliferation (CitationRisitano et al 2005). In addition, according to a recent report, these effector T cells overexpress the activating isoforms of inhibiting superfamily receptors which elicit a powerful cytolytic activity (CitationPoggi et al 2005).
Gene expression profiling has been employed to investigate putative differences between normal and PNH HSCs: when CD34+ cells from PNH patients were separated according to the presence or absence of GPI-AP on their surface, distinct patterns of gene expression were identified. Phenotypically normal (GPI-AP positive) CD34+ cells harbored diffuse abnormalities of their transcriptome, with over-expression of genes involved in apoptosis and immune activity, paralleling the findings seen in CD34+ cells of AA patients. By contrast, phenotypically abnormal PNH CD34+ (GPI-AP negative) showed a gene expression profiling closer to that obtained in CD34+ cells from healthy individuals (CitationChen et al 2005). The presence of sublethal damage in phenotypically normal HSCs but not in their PIG-A mutated counterpart strongly supports the presence of an immune attack to the hematopoietic stem/progenitor cells that spares PNH cells; thus, they ultimately expand as a result of a selective pressure negatively acting on normal hematopoiesis. The “escape” of PNH cells may be interpreted in various manners. Contradictory data have been produced on a putative differential sensitivity to inhibitory stimuli between normal and PNH cells; susceptibility to apoptosis has been reported to be increased, normal, or decreased in different models. Recently, it has been shown that human cell lines carrying the PIG-A mutation are less susceptible to NK-mediated killing compared to their normal counterpart (CitationNagakura et al 2002). In a more sophisticated model, GPI-deficient cells were not able to induce primary and secondary stimulation of both antigen-specific and alloreactive T cells, providing experimental support to the hypothesis that the PNH clone could inefficiently interact with the immune system (CitationMurakami et al 2002). However, the actual mechanisms causing the escape are still elusive. They may include the absence of specific GPI-APs directly targeted by effector immune cells, or a protection due to the absence of important molecules involved in cell-cell interaction (eg, accessory molecules). Alternatively, a broader impaired sensitivity to common effector mechanisms may be hypothesized, possibly due to the lack of GPI-APs or to non-specific structural changes of the raft structure in the outer surface.
Clinical features and natural history
Diagnosis
PNH has to be suspected in patients showing mild to severe anemia with moderate reticulocytosis, elevated serum lactate dehydrogenase (LDH) and possibly mild jaundice, with negative Coombs test; all these clues suggest a non-immune hemolytic anemia. The occurrence of dark urine and urinary hemosiderin, both evidence of intravascular hemolysis, strongly suggest PNH. Additional signs to be considered are the presence of mild to severe leucothrombocytopenia and/or a history of thromboembolic events of unknown origin, including cerebrovascular accidents. In specific conditions, PNH may be considered even in the absence of clinically evident hemolytic anemia, such as in patients with AA or those showing recurrent thromboembolic events in the absence of documented risk factors; all these patients may deserve a careful screening for PNH.
Until the eighties, the diagnosis was confirmed by the Ham test, which assesses the lysis of RBCs in acidified serum; less specific but simpler tests were the sucrose and the sugar water test (both based upon lysis in serum with reduced ionic strength). However, at present the diagnosis of PNH is based on flow cytometry analysis of blood cells (CitationParker et al 2005; CitationRotoli et al 2006); the high sensitivity and specificity of this analysis made the Ham and similar tests obsolete. In fact, fluorochrome-conjugated monoclonal antibodies specific to several GPI-APs expressed on the various blood cell lineages are available for routine testing; thus, simultaneous multi-parameter analysis allows accurate detection of GPI-AP deficient populations, measuring their extent within each cell lineage. By this technique, one or two RBC populations with abnormal expression of GPI-APs may be shown in PNH patients: one completely lacking GPI-AP expression (type III PNH cells), and another characterized by GPI-AP faint (dim) expression (type II PNH cells). These findings match the observation initially made by Rosse (CitationRosse 1990), demonstrating the presence of distinct subpopulations of RBCs with different sensitivity to complement-mediated lysis; as discussed above, this may also imply that different populations may be genetically different, reflecting the expansion of two or more clones (as a result of a selective pressure for PIG-A mutated HSCs). The finding of GPI-AP deficient populations may be obtained for each cell lineage, even if discrimination between type II and type III PNH cells is more difficult for white blood cells; antibodies specific for GPI-APs selectively expressed by different cell lineage may render the test more sensitive and specific. The simultaneous absence of different GPI-linked proteins on the same cells validates the specificity of the test; using this method, flow cytometry analysis detects even very small PNH clones (below 1% of the tested cell population) (CitationAlfinito et al 1996; CitationBoccuni et al 2000; CitationRichards et al 2000). More recently, the novel fluorescent reagent aerolysin, which specifically binds the GPI anchor, showed even greater sensitivity, with detection limits dropping by 1–2 logs compared with standard flow cytometry (CitationBrodsky et al 2000; CitationSutherland et al 2007). Molecular studies on DNA or mRNA, aimed to identify the causative specific mutation within the PIG-A gene, are clinically unnecessary (even normal individuals may harbor a few PNH cells); in fact, they do not add clinically relevant data and may be even misleading, so they are now usually limited to research purposes.
Classification of PNH
Recently, a group of experts has classified PNH into distinct clinical entities, based on the improved information coming from the use of flow cytometry. According to this classification (CitationParker et al 2005), three subgroups have been proposed: i. classic PNH; ii. PNH in the setting of another bone marrow disorder (eg, aplastic anemia, myelodysplastic syndrome, or myelofibrosis); iii. PNH-subclinical, in the setting of another bone marrow disorder (as above). However, this classification may be argued, and some concerns need to be addressed. There is overlap between classic PNH and PNH in the setting of another bone marrow disorder, given that hemolysis and some degree of bone marrow failure are hallmarks in both groups. Indeed, the presence of signs of marrow failure (ie, leukopenia, thrombocytopenia, reduced number of circulating and bone marrow colony forming cells) cannot be considered a discriminator between different groups of patients. In our opinion, some degree of marrow failure is present in all classic PNH patients, consistent with the pathophysiology of the disease, as described by the dual theory discussed above. Patients showing severe marrow failure may have various degrees of hemolysis, from the massive type as in classic PNH, to the subclinical one, with all the intermediate forms; thus it is difficult to identify precise cut-offs. Similarly, some degree of morphologic abnormalities definable as dysplasia, mostly of the erythroid lineage, may be consistent with stressed hematopoiesis; thus, the difference between PNH and PNH in the context of myelodysplastic syndromes is not clear-cut. Even the presence of karyotypic abnormalities may be an epiphenomenon resulting from clonal hematopoiesis and fixation of possible mutations, rather than an indicator of the presence of a true myelodysplastic (preleukemic) clone (CitationAraten et al 2001). Finally, PNH-subclinical refers to the presence of very small GPI-deficient clones deprived of any clinical significance; in this context, the term PNH itself is misleading and should not be used, since none of the clinical manifestations of this condition are present. The term “acquired GPI-AP deficiency” seems to be more appropriate, and might be extended to the entire classification (possibly including other acquired GPI-AP deficiency, such as those emerging within lymphocyte subsets after treatment by alemtuzumab) (CitationHertenstein et al 1995). In addition, this definition also better applies to the rare patients showing GPI-AP deficient neutrophils and monocytes, with no PNH erythrocytes; such patients, who possibly reflect a PIG-A mutation occurring in committed hematopoietic progenitors rather than in a multipotent HSC, remain often undiagnosed and are excluded from the proposed classification.
Natural history
As recalled above, PNH is dominated by the clinical triad of chronic intravascular haemolytic anemia, propensity to develop thromboembolic events and stigmata of bone marrow failure. While intravascular hemolysis is constant in all typical PNH patients, thrombosis and cytopenia have a variable prevalence within the PNH population and may be undetectable in single patients. This is mostly true for thromboembolic events, which are erratic and unpredictable, and may not affect all individuals, although recurrence is frequent after the first episode (CitationHillmen et al 1995; CitationSocie et al 1996).
