Abstract
Single agent gemcitabine has been the mainstay of therapy for advanced pancreatic cancer over the past decade. Multiple trials of newer chemotherapeutic agents both alone and in combination have yielded disappointing results, spurring the ongoing search for new agents and combinations in this aggressive malignancy. Inhibitors of the epidermal growth factor receptor (EGFR) have shown promising activity in multiple solid tumors types, and preclinical data support a role for EGFR inhibition in pancreatic cancer. A recent phase III study by the National Cancer Institute of Canada Clinical Trials Group (NCIC-CTG) demonstrated a significant survival benefit with the addition of the EGFR tyrosine kinase inhibitor, erlotinib, to gemcitabine chemotherapy for the first-line treatment of patients with advanced pancreatic cancer, becoming the first phase III study to demonstrate a survival benefit of combination therapy as well as targeted therapy in this disease. This article reviews the evidence supporting EGFR inhibition and the use of erlotinib in advanced pancreatic cancer as well as future implications of targeted therapy in this challenging malignancy.
Keywords:
Introduction
Pancreatic carcinoma is the fourth leading cause of cancer death in the United States with 37,170 new cases and 33,370 deaths anticipated in 2007. More than half of patients have metastatic disease at the time of diagnosis, with a median 5-year survival of 2%. Locally advanced, unresectable disease likewise accounts for over 25% of patients at the time of presentation and confers a median 5-year survival of 8% (CitationJemal et al 2007). Locally advanced tumors together with metastatic disease are often collectively considered as advanced pancreatic cancer for the purposes of clinical research, although there are subtle differences in prognosis (making stratification in such studies a necessity) as well as in treatment options for each.
Current standards of care for advanced pancreatic cancer have evolved from a 1997 study by Burris et al that resulted in approval of gemcitabine for the treatment of this disease (CitationBurris et al 1997). This study demonstrated a clinical benefit response as well as a modest survival benefit with the use of gemcitabine compared to fluorouracil (5-FU) monotherapy. Clinical benefit response was defined as a sustained improvement in pain, Karnofsky performance status, or weight for at least 4 weeks on therapy. In patients treated with gemcitabine, clinical benefit response was achieved in 23.8% of patients, versus 4.8% of those treated with 5-FU (p = 0.0022). Median survival durations were 5.65 months versus 4.41 months in the gemcitabine versus 5-FU groups, respectively (p = 0.0025).
Multiple ensuing studies have evaluated gemcitabine in combination with other cytotoxic agents for treatment of advanced pancreatic cancer. Over the past decade, a number of large, randomized phase III trials have failed to demonstrate a survival advantage for any gemcitabine-containing combination, including gemcitabine plus fluoropyrimidines, platinum analogues, and topoisomerase inhibitors (CitationBerlin et al 2002; CitationRocha Lima et al 2004; CitationLouvet et al 2005; CitationAbou-Alfa et al 2006; CitationHerrmann et al 2007), although a recent pooled analysis did suggest a survival benefit for the combination of gemcitabine and a platinum analogue (CitationHeinemann et al 2007). One positive phase III study was reported in Europe in 2005 with the combination of gemcitabine and capecitabine, but these data have yet to be presented in mature form (CitationCunningham et al 2005). In 2006, a Cochrane review of data from 50 trials concluded that combination cytotoxic chemotherapy did not improve overall survival beyond monotherapy (CitationYip et al 2006). Conversely, a recent meta-analysis of 15 randomized trials suggests there may be a small but significant survival benefit of gemcitabine-based combination therapy, with greatest benefit in patients with good performance status (CitationHeinemann et al 2007).
Given the modest survival benefits of traditional chemotherapy, the search for new active agents in this aggressive malignancy is an area of avid investigation. In addition to cytotoxic chemotherapy, multiple targeted agents have been and are being studied for efficacy in pancreatic cancer. Agents that have been rigorously evaluated in phase III study designs include matrix metalloproteinase inhibitors, farnesyl transferase inhibitors, angiogenesis inhibitors, and inhibitors of the epidermal growth factor receptor (EGFR), with myriad other targeted therapies under active investigation in earlier phases of testing.
Preclinical studies show that EGFR-mediated signaling is involved in tumorigenesis of pancreatic cancer. Erlotinib (OSI-774, Tarceva®; OSI Pharmaceuticals, Melville, NY, in collaboration with Genentech and Roche Pharmaceuticals) is a small molecule tyrosine kinase inhibitor (TKI) with specificity for EGFR. Based on the findings of preclinical studies and early phase clinical trials, the National Cancer Institute of Canada Clinical Trials Group (NCIC-CTG) conducted a phase III study (PA.3) comparing single-agent gemcitabine to gemcitabine in combination with erlotinib as first-line therapy for advanced pancreatic cancer. The PA.3 study became the first phase III randomized study to demonstrate a significant survival benefit with combination therapy as well as with a targeted agent in advanced pancreatic cancer (CitationMoore et al 2005). On this basis, the FDA approved erlotinib in combination with gemcitabine chemotherapy for first-line use in locally advanced and metastatic pancreatic carcinoma in 2005. This article will review the molecular mechanism of EGFR inhibition as a therapeutic approach in solid tumors; the preclinical data supporting EGFR inhibition in solid tumors including pancreatic cancer; and finally, the clinical trials that have established the small molecule TKI erlotinib as an approved therapy for advanced pancreatic cancer.