Hemolysis is present in all PNH patients, but anemia occurs with variable severity; this results from the degree of complement activation and subsequent hemolysis, as well as from the capacity of compensatory erythropoiesis, which depends in turn on the efficiency of the residual marrow function (indicated by reticulocyte count). Indeed, the size of the PNH clone within the total RBCs and the type of PIG-A molecular lesion accounting for the different sensitivity to complement-mediated lysis (ie, type II and type III PNH RBCs) shape the clinical picture, with some patients having an almost normal hemoglobin level and others requiring massive transfusional support. Exacerbations of hemolysis are typical, and account for the term paroxysmal nocturnal hemoglobinuria; they are unpredictable in most cases, although some patients experience quite regular crises. Paroxysms likely reflect transient complement activation above the physiological low-level (which is due to the alternative pathway), and are often associated with external triggers such as infections and inflammatory responses, which massively activate both the classical and alternative complement pathways. Beside the paroxysms, anemia may significantly vary during the clinical course of individual patients, with cases suddenly worsening and others unexpectedly improving in the absence of any known reason; anecdotal cases showing spontaneous remission of the disease have also been reported (CitationHillmen et al 1995; CitationSocie et al 1996; CitationPeffault de Latour et al 2006). Signs of intravascular hemolysis are jaundice, hemoglobinuria, increased LDH, and increased unconjugated bilirubinemia; the symptoms are those common to other types of hemolytic anemia, such as asthenia, fatigue to the extent of lethargy, with significant quality of life impairment. In addition, some specific symptoms have been described, mostly in association with paroxysms, including dysphagia, painful abdominal crises and erectile dysfunction; these symptoms have been linked to smooth muscle dystonia possibly resulting from local nitric oxide (NO) deficiency subsequent to NO consumption by excess free hemoglobin (Rosse 2000; CitationMoyo et al 2004; CitationRother et al 2005). All these symptoms significantly affect the well-being of PNH patients, even if they are often underestimated by physicians because they are not life-threatening.
Cytopenias are frequent, and may be anything from mild to severe, depending on the underlying bone marrow defect; some patients may develop true aplastic anemia during the course of the disease, and may require specific treatment. The consequences of cytopenia include increased risk of infection and hemorrhage; their management depends on the severity of the cytopenia and on the clinical picture, paralleling that of aplastic patients. In addition, a specific infectious risk may be present as a result of impaired function of PNH neutrophils and monocytes, mostly affecting microorganism destruction through the radical oxygen species pathway (CitationCacciapuoti et al 2007).
Thrombosis is the most life-threatening clinical manifestation. It usually affects veins, and occurs in sites unusual for patients with other types of thrombophilia; typical sites are the suprahepatic veins, leading to the Budd-Chiari syndrome, mesenteric veins, the splenic vein, and cerebral veins. The incidence of thrombosis seems to be different according to ethnicity, with Asian patients showing lower risk than Europeans or Americans, or particularly Afro-Americans and Hispanics (CitationNishimura et al 2004). Maybe this results from the multifactorial pathophysiology of thrombosis in PNH as previously discussed, and from the possible contribution of genetic factors predisposing to thrombosis. In various observations thromboses have been reported associated with massive intravascular hemolysis and larger WBC PNH clones (CitationHall et al 2003; CitationMoyo et al 2004); even if this statement has to be confirmed, it has to be remarked that even patients with small clones have a significant thrombosis risk compared to healthy individuals (CitationHall et al 2003). Thromboembolic events may significantly affect survival; as a whole, thrombosis may affect half of PNH patients, accounting for at least one third of mortality.
PNH is a clonal but non-malignant disease; even if progression to acute leukemia has been occasionally reported (sometimes leading to overestimation as a result of preferential reporting), it has been estimated in the range of 2%–3%, similar to that reported for AA patients. PNH is a very rare disease; information on large series are few and somewhat dated; however, the estimated median survival may be over 10 years, with a quarter of patients surviving longer than 25 years (CitationSocié et al 1996; Hillmen et al 2005; CitationPeffault de Latour 2006). These data may have a bias due to a negative selection of patients presenting with early complications (eg, those with lethal thrombosis), and do not consider changes in the management of PNH, including transplant procedures and, more recently, eculizumab treatment. As discussed, the major causes of death are thrombosis, followed by infectious complications and, lastly, hemorrhage; thus, an aplastic presentation significantly affects the prognosis. Notwithstanding the relatively long survival, the course of the disease may be stormy, with frequent hospitalizations, possible comorbidities, and subsequent poor quality of life.
Management: the pre-eculizumab era
Supportive treatment
The treatment of PNH is mainly supportive, aiming to control the clinical manifestations of the disease. Thus, it may be divided into management of hemolysis and anemia, management of thrombophilia, and management of bone marrow failure.
Management of hemolysis and anemia
Hemolysis and the subsequent anemia are the hallmarks of PNH, and usually require specific treatment. Unfortunately, until the new millennium there were no clinically satisfactory options for controlling hemolysis. Steroids were broadly used as chronic administration or for acute hemolytic crises. Indeed, some investigators claim that steroids are useful in controlling chronic complement-mediated hemolysis (Issaragrisil et al 1987; CitationBourantas 1994) and, even more, paroxysmal crises, likely interfering with complement activation and/or underlying conditions triggering the complement cascade; however, so far no conclusive data or convincing mechanisms of action have been provided. Clinically, steroids seem to ameliorate patient well-being even in the absence of complete control of hemolysis (CitationParker et al 2005); in particular, they may be effective in improving symptoms associated with paroxysmal crises, such as dysphagia and abdominal pain (possibly as a result of less severe crises). However, since the long-term toxicity of chronic steroidal therapy, in the range of 0.5–1 mg/kg, advises against its systematic use in PNH patients, it is currently not recommended.
Androgens have been utilized, too, with limited benefit (CitationHarrington et al 1997); however, given their potential utility in stimulating erythropoiesis and megakaryocytopoiesis (CitationKatayama et al 2001), they are primarily indicated in the presence of signs of marrow impairment rather than to control hemolysis. Once again, a risk-benefit evaluation should be specifically made for all individual cases, given that liver toxicity, virilizing action, and other side effects have to be considered; in addition, some concerns about a potential increase in the Budd-Chiari syndrome have been raised by some physicians (CitationParker et al 2005). A few patients in our series benefited from danazol, a non-virilizing anabolic steroid, given at doses of 400–600 mg/day.
Thus, given the impossibility of blocking intravascular hemolysis, the main strategy to control anemia was transfusions. RBC transfusions are given to PNH patients according to their hemoglobin level, which may be highly variable even in the same patient; as for other anemic patients, transfusions should be administered to maintain hemoglobin level above 8 gm/dL. Transfusions transiently improve anemia-related symptoms; however, refractoriness to transfusion may develop, and is considered a severe complication requiring more aggressive treatment – namely stem cell transplantation. In contrast to other transfusion-dependent patients, those with PNH have a low risk of iron overload, given the massive iron loss due to hemoglobinuria and hemosiderinuria. Thus, strategies for iron chelation are not necessary; in fact these patients may sometimes even require iron supplementation to sustain the compensatory erythropoiesis (CitationRosse 1982). Similarly, vitamin B12 and folate supplementation are usually indicated to sustain the enhanced erythropoiesis (CitationRosse 1982); furthermore, endogenous erythropoiesis may also be increased by recombinant erythropoietin, mostly in cases with inadequate production (CitationStebler et al 1990; CitationBourantas 1994; CitationMcMullin et al 1996). Paradoxically, all these strategies, including iron replacement, may lead to increased hemolysis as a result of increased PNH hematopoiesis, contrary to what is seen following suppression of erythropoiesis by transfusions.
Management of thrombophilia
The management of the propensity to develop thrombosis is a hot issue in PNH, given that this complication represents the first cause of death. Unfortunately, there are no controlled prospective clinical trials on primary and secondary thrombosis prophylaxis, nor on acute treatment of the event. The issue of primary prophylaxis is controversial, and no consensus exists; some physicians advocate the use of warfarin for all newly diagnosed PNH patients, while others do not use any prophylaxis. Both approaches are reasonable, given the unpredictability of thromboembolic events and the lack of evidence supporting any of these strategies; possible benefits are counterbalanced by the risk of hemorrhage from warfarin therapy, which may be considerable in PNH patients with low platelet count. With the recent attempt to identify patients at higher risk of thrombosis, a reasonable compromise may be the adoption of prophylaxis for patients with WBC PNH clones larger than 50%, or those with additional genetic risk for thrombosis, including ethnicity. Using such and approach, Hall and colleagues (CitationHall et al 2003) demonstrated a very low incidence of thromboembolic events compared with historical controls; however, these data need to be confirmed in larger series and possibly in prospective randomized trials that include alternative, more manageable, agents such as antiplatelet drugs or the new thrombin inhibitors.
The opinion on secondary prophylaxis is less controversial, with a general agreement that all PNH patients experiencing any thromboembolic event should remain life-long on anticoagulants; however, even in this setting, no consensus exists on the best strategy. Low-molecular-weight heparin, as well as warfarin at different therapeutic ranges, are both utilized, with some physicians even considering the addition of anti-platelet agents. However, despite this extended pro-phylaxis, recurrence of thrombosis (either as new events or progression of the existing ones) is frequent and affects the survival of PNH patients (CitationAudebert et al 2005). Moreover, life-threatening hemorrhagic events are quite frequent in this cohort of patients, mostly when concomitant thrombocytopenia is present (CitationHall et al 2003; CitationMoyo et al 2004).
Finally, the management of an acute thromboembolic disease may require an intensive therapy similar to that for myocardial infarct; fibrinolytic therapies using tissue plasminogen activator have been exploited, showing efficient clearance of the thrombus in individual cases (CitationMcCullin et al 1994; CitationHauser et al 2003).
Management of marrow failure
The management of marrow failure in PNH patients is the same as that for AA patients. Indeed, in addition to supportive strategies such as anti-infectious, anti-thrombotic, and anti-hemorrhagic prophylaxis and/or treatment, etiologic therapies can also be attempted. According to the pathogenic mechanisms and the dual hypothesis described above, an immune-mediated inhibition of hematopoiesis is postulated in PNH, similar to that demonstrated in AA. Thus, immunosuppressive strategies have been reasonably attempted in PNH patients, using regimens with antilymphocyte or antithymocyte globulin associated with high dose prednisone and cyclosporin A, as those recommended in AA (CitationTichelli et al 1992); the results have been quite variable (CitationSanchez-Valle et al 1993), even if extensive studies are still lacking. Newer immunosuppressive agents such as the anti-CD52 monoclonal antibody alemtuzumab may be an alternative candidate as immunosuppressant; in this setting, there is no concern about the potential risk of selecting for PNH hematopoiesis, given that the GPI-linked CD52 is not expressed on HSCs.
So, after 3 decades of efforts the conclusion is that no therapy adequate to the patients’ medical needs had been developed, and most patients had to face every day the symptoms of their disease.
Curative strategies
The only curative approach for PNH is allogeneic hematopoietic stem cell transplantation (SCT); alternative strategies include cell therapy (insertion of molecules on the outer surface of blood cells) (CitationHill et al 2006) and gene therapy (insertion of a functional PIG-A gene in early hematopoietic progenitors), but all experimental efforts have so far been unfruitful and seem to be far away from clinical applications. In addition, if the escape theory is correct, the latter approach may not result in clinical benefit in PNH, since a repair of the damaged cell should result in cell destruction, as is believed to occur for normal hematopoiesis in PNH patients.
SCT has been exploited since the late 1980s, with very positive long-term results; however, SCT is burdened by significant early transplant-related mortality. Most reports in the literature refer to single cases or small series from single institutions (CitationBemba et al 1999; CitationSaso et al 1999; CitationRaiola et al 2000), while large prospective studies are lacking. An overview has been recently published by Parker et al (CitationParker et al 2005), who collected data from 67 patients transplanted from different types of donors (syngeneic, sibling, or HLA-identical unrelated) and using different types of conditioning (myeloablative or reduced intensity). Data from the entire group showed a 75% long-term survival, which is higher than that reported in a majority of individual series, likely as a result of a reporting bias. A recent survey from the Italian Transplant Group (GITMO) on 23 PNH patients transplanted between 1988 and 2006 showed a 6-month treatment-related mortality of 26% and an overall survival of 70%, with a median follow-up of 107 months (CitationSantarone et al 2008). A similar retrospective study is currently ongoing within the European Blood and Marrow Transplantation (EBMT) group, and promises to provide useful information for evidence-based decision making; preliminary results confirm an overall survival in the range of 55%–65%, with older age and acute GvHD significantly affecting the outcome, and apparently comparable results between sibling and unrelated transplants.
Given these results, guidelines for SCT in PNH are hard to define; the procedure should be restricted to young patients with a suitable HLA matched donor, even if the improvement in transplant procedures may extend SCT even to older recipients, once the indication is established. A clear indication may be an underlying bone marrow failure unresponsive to immunosuppression; thrombosis and refractoriness to transfusion may be additional indications, even if they should be re-evaluated in the present era of biological agents, namely eculizumab. Specific procedures to achieve the best outcome will, we hope, result from the ongoing retrospective and prospective studies, mostly investigating whether the new reduced-intensity conditioning regimens may further reduce transplant-related mortality as well as long-term complications.
Management: the eculizumab era
Mode of action and pharmacokinetics
Eculizumab (h5G1.1-mAb, Soliris®, Alexion Pharmaceuticals) is a humanized monoclonal antibody (mAb) () derived from the murine anti-human C5 mAb (CitationRother et al 2007). It specifically binds the terminal complement fraction 5, thereby inhibiting the cleavage to C5a and C5b () (CitationMatis and Rollins 1995), thus blocking the formation of the membrane attack complex (MAC), which is the terminal effector mechanism leading to intravascular hemolysis of PNH RBCs. In addition, eculizumab prevents the release of pro-inflammatory mediators resulting from C5a cleavage. The blockade of the complement cascade at the level of C5 does not affect early complement components (such as C3 and its cleavage products), preserving pivotal functions leading to clearance of immune complexes and microorganisms (CitationMatis and Rollins 1995). Eculizumab was initially investigated in patients suffering from rheumatoid arthritis, psoriasis, systemic lupus erythematosus, dermatomyositis, and idiopathic membranous glomerulopathy; however, PNH appeared the best candidate disease to benefit from eculizumab treatment. In fact, the lack of CD59 on red cell membranes accounts for uncontrolled C5b-9 (MAC) assembly, finally resulting in intravascular hemolysis (); theoretically, eculizumab may compensate for the absence of CD59 blocking the MAC formation, thus rendering irrelevant the absence of CD59 on PNH RBCs ().
Figure 4 Eculizumab: the molecule. The structure of the anti-complement 5 humanized monoclonal antibody eculizumab (h5G1.1 mAb).
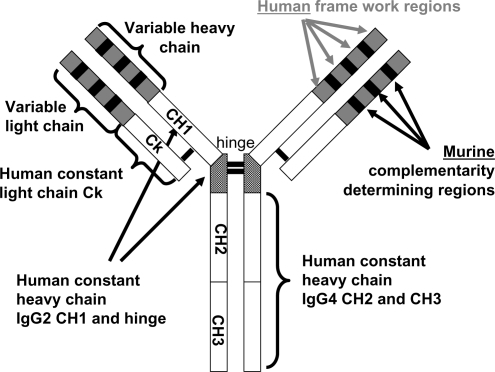
Figure 5 The complement cascade and the C5 blockade by eculizumab. Eculizumab blocks the cleavage of C5 to C5a and C5b; all earlier steps of the complement cascade are preserved, including C3 cleavage.