EGF receptor: structure, function, and transforming potential
The EGF receptor is a ubiquitous 170-kd transmembrane glycoprotein in the HER (erbB) family of receptors (CitationArteaga 2001; CitationYarden and Sliwkowski 2001; CitationGrunwald and Hidalgo 2003; CitationKolb et al 2006; CitationCohenuram and Saif 2007). Also known as HER-1 or erbB1, this receptor includes an N-terminal extracellular ligand-binding domain, a transmembrane region, and an intracellular domain with tyrosine kinase function and C-terminal regulatory motifs. The extracellular ligand-binding domain binds specific ligands including EGF, transforming growth factor alpha (TGFα), heparin-binding growth factor, amphiregulin, betacellulin, and epiregulin. The existence of multiple ligands may confer redundancy in activation of EGFR signaling as well as enable differential signaling patterns specific to each ligand.
Upon ligand binding, the EGFR undergoes homodimerization or heterodimerization. Coreceptors in heterodimer formation include the other members of the HER family of receptors: HER2/neu, HER3, and HER4 (CitationArteaga 2001; CitationYarden and Sliwkowski 2001; CitationGrunwald and Hidalgo 2003). Dimerization results in autophosphorylation of EGFR C-terminal regulatory motifs and subsequent activation of downstream signaling. Multiple combinatorial receptor interactions may influence the specificity and potency of EGFR-mediated signal transduction. EGFR activation has been shown to activate multiple downstream signal transduction pathways including the Ras-Raf-MAPK and PI3K/Akt pathways, thereby influencing cell cycle progression, angiogenesis, growth, and survival.
In the context of this role in regulation of cell proliferation, EGFR signaling pathways have been linked to the pathogenesis of multiple human malignancies. In tissue culture, overexpression of members of the EGFR family as well as activation of EGFR signaling can cause malignant transformation (CitationGrunwald and Hidalgo 2003). EGFR may be overexpressed in malignant tumor specimens arising from the upper aerodi-gestive tract, pancreas, colon, rectum, breast, ovary, bladder, kidney, prostate, and central nervous system (CitationGrandis and Sok 2004). This overexpression may occur by gene amplification or by upregulation of autocrine and paracrine growth factors that subsequently induce EGFR expression.
In addition to elevated transcript and/or protein levels of EGFR, activating mutations within the EGFR gene have also been implicated in several malignancies. In non-small-cell lung cancers (NSCLC), activating mutations have been identified within exons 18–21 of the tyrosine kinase domain (CitationGrunwald and Hidalgo 2003; CitationPaez et al 2004; CitationLynch et al 2004; CitationPao and Miller 2005; CitationTakano et al 2005). Mutations in the N-terminal extracellular ligand binding domain including the EGFRvIII variant are found in gliomas (CitationPao and Miller 2005). However, no activating mutations have been identified in pancreatic cancer to date (CitationKwak et al 2006; CitationLee et al 2007; CitationTzeng et al 2007a). Other potential mechanisms of inappropriate EGFR activation include ligand overexpression, coreceptor over-expression or mutations leading to altered dimerization, and abnormalities in regulatory mechanisms including receptor internalization, degradation, and recycling.
Blockade of the EGFR pathway by various strategies has been shown to inhibit proliferation of tumor cell lines in vitro, leading to clinical trials of EGFR inhibitors in a wide array of human malignancies. A therapeutic role has now been established for EGFR inhibitors in carcinomas of the head and neck, lung, breast, and colon. With the approval of erlotinib for treatment of pancreatic cancer in 2005, EGFR inhibition has now become an appropriate strategy for pancreatic cancer as well.