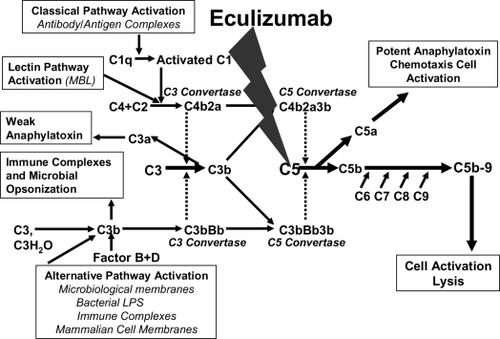
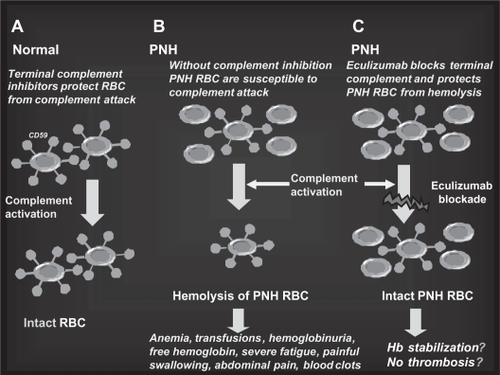
Figure 6 The rationale for eculizumab in paroxysmal nocturnal hemoglobinuria (PNH). A. Normal red blood cells (RBCs) are protected from complement attack by CD59. B. PNH RBCs are susceptible to complement attack due to lack of CD59, resulting in hemolysis and consequent clinical manifestations. C. Eculizumab blocks the complement cascade inhibiting MAC formation; thus, even upon complement activation, PNH RBCs are protected from hemolysis, thus resulting in hemoglobin stabilization and reduction of thromboembolisms.
At a more detailed analysis of the properties of eculizumab, it appears that the antibody has a human kappa light chain constant region, while the heavy constant region results from the combination of IgG2 CH1 and IgG4 CH2 and CH3 domains; thus eculizumab does not activate the complement by itself. The specificity for human C5 is quite stringent, as demonstrated by the almost total absence of species cross-reactivity. Pharmacodynamic (PD) and pharmacokinetic (PK) profiles of eculizumab have been assessed by a serum complement hemolysis in vitro assay and an ELISA analysis of serum concentration, using different dosing schedules. Hemolysis was considered inhibited if lysis of antibody-sensitized chicken erythrocytes after sample serum incubation was <20% of that with normal serum; eculizumab through levels above 35 μg/mL were demonstrated to saturate the C5 target and to block hemolysis in more than 80% of samples. Several loading doses were evaluated, and a fixed 600 mg weekly dose was shown to be effective in achieving pharmacodynamically effective concentrations of >35 μg/mL; the fixed dose was exploited after demonstrating that the PK profile did not correlate with weight or other patient-specific factors. The PK profile demonstrated a non-linear behavior; further complex mathematical models describing PK parameters (ie, an equation relating the volume of the central compartment to the amount of drug in that compartment) yielded an estimated half-life of 271 hours, with the onset of steady-state concentrations predicted to be reached after 57 days of therapy. Eculizumab is administered intravenously, thus its bioavailability is 100%. The drug is distributed within vascular and extracellular spaces as other endogenous antibodies; the effects of non-C5 plasma proteins on eculizumab distribution have not been determined. The elimination of eculizumab is expected to involve uptake into endothelial pinocytotic vesicles containing Fc receptors and other mechanisms active in native antibody recycling; given its size, elimination through the kidneys, the liver, the lungs, and the gastrointestinal system is not expected (and even experimentally proven for the kidneys). Based on these findings, eculizumab therapy was designed to rapidly reach PD levels using an induction regimen, followed by a maintenance dosing schedule to avoid concentration drop below 35 μg/mL. In all PNH studies eculizumab has been administered as 4-weekly doses of 600 mg (induction regimen), followed by 900 mg doses every other week (maintenance regimen), starting 1 week after induction (week 5); this is the standard schedule, recently approved by the FDA for the treatment of PNH.
Efficacy
Eculizumab has been evaluated in a number of clinical trials enrolling PNH patients with hemolytic disease and transfusion dependence; overall, almost 200 patients have been enrolled. The first open-label pilot study included 11 patients and provided the initial evidence of efficacy, proving the principle that blocking C5 may reduce hemolysis in PNH patients (CitationHillmen et al 2004; CitationHill et al 2005a). Then, the definitive evidence of efficacy was provided by 2 large multi-center trials, the TRIUMPH and the SHEPHERD studies.
The TRIUMPH study (CitationHillmen et al 2006) was a double-blind, placebo-controlled study which randomized 87 patients to receive intravenously either placebo or eculizumab at 4-weekly doses of 600 mg (induction period), followed by 900 mg every other week (maintenance period). Inclusion criteria were transfusion-dependent PNH patients with no clinically significant marrow failure (no neutropenia or thrombocytopenia); the treatment period was 6 months. Eculizumab was able to inhibit in vivo the intravascular hemolysis of PNH patients, as confirmed by rapid and sustained reduction of LDH levels, measured as area under the curve, in patients receiving the drug compared with the control group. Eculizumab was more effective than placebo in improving anemia, with hemoglobin stabilization in 48.8% of patients versus 0% in the placebo group. As a result, 51.2% of eculizumab-treated patients no longer required transfusions compared with 0% of those receiving placebo. As a group, the median number of red cell units transfused was 0 in the eculizumab group and 10 in the placebo group, globally indicating a significant reduction in transfusion requirement; even patients who still required transfusions while on eculizumab showed a significantly reduced need for transfusion than before treatment. The amelioration of anemia translated into a significant improvement of fatigue and quality of life (see below).
These data were confirmed in the subsequent SHEPHERD trial (CitationBrodsky et al 2008), an open-label study designed to test eculizumab in a broader PNH population, which included patients with minimal transfusion requirement and even those with thrombocytopenia. This international study enrolled 97 patients, who received a 52-week treatment; globally, the patients showed a significant blockade of hemolysis compared with the pre-treatment period, as assessed by an 87% reduction of LDH level. As predicted, the reduced hemolysis improved the condition of anemia, to the extent that 51% of patients did not require transfusion.
Patients originally enrolled in the TRIUMPH and the SHEPHERD studies joined the Extension study, an ongoing non-placebo-controlled trial involving patients coming from any of the previous eculizumab-based trials (including the initial pilot study). The Extension study was mostly designed to assess the effect of long-term treatment in terms of both efficacy and safety; however, among the endpoints there was the prospective evaluation of thromboembolic events (TE) in comparison to that of the pre-treatment period. Such endpoint has been recently evaluated in an ad interim analysis, and data have been recently published; the analysis included 195 patients who had been exposed to eculizumab (CitationHillmen et al 2007). The TE rate in patients on eculizumab was 1.07/100 patient-years, which represents an 87% reduction compared with that inferred from the pre-treatment period (7.37/100 patient-years). Such a reduction seemed even more striking when the pre-treatment observation period was limited to the 12 months before starting eculizumab. The protection from thrombosis conferred by eculizumab in high risk patient is confirmed by data in patients on antithrombotics because of a previous TE event: eculizumab associated with antithrombotics resulted in a 0.62/100 patient-years TE rate, which is significantly lower than that on antithrombotics alone, before eculizumab treatment (10.61/100 patient-years). Thus, eculizumab seems to reduce thrombosis in PNH patients, even in patient cohorts where common antithrombotics usually fail to show any efficacy.
Globally, the clinical efficacy of eculizumab in PNH patients consists of a rapid and sustained reduction of intra-vascular hemolysis, leading to increased hemoglobin level, and to reduced need – or not need at all – for transfusions; in addition, all symptoms related to hemolysis, such as abdominal pain and dysphagia, completely remitted (CitationHill et al 2005b). Furthermore, a significant reduction in the TE risk emerges, which may likely translate into increased life-expectancy. Such benefits were demonstrated to be maintained in long-term treatment, as no reduced efficacy has been documented even in patients receiving the drug for 3 years or longer.
Safety and tolerability
The safety profile of eculizumab was assessed in the 4 studies involving PNH patients, as well as on 11 studies utilizing eculizumab for different indications; the cumulative exposure was 147.44 and 492.20 patient-years in the two populations, respectively. Three deaths were reported in the PNH studies, all considered unrelated to the investigational drug; fatal events were related to the underlying disease in 2 cases (1 cerebral vascular accident and 1 progression to chronic myelomonocytic leukemia) and to an unrelated accident in the third (cerebral herniation). The main concern was a putative increased risk of infection, mostly by encapsulated bacteria, namely Neisseria spp. A case of meningitis by Neisseria meningitidis was initially described within the non-PNH cohort; since then, all patients exposed to eculizumab are vaccinated against Neisseria meningitidis using a polyvalent vaccine. In addition, all patients received a warning on meningitis and infectious symptoms, as well as a rescue antibiotic prescription. No case of meningitis has been documented among the 195 PNH patients receiving eculizumab; however, 3 patients developed a Neisseria meningitidis infection, with sepsis in 2 cases. None of these patients developed meningitis or other complications, and all recovered promptly as a result of early diagnosis and treatment. Furthermore, at least 2 of the 3 cases seemed to be due to the Neisseria meningitidis groups, which are not covered by the prescribed vaccine. The incidence of serious adverse events was similar in eculizumab-treated patients and in those receiving the placebo within the TRIUMPH trial; furthermore, none of the serious adverse events was considered as possibly, probably, or definitely related to eculizumab. The overall rate of infectious events did not increase compared with the placebo group; however, herpes simplex and some other site-specific infections (nasopharyngitis, upper respiratory tract infection, urinary tract infection, and sinusitis) appeared to be more frequent within the eculizumab-treated population. However, in all cases the intensity was mild and the clinical resolution prompt.
Globally, the most common adverse events were headache, followed by pyrexia, nasopharyngitis, herpes simplex, nausea, fatigue, and myalgia; all these events were generally mild and resolved quickly. No discontinuation due to side effects of eculizumab was observed. There was some concern about possibly severe hemolysis episodes following withdrawal or non-compliance with the eculizumab dosage regimen; however, there was no evidence of life-threatening or serious hemolysis, even in the few patients who discontinued treatment and progressively returned to their pre-treatment levels of hemolysis. The occurrence of immunogenity was assessed, and was demonstrated to be very infrequent, if present at all, and without consequence on drug efficacy. Treatment was discontinued in 3 patients due to pregnancy, as per protocol; of these, the two with adequate follow up gave birth to a normal baby, with no maternal complications.
In summary, a wide variety of data generated from studies of patients with PNH, supported by safety data from studies of patients diagnosed with other diseases, demonstrate that treatment with eculizumab is safe and well-tolerated for the treatment of PNH. Long-term treatment has not demonstrated deviation from this safety profile; however, anti-meningococcal vaccination and warning for symptoms of meningitis remain mandatory.
Patient-focused perspectives
Patient-reported outcomes were systematically assessed in all eculizumab-based studies (CitationHillmen et al 2006; CitationHillmen et al 2007; CitationBrodsky et al 2008); fatigue is a common symptom of PNH patients, and significantly affects their quality of life. In addition, painful and disabling hemolysis-specific symptoms may be present, finally resulting in global health status and patient functioning impairment (CitationHill et al 2005b). Eculizumab treatment was associated with significant improvement in fatigue, as measured by both the FACIT and the EORTC QLQ-C30 fatigue scales. In addition, eculizumab-treated patients reported a significant improvement in a number of EORTC QLQ-C30 subscales, including global health status, role, social, cognitive, physical and emotional functioning, pain, dyspnea, and appetite loss. This unequivocally demonstrated that eculizumab improves the quality of life and the well-being of PNH patients improving both anemia and anemia-related symptoms, as well as other disease-related symptoms. As a result, patient’s acceptance of the treatment is enthusiastic, even if the treatment schedule requires frequent and possibly tiresome journeys and hospitalizations, usually in day hospital, to receive the drug.
Future directions
Eculizuamb was effective in controlling intravascular hemolysis in all PNH patients, as demonstrated by their LDH levels, with the exception of a few patients who experienced breakthrough hemolysis on eculizumab with the standard 14-day schedule. These patients usually show recurrence of hemolysis toward the end of the interval, ie, 1 or 2 days before receiving the following dose, and may be rescued by shortening to 12 days the interval between administrations. Beside this PK exception, all patients on eculizumab show a complete and sustained block of intravascular hemolysis; however, some of them do not achieve normalization of their hemoglobin level and show persistent high reticulocyte count and increased serum bilirubin in presence of a normal marrow function, and even after stimulation by erythropoietin (CitationHill et al 2007b). Thus, residual low-titer hemolysis can be suspected in most PNH patients on eculizumab (CitationHillmen et al 2004); the hemolysis may be due to pathophysiological mechanisms other than those so far described in PNH (CitationRosenfeld et al 1985). Based on personal ongoing investigation (CitationRisitano et al 2006), we suggest that eculizumab may induce substantial changes in PNH biology, leading to the possible transformation of a disease with typical intravascular hemolysis into a new one characterized by prevalent extravascular hemolysis. As discussed above, eculizumab blocks C5 cleavage and subsequent intravascular hemolysis, but does not affect C3 deposition and cleavage on RBC membrane, which is not controlled given the absence of CD55 (). Thus the accumulation of C3 allowed by the increased PNH RBC survival on eculizumab treatment may lead to unexpected extravascular hemolysis in the reticuloendothelial system via complement receptors. This is confirmed by preliminary data from Italian patients, showing that in vivo RBC survival, as assessed by 51-Cr study, remains reduced in PNH patients on eculizumab, who have no evidence of residual intravascular hemolysis (CitationRisitano et al 2007). In addition, C3 coating was demonstrated by direct anti-globulin test (C3d only) and flow cytometry in all PNH patients on eculizumab; these harbor a characteristic C3-positive RBC population with the PNH phenotype, in addition to the two expected populations of C3-negative phenotypically normal RBCs and C3-negative PNH RBCs. These findings were not found in untreated PNH patients, who only showed the two latter RBC populations (CitationRisitano et al 2007). This intriguing new mechanism of disease may become apparent only upon eculizumab treatment, and is currently under investigation to assess its actual importance in vivo and explore future clinical implications, namely possible strategies to optimize the clinical efficacy of eculizumab.
Conclusion
Even if PNH is a rare condition, it represents an intriguing model of disease and has attracted the efforts of several generations of investigators in the last three decades. While the ’80s unraveled the GPI-anchor and the functional defect of the PNH clone, and the 1990s revealed the PIG-A gene and its role in the pathophysiology of the disease, this new century is finally bringing forth innovative therapeutic approaches. Mechanisms of disease in PNH have been almost completely elucidated in recent years and now, in the era of biological agents, have led to the development of targeted therapy through the terminal complement inhibitor eculizumab. Eculizumab showed great efficacy in blocking complement-mediated intravascular hemolysis in PNH patients, leading to substantial improvements in anemia, fatigue, hemolysis-related symptoms and quality of life. In addition, preliminary data suggest that eculizumab may significantly reduce the risk of thrombosis in PNH patients, likely as a result of controlling free hemoglobin level and inhibiting the subsequent NO depletion. Thus, the new millennium of PNH starts with the first treatment able to specifically control most symptoms of the disease, and likely reduce the risk of life-threatening complications, leading to a substantial change in the algorithm of the clinical management of PNH patients (). Whether and how this new drug will affect the biology of PNH, possibly changing the paradigm of known pathogenic mechanisms, and, more importantly, affecting patient survival are the new challenge for the next few years.