Mechanisms of pharmacologic EGFR inhibition
Two principal mechanisms of therapeutic EGFR inhibition have been developed: monoclonal antibodies (mAbs) that bind to the extracellular domain of EGFR, and TKIs that inhibit the intracellular tyrosine kinase domain. MAbs, by blocking ligand binding to the receptor, interfere with dimerization, phosphorylation, and signal transduction. Antibody binding can also cause receptor endocytosis and downregulation from the cell surface (CitationArteaga 2001; CitationGrunwald and Hidalgo 2003; CitationMarshall 2006). Additionally, bound antibody may recruit Fc receptor effector cells leading to antibody-dependent cell-mediated cytotoxicity (CitationArteaga 2001). In contrast, TKIs, by competitively binding to the ATP binding site of the intracellular domain of EGFR, inhibit EGFR autophosphorylation and impair downstream signaling, leading to cell cycle arrest and apoptosis in vitro (CitationMoyer et al 1997; CitationPollack et al 1999; CitationArteaga 2001; CitationGrunwald and Hidalgo 2003; CitationPao and Miller JCO 2005; CitationMarshall 2006). In addition to these two major classes of agents, other strategies targeting the epidermal growth factor (EGF) pathway are in preclinical and early clinical phases of development. These include immunotoxin conjugates, antisense oligonucleotides, and agents that target downstream signaling effectors of EGFR, such as farnesyl transferase inhibitors, angiogenesis inhibitors, and the multikinase inhibitor sorafenib.
Both mAbs and TKIs designed to block EGFR signaling have been extensively studied in a variety of malignancies, including pancreatic cancer. While the clinical focus of these agents has been on solid tumors with high levels of EGFR expression whose malignant phenotypes are known to be dependent on EGFR signaling, EGFR expression in and of itself may not be a reliable indicator of the potential antitumor activity of either class of agents, as will be discussed later. Moreover, although mAbs and TKIs may putatively have the same primary target, their activity profiles may in fact be quite different. TKIs have the advantage of being orally bioavailable and are more rapidly distributed throughout the body than mAbs; on the other hand, they may be less specific for their target than mAbs, and exhibit dose-limiting toxicities not generally observed with antibody therapy (CitationMendelsohn and Baselga 2003).
Cetuximab (Erbitux®, Imclone/Bristol-Meyers Squibb, New York), is a chimeric anti-EGFR mAb now approved for use in both head and neck and advanced colorectal cancers. The fully humanized anti-EGFR mAb panitumumab (Vectibix®, Amgen, Thousand Oaks, CA, USA) has also recently received approval in the salvage setting for advanced colorectal cancer. Other anti-EGFR mAbs under investigation include matuzumab and nimotuzumab (CitationCohenuram and Saif 2007). Evidence of promising activity of cetuximab in pancreatic cancer led to a large phase III study conducted by the Southwest Oncology Group (S0205), in which patients with locally advanced or metastatic pancreatic cancer were randomized to receive gemcitabine alone or in combination with cetuximab (CitationPhilip et al 2007). However, this 735-patient study showed no significant difference in median survival, progression-free survival (PFS), or response rate in patients on the cetuximab-containing arm.
The TKI erlotinib was first approved for use in 2004 in patients with advanced or metastatic NSCLC who had progressed on a platinum-based chemotherapy regimen. Another TKI, gefitinib (ZD1839, Iressa®; AstraZeneca, Wilmington, DE, USA), was also originally approved for advanced NSCLC, but its use has since been restricted to patients who had achieved documented clinical benefit from this agent or who were enrolled in selected clinical trials. More recently, lapatinib (Tykerb®, GlaxoSmithKline, Philadelphia, PA, USA), an oral TKI directed against both EGFR and the associated HER family member HER-2, has been approved for use in women with advanced HER-2 (+) breast cancer.
As noted above, the FDA approved erlotinib for use in pancreatic cancer in 2005. The remainder of this review will discuss the preclinical and clinical data supporting a role for targeted anti-EGFR therapy in advanced pancreatic cancer, with specific attention to this particular agent.
Rationale for EGFR as a therapeutic target in pancreatic cancer
In the normal pancreas, EGFR expression is limited to ductal cells and the islets of Langerhans (CitationUeda et al 2004). Similar to a number of other malignancies, EGFR has been shown to be overexpressed in pancreatic cancers (CitationYamanaka et al 1993; CitationArteaga 2001; CitationTobita et al 2003; CitationGrandis and Sok 2004; CitationMarshall 2006). EGFR overexpression with resultant increase in EGFR signaling may be associated with metastatic potential and decreased survival in patients with pancreatic cancer. Tobita et al studied EGFR expression by immunohistochemistry in 77 cases of invasive ductal adenocarcinoma of the pancreas and found EGFR expression to be significantly associated with metastatic potential (CitationTobita et al 2003). Likewise, Ueda et al demonstrated that cytoplasmic EGFR overexpression by immunohistochemistry was associated with higher grade tumors and shorter survival in a study of 76 archived cases of pancreatic ductal carcinoma (CitationUeda et al 2004). In contrast, membranous EGFR staining did not correlate with tumor grade or survival in a study of 71 cases of pancreatic adenocarcinoma by CitationBloomston et al (2006).