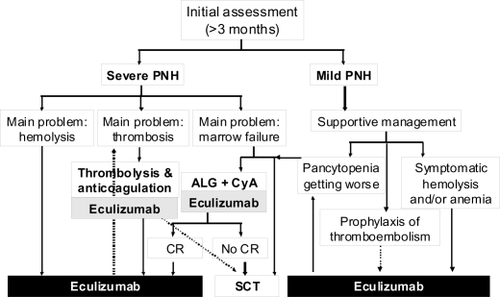
Figure 7 Algorithm of the clinical management of paroxysmal nocturnal hemoglobinuria (PNH) in the eculizumab era. Once PNH is confirmed, all patients suffering from its severe form should receive eculizumab to control intravascular hemolysis; in case of concomitant thromboembolic manifestations an appropriate anticoagulant and/or antithrombotic therapy should be added. In the presence of concomitant severe marrow failure, the indication of an immunosuppressive regimen should be discussed, and programmed possibly before eculizumab treatment (no data on concomitant therapy are available so far). Patients suffering from mild PNH should receive supportive therapy according to the main clinical manifestations: symptomatic hemolysis and/or anemia should be treated by eculizumab, signs of marrow failure may require immunosuppression. Even in this subset the use of eculizumab is reasonable as prophylaxis of thromboembolic events. Indications to stem cell transplantation remain severe marrow failure not responding to immunosuppression (or even as first line in young patients, if a low risk transplant procedure is possible) and possibly refractory thromboembolic disease (but the risk-benefit ratio in comparison to eculizumab has to be demonstrated in this setting).
Disclosures
AMR and BR have received research grants and lecture fees from Alexion Pharmaceuticals; they have also served as consultants and on advisory committees for the same company.
References
- AlfinitoFDel VecchioLRoccoS1996Blood cell flow cytometry in paroxysmal nocturnal hemoglobinuria: a tool for measuring the extent of the PNH cloneLeukemia101326308709638
- AlmeidaAMMurakamiYBakerA2007Targeted therapy for inherited GPI deficiencyN Engl J Med3561641717442906
- AlmeidaAMMurakamiYLaytonDM2006Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiencyNat Med128465116767100
- AratenDJBesslerMMcKenzieS2002Dynamics of hematopoiesis in paroxysmal nocturnal hemoglobinuria (PNH): no evidence for intrinsic growth advantage of PNH clonesLeukemia162243812399968
- AratenDJNafaKPakdeesuwanK1999Clonal populations of hematopoietic cells with paroxysmal nocturnal hemoglobinuria genotype and phenotype are present in normal individualsProc Natl Acad Sci USA9652091410220445
- AratenDJSwirskyDKaradimitrisA2001Cytogenetic and morphological abnormalities in paroxysmal nocturnal haemoglobinuriaBr J Haematol115360811703336
- AudebertHJPlanckJEisenburgM2005Cerebral ischemic infarction in paroxysmal nocturnal hemoglobinuria: report of 2 cases and updated review of 7 previously published patientsJ Neurol25213798616021362
- BembaMGuardiolaPGarderetL1999Bone marrow transplantation for paroxysmal nocturnal haemoglobinuriaBr J Haematol105366810233405
- BesslerMHillmenPLongoL1994Genomic organization of the X-linked gene (PIG-A) that is mutated in paroxysmal nocturnal haemoglobinuria and of a related autosomal pseudogene mapped to 12q21Hum Mol Genet375178081362
- BoccuniPDel VecchioLDi NotoR2000Glycosyl phosphatidylinositol (GPI)-anchored molecules and the pathogenesis of paroxysmal nocturnal hemoglobinuriaCrit Rev Oncol Hematol33254310714960
- BourantasK1994High-dose recombinant erythropoietin and low-dose corticosteroids for treatment of anemia in paroxysmal nocturnal hemoglobinuriaActa Haematol916258023644
- BrodskyRAMukhinaGLLiS2000Improved detection and characterization of paroxysmal nocturnal hemoglobinuria using fluorescent aerolysinAm J Clin Pathol1144596610989647
- BrodskyRAYoungNSAntonioliE2008Multicenter phase III study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuriaBlood1111840718055865
- BurgeJNicholson-WellerAAustenKF1981Isolation of C4-binding protein from guinea pig plasma and demonstration of its function as a control protein of the classical complement pathway C3 convertaseJ Immunol12623256778916
- CacciapuotiCTerrazzanoGBaroneL2007Glycosyl-phosphatidyl-inositol-defective granulocytes from paroxysmal nocturnal haemoglobinuria patients show increased bacterial ingestion but reduced respiratory burst inductionAm J Hematol829810717013814
- ChenGZengWMaciejewskiJP2005Differential gene expression in hematopoietic progenitors from paroxysmal nocturnal hemoglobinuria patients reveals an apoptosis/immune response in ‘normal’ phenotype cellsLeukemia19862815759038
- ChristmasSEde la Mata EspinosaCTHallidayD2006Levels of expression of complement regulatory proteins CD46, CD55 and CD59 on resting and activated human peripheral blood leucocytesImmunology119522816999828
- DufourCCorcioneASvahnJ2001Interferon gamma and tumour necrosis factor alpha are overexpressed in bone marrow T lymphocytes from paediatric patients with aplastic anaemiaBr J Haematol11510233111843845
- DunnDELiuJMYoungNS2000Paroxysmal nocturnal hemoglobinuriaYoungNSBone Marrow Failure SyndromesPhiladelphiaWB Saunders Company99121
- EndoMWareREVreekeTM1996Molecular basis of the heterogeneity of expression of glycosyl phosphatidylinositol anchored proteins in paroxysmal nocturnal hemoglobinuriaBlood872546578630422
- GargiuloLLastraioliSCerrutiG2007Highly homologous T-cell receptor beta sequences support a common target for autoreactive T cells in most patients with paroxysmal nocturnal hemoglobinuriaBlood10950364217284529
- GoebelJForrestKFlynnD2002Lipid rafts, major histocompatibility complex molecules, and immune regulationHum Immunol638132012368033
- HallCRichardsSHillmenP2003Primary prophylaxis with warfarin prevents thrombosis in paroxysmal nocturnal hemoglobinuria (PNH)Blood10235879112893760
- HarringtonWJSrKolodnyRHorstmannLL1997Danazol for paroxysmal nocturnal hemoglobinuriaAm J Hematol54149549034290
- HauserACBrichtaAPabinger-FaschingI2003Fibrinolytic therapy with rt-PA in a patient with paroxysmal nocturnal hemoglobinuria and Budd-Chiari syndromeAnn Hematol8229930212707719
- HertensteinBWagnerBBunjesD1995Emergence of CD52-, phosphatidylinositolglycan-anchor-deficient T lymphocytes after in vivo application of Campath-1H for refractory B-cell non-Hodgkin lymphomaBlood861487927632956
- HillAHillmenPRichardsSJ2005aSustained response and long-term safety of eculizumab in paroxysmal nocturnal hemoglobinuriaBlood10625596515985537
- HillARidleySHEsserD2006Protection of erythrocytes from human complement-mediated lysis by membrane-targeted recombinant soluble CD59: a new approach to PNH therapyBlood1072131716322479
- HillARotherRPHillmenP2005bImprovement in the symptoms of smooth muscle dystonia during eculizumab therapy in paroxysmal nocturnal hemoglobinuriaHaematologica90ECR4016464755
- HillARichardsSJHillmenP2007aRecent developments in the understanding and management of paroxysmal nocturnal haemoglobinuriaBr J Haematol1371819217408457
- HillARichardsSJRotherRP2007bErythopoietin treatment during complement inhibition with eculizumab in a patient with paroxysmal nocturnal hemoglobinuriaHaematologica92ECR14
- HillmenPHallCMarshJC2004Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuriaN Engl J Med350552914762182
- HillmenPLewisSMBesslerM1995Natural history of paroxysmal nocturnal hemoglobinuriaN Engl J Med333125387566002
- HillmenPMuusPDuhrsenU2007Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuriaBlood1104123817702897
- HillmenPYoungNSSchubertJ2006The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuriaN Engl J Med35512334316990386
- HiroseSRaviLPrinceGM1992Synthesis of mannosylglucos-aminylinositol phospholipids in normal but not paroxysmal nocturnal hemoglobinuria cellsProc Natl Acad Sci USA89602591378620
- HolguinMHWilcoxLABernshawNJ1989Relationship between the membrane inhibitor of reactive lysis and the erythrocyte phenotypes of paroxysmal nocturnal hemoglobinuriaJ Clin Invest841387942478585
- HuRMukhinaGLPiantadosiS2005PIG-A mutations in normal hematopoiesisBlood10538485415687243
- HugelBSociéGVuT1999Elevated levels of circulating procoagulant microparticles in patients with paroxysmal nocturnal hemoglobinuria and aplastic anemiaBlood933451610233897
- IssaragrisilSPiankijagumATang.NaitrisoranaYCorticosteroid therapy in paroxysmal nocturnal hemoglobinuriaAm J Hematol2577833578265
- JasinskiMKellerPFujiwaraY2001GATA1-Cre mediates Piga gene inactivation in the erythroid/megakaryocytic lineage and leads to circulating red cells with a partial deficiency in glycosyl phosphatidylinositol-linked proteins (paroxysmal nocturnal hemoglobinuria type II cells)Blood9822485511568013
- KaradimitrisAManavalanJSThalerHT2000Abnormal T-cell repertoire is consistent with immune process underlying the pathogenesis of paroxysmal nocturnal hemoglobinuriaBlood9626132011001919
- KatayamaYHiramatsuYKohriyamaH2001Monitoring of CD59 expression in paroxysmal nocturnal hemoglobinuria treated with danazolAm J Hematol68280311754418
- KellerPPayneJLTremmlG2001FES-Cre targets phosphatidylinositol glycan class A (PIGA) inactivation to hematopoietic stem cells in the bone marrowJ Exp Med194581911535627
- KookHRisitanoAMZengW2002Changes in T-cell receptor VB repertoire in aplastic anemia: effects of different immunosuppressive regimensBlood9936687511986222
- LuzzattoLBesslerM1996The dual pathogenesis of paroxysmal nocturnal hemoglobinuriaCurr Opin Hematol3101109372059
- LuzzattoLBesslerMRotoliB1997Somatic mutations in paroxysmal nocturnal hemoglobinuria: a blessing in disguise?Cell88149019395
- MaciejewskiJPSloandEMSatoT1997Impaired hematopoiesis in paroxysmal nocturnal hemoglobinuria/aplastic anemia is not associated with a selective proliferative defect in the glycosylphosphatidylinositol-anchored protein-deficient cloneBlood891173819028939
- MahoneyJFUrakazeMHallS1992Defective glycosylphosphatidylinositol anchor synthesis in paroxysmal nocturnal hemoglobinuria granulocytesBlood79140031372185
- MatisLARollinsSA1995Complement-specific antibodies: designing novel anti-inflammatoriesNat Med1839427585191
- McMullinMFHillmenPElderGE1996Serum erythropoietin level in paroxysmal nocturnal hemoglobinuria: implications for therapyBr J Haematol9281578616071
- McMullinMFHillmenPJacksonJ1994Tissue plasminogen activator for hepatic vein thrombosis in paroxysmal nocturnal haemoglobinuriaJ Intern Med2358598283166
- MedofMEKinoshitaTNussenzweigV1984Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (DAF) into their membranesJ Exp Med1601558786238120
- MiyataTTakedaJIidaY1993The cloning of PIG-A, a component in the early step of GPI-anchor biosynthesisScience2591318207680492
- MoyoVMMukhinaGLGarrettES2004Natural history of paroxysmal nocturnal haemoglobinuria using modern diagnostic assaysBr J Haematol126133815198744
- MurakamiYKosakaHMaedaY2002Inefficient response of T lymphocytes to glycosylphosphatidylinositol anchor-negative cells: implications for paroxysmal nocturnal hemoglobinuriaBlood10041162212393537
- NafaKBesslerMDeegHJ1998New somatic mutation in the PIG-A gene emerges at relapse of paroxysmal nocturnal hemoglobinuriaBlood92342279787183
- NafaKBesslerMMasonP1996Factor V Leiden mutation investigated by amplification created restriction enzyme site (ACRES) in PNH patients with and without thrombosisHaematologica8154029009443
- NagakuraSIshiharaSDunnDE2002Decreased susceptibility of leukemic cells with PIG-A mutation to natural killer cells in vitroBlood1001031712130519
- NakaoSTakamiATakamatsuH1997Isolation of a T-cell clone showing HLA-DRB1*0405-restricted cytotoxicity for hematopoietic cells in a patient with aplastic anemiaBlood89369199160674
- Nicholson-WellerAMarchJPRosenfeldSI1983Affected erythrocytes of patients with paroxysmal nocturnal hemoglobinuria are deficient in the complement regulatory protein, decay accelerating factorProc Natl Acad Sci USA805066706576376
- Nicholson-WellerASpicerDBAustenKF1985Deficiency of the complement regulatory protein, “decay-accelerating factor,” on membranes of granulocytes, monocytes, and platelets in paroxysmal nocturnal hemoglobinuriaN Engl J Med312109173845321
- NinomiyaHHasegawaYNagasawaT1997Excess soluble urokinase-type plasminogen activator receptor in the plasma of patients with paroxysmal nocturnal hemoglobinuria inhibits cell-associated fibrinolytic activityInt J Hematol65285919114600
- Nishimura JiJHirotaTKanakuraY2002Long-term support of hematopoiesis by a single stem cell clone in patients with paroxysmal nocturnal hemoglobinuriaBlood9927485111929762
- NishimuraJKanakuraYWareRE2004Clinical course and flow cytometric analysis of paroxysmal nocturnal hemoglobinuria in the United States and JapanMedicine (Baltimore)8319320715118546
- OkadaNHaradaROkadaH1990Erythrocytes of patients with paroxysmal nocturnal haemoglobinuria acquire resistance to complement attack by purified 20-kD homologous restriction factorClin Exp Immunol80109131691059
- OlsenSBTangDBJacksonMR1996Enhancement of platelet deposition by cross-linked hemoglobin in a rat carotid endarterectomy modelCirculation93327328548906
- OniBSOsunkoyaBOLuzzattoL1970Paroxysmal nocturnal hemoglobinuria: evidence for monoclonal origin of abnormal red cellsBlood36145525448520
- ParkerCOmineMRichardsSInternational PNH Interest Group2005Diagnosis and management of paroxysmal nocturnal hemoglobinuriaBlood106369970916051736
- Peffault de LatourRMaryJYSalanoubatC2006Paroxysmal Nocturnal Hemoglobinuria (PNH): Long-Term Epidemiological StudyBlood108291a
- PlasilovaMRisitanoAMaciejewskiJP2003Application of the molecular analysis of the T-cell receptor repertoire in the study of immune-mediated hematologic diseasesHematology81738112745651
- PlasilovaMRisitanoAMO’KeefeCL2004Shared and individual specificities of immunodominant cytotoxic T cell clones in Paroxysmal Nocturnal Hemoglobinuria as determined by molecular analysisExp Hematol32261915003311
- PoggiANegriniSZocchiMR2005Patients with paroxysmal nocturnal hemoglobinuria have a high frequency of peripheral-blood T cells expressing activating isoforms of inhibiting superfamily receptorsBlood106239940815956278
- PramoonjagoPPakdeesuwanKSiripanyaphinyoU1999Genotypic, immunophenotypic and clinical features of Thai patients with paroxysmal nocturnal haemoglobinuriaBr J Haematol10549750410233427
- RaiolaAMVan LintMTLamparelliT2000Bone marrow transplantation for paroxysmal nocturnal hemoglobinuriaHaematologica85596210629593
- RichardsSJRawstronACHillmenP2000Application of flow cytometry to the diagnosis of paroxysmal nocturnal hemoglobinuriaCytometry422233310934341
- RisitanoAMHillARicciP2007Paroxysmal nocturnal hemoglobinuria (PNH) in the eculizumab era: the bedside and beyondHaematologica92s389a
- RisitanoAMHillmenPBarbanoF2006The complement inhibitor eculizumab to treat paroxysmal nocturnal hemoglobinuria patients: a single center experience within the Triumph, Shepherd and Extension studiesHaematologica91s3135a
- RisitanoAMKookHZengW2002Oligoclonal and polyclonal CD4 and CD8 lymphocytes in aplastic anemia and paroxysmal nocturnal hemoglobinuria measured by Vβ CDR3 spectratyping and flow cytometryBlood1001788312070025
- RisitanoAMMaciejewskiJPGreenS2004In vivo dominant immune responses in aplastic anemia patients: molecular tracking of putatively pathogenic T cells by TCRβ-CDR3 sequencingLancet36435363
- RisitanoAMMaciejewskiJPMuranskiP2005Large granular lymphocyte (LGL)-like clonal expansions in paroxysmal nocturnal hemoglobinuria (PNH) patientsLeukemia192172215668701
- RosenfeldSIJenkinsDEJrLeddyJP1985Enhanced reactive lysis of paroxysmal nocturnal hemoglobinuria erythrocytes by C5b-9 does not involve increased C7 binding or cell-bound C3bJ Immunol134506113964820
- RosseWF1982Treatment of paroxysmal nocturnal hemoglobinuriaBlood602037044450
- RosseWF1990Phosphatidylinositol-linked proteins and paroxysmal nocturnal hemoglobinuriaBlood7515956012183885
- RosseWF1996Epidemiology of PNHLancet34857378774569
- RosseWFDacieJV1966Immune lysis of normal human and paroxysmal nocturnal hemoglobinuria (PNH) red blood cells. I. The sensitivity of PNH red cells to lysis by complement and specific antibodyJ Clin Invest45736485935361
- RostiVTremmlGSoaresV1997Murine embryonic stem cells without pig-a gene activity are competent for hematopoiesis with the PNH phenotype but not for clonal expansionJ Clin Invest1001028369276719
- RotherRPBellLHillmenP2005The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human diseaseJAMA29316536215811985
- RotherRPRollinsSAMojcikCF2007Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuriaNat Biotechnol2512566417989688
- RotoliBBesslerMAlfinitoF1993Membrane proteins in paroxysmal nocturnal haemoglobinuriaBlood Rev775868369662
- RotoliBLuzzattoL1989Paroxysmal nocturnal haemoglobinuriaBaillieres Clin Haematol2113382645958
- RotoliBRobledoRLuzzattoL1982Decreased number of circulating BFU-Es in paroxysmal nocturnal hemoglobinuriaBlood6015797082835
- RotoliBRobledoRScarpatoN1984Two populations of erythroid cell progenitors in paroxysmal nocturnal hemoglobinuriaBlood64847516478058
- RotoliBNafaDRisitanoAM2006Paroxysmal nocturnal hemoglobinuriaRungeMDPattersonCPrinciples of Molecolar Medicine2nd editionPhiladelphiaHumana Press83847
- Sanchez-ValleEMorales-PolancoMRGomez-MoralesE1993Treatment of paroxysmal nocturnal hemoglobinuria with antilymphocyte globulinRev Invest Clin45457618134727
- SantaroneSDi BartolomeoEBacigalupo2008Long-term results of a GITMO retrospective study on haematopoietic stem cell transplantation for paroxysmal nocturnal hemoglobinuraBone Marrow Transplant41S11917934531
- SasoRMarshJCevreskaL1999Bone marrow transplants for paroxysmal nocturnal haemoglobinuriaBr J Haematol104392610050724
- SatoTSelleriCAndersonS1997Expression and modulation of cellular receptors for interferon-gamma, tumour necrosis factor, and Fas on human bone marrow CD34+ cellsBr J Haematol97356659163602
- SchaferAWiesmannFNeubauerS2004Rapid regulation of platelet activation in vivo by nitric oxideCirculation10918192215066953
- SelvarajPRosseWFSilberR1988The major Fc receptor in blood has a phosphatidylinositol anchor and is deficient in paroxysmal nocturnal haemoglobinuriaNature33356572967435
- ShichishimaTSaitohYTerasawaT1999Complement sensitivity of erythrocytes in a patient with inherited complete deficiency of CD59 or with the Inab phenotypeBr J Haematol104303610050712
- ShinMLHänschGHuVW1986Membrane factors responsible for homologous species restriction of complement-mediated lysis: evidence for a factor other than DAF operating at the stage of C8 and C9J Immunol1361777822419414
- SimakJHoladaKRisitanoAM2004Elevated counts of circulating endothelial membrane microparticles in paroxysmal nocturnal hemoglobinuria indicate inflammatory status and ongoing stimulation of vascular endotheliumBr J Hemat12580413
- SloandEMaciejewskiJPTisdaleJ2002Intracellular Interferon-γ (IFN-γ) in circulating and marrow T cells detected by flow cytometry and the response to immunosuppressive therapy in patients with aplastic anemiaBlood10031293511964274
- SloandEMMoreKShahS2006Soluble urokinase plasminogen activator receptor is increased in patients with paroxysmal nocturnal hemoglobinuria (PNH) and thrombosis and inhibits plasmin generation in vitro and promotes thrombosis in the mouse modelBlood108422a
- SocieGMaryJYde GramontA1996Paroxysmal nocturnal haemoglobinuria: long-term follow-up and prognostic factors. French Society of HaematologyLancet34857378774569
- SteblerCTichelliADazziH1990High-dose recombinant human erythropoietin for treatment of anemia in myelodysplastic syndromes and paroxysmal nocturnal hemoglobinuria: a pilot studyExp Hematol18120482226680
- SutherlandDRKuekNDavidsonJ2007Diagnosing PNH with FLAER and multiparameter flow cytometryCytometry B Clin Cytom721677717285629
- TakahashiMTakedaJHiroseS1993Deficient biosynthesis of N-acetylglucosaminyl-phosphatidylinositol, the first intermediate of glycosyl phosphatidylinositol anchor biosynthesis, in cell lines established from patients with paroxysmal nocturnal hemoglobinuriaJ Exp Med177517218426120
- TakedaJMiyataTKawagoeK1992Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuriaCell73703118500164
- TichelliAGratwohlANissenC1992Morphology in patients with severe aplastic anemia treated with antilymphocyte globulinBlood80337451627795
- van der SchootCEHuizingaTWvan ‘t Veer-KorthofET1990Deficiency of glycosylphosphatidylinositol-linked membrane glycoproteins of leukocytes in paroxysmal nocturnal hemoglobinuria, description of a new diagnostic cytofluorometric assayBlood76185392145990
- WiedmerTHallSEOrtelTL1993Complement-induced vesiculation and exposure of membrane prothrombinase sites in platelets of paroxysmal nocturnal hemoglobinuriaBlood82119267688991
- WilcoxLAEzzellJLBernshawNJ1991Molecular basis of the enhanced susceptibility of the erythrocytes of paroxysmal nocturnal hemoglobinuria to hemolysis in acidified serumBlood7882091713516
- YamashinaMUedaEKinoshitaT1990Inherited complete deficiency of 20-kilodalton homologous restriction factor (CD59) as a cause of paroxysmal nocturnal hemoglobinuriaN Engl J Med323118491699124
- YoungNSMaciejewskiJP2000Genetic and environmental effects in paroxysmal nocturnal hemoglobinuria: this little PIG-A goes “Why? Why? Why?”J Clin Invest1066374110974016
- ZalmanLSWoodLMMüller-EberhardHJ1986Isolation of a human erythrocyte membrane protein capable of inhibiting expression of homologous complement transmembrane channelsProc Natl Acad Sci USA83697592428045
- ZalmanLSWoodLMFrankMM1987Deficiency of the homologous restriction factor in paroxysmal nocturnal hemoglobinuriaJ Exp Med16557272434597
- ZengWMaciejewskiJPChenG2001Limited heterogeneity of T-cell receptor VB usage in aplastic anemiaJ Clin Invest1087657311544283
- ZoumbosNGasconPTrostS1985Circulating activated suppressor T lymphocytes in aplastic anemiaN Engl J Med312257652981406