In addition to overexpression of EGFR, autocrine stimulation of the receptor may also play a role in the pathogenesis of pancreatic cancer. In a pancreatic tumor cell line, Murphy et al showed that autocrine stimulation of EGFR in vitro results in sustained cell proliferation that can be abrogated by inhibiting EGFR tyrosine kinase activity, by administering inhibitors of downstream MAPK signaling, or by frequently changing the culture medium to remove potential autocrine growth factors (CitationMurphy et al 2001).
Though EGFR activating mutations have been demonstrated in certain malignancies including NSCLC and gliomas, similar mutations have not been identified in pancreatic cancer to date. The EGFR sequence appears to be highly conserved in pancreatic cancer (CitationLee et al 2007; CitationTzeng et al 2007a). A deletion in exon 19 was identified in 3.6% (2 of 55) patients with pancreatic cancer in a study by Kwak et al although the small sample size precluded interpretation of the clinical significance of this finding (CitationKwak et al 2006). In another small study of 30 pancreatic surgical specimens and 9 pancreatic cancer cell lines, investigators identified a polymorphism in EGFR intron 1 that was associated with shorter survival (CitationTzeng et al 2007b). Though hypothesized to increase transcriptional efficiency of EGFR and thus lead to overexpression, the polymorphism was not associated with any increased expression of downstream molecular markers of EGFR pathway activation.
At this time, the mechanism of EGFR activation in pancreatic cancer remains poorly understood. Gene amplification, autocrine or paracrine upregulation, altered receptor processing, ligand overexpression, and/or dysregulation of co-receptor dimerization each may represent a potential contributing factor. Despite the current gaps in our knowledge, the evidence of EGFR overexpression in pancreatic cancer specimens, combined with convincing in vitro data demonstrating the oncogenic potential of EGFR activation, together support a role for EGFR activation in the progression of this malignancy. Preclinical and clinical data of pharmacologic EGFR inhibition in pancreatic cancer further support this principle and are presented below.
Preclinical data of EGFR inhibition by TKIs in pancreatic cancer
Preclinical studies in a variety of solid tumor cell lines have demonstrated anti-tumor effect of EGFR inhibition by TKIs, including several studies specific to pancreatic cancer. In two pancreatic cancer mouse xenograft models, erlotinib alone and in combination with gemcitabine significantly inhibited EGFR phosphorylation and increased apoptosis (CitationNg et al 2002). Treatment of the human pancreatic cell line HPAC with erlotinib reduced cell proliferation in tissue culture (CitationDurkin et al 2006). When these HPAC cells were implanted into athymic/nude mice, the tumors that formed both grew more slowly and exhibited decreased metastatic potential when the mice were treated with erlotinib. A separate study showed that the TKI gefitinib completely inhibited EGF-induced cell proliferation as well as EGFR autophosphorylation and phosphorylation of MAPK in pancreatic cancer cell lines (CitationLi et al 2004). PKI-166, another TKI in development, was shown to have activity alone and in combination with gemcitabine in a mouse xenograft model of pancreatic cancer (CitationBruns et al 2000).
Erlotinib is a low molecular weight quinazolin derivative which acts to selectively and reversibly inhibit of EGFR tyrosine kinase activity (CitationGrunwald and Hidalgo 2003; CitationPao and Miller 2005). Molecular studies show that erlotinib selectively inhibits EGFR tyrosine kinase activity at concentrations of 2nM in purified assays and 20nM in whole-cell assays; nonspecific inhibition of unrelated tyrosine kinases occurs at 1000-fold higher concentrations (CitationGrunwald and Hidalgo 2003). In pharmacokinetic analyses using the head and neck tumor cell line HN5, maximum inhibitory effects as measured by EGFR-associated phosphotyrosine and tumor growth were obtained 1 hour post treatment with erlotinib, with 75%–85% inhibition maintained for at least 12 hours; recovery to baseline activity occurred by 24 hours (CitationPollack et al 1999). Based on preclinical animal data, human plasma concentrations of 0.5 μg/mL have been estimated to inhibit EGFR adequately to achieve antitumor effect (CitationHidalgo et al 2001). A direct relationship exists between degree of EGFR inhibition and antitumor effect, suggesting the need for adequate dosing (CitationGrunwald and Hidalgo 2003; CitationMarshall 2006).
Clinical trials of erlotinib in pancreatic cancer
Based on the above preclinical findings, erlotinib has undergone extensive evaluation in numerous clinical trials and has gained approval for use in both NSCLC and pancreatic caricnoma. The original phase I trials of this agent were conducted in previously treated patients with a broad range of solid tumors and allowed for the establishment of optimal dosing and toxicity profiles. In one such study, Hidalgo et al evaluated erlotinib as a single agent in 40 patients with advanced solid malignancies in doses ranging from 25 to 200 mg daily, in continuous and intermittent schedules, and established a maximum tolerated daily dose of 150 mg (CitationHidalgo et al 2001). 59% of patients (23/39) on study developed grade 1–2 dermatologic toxicity, including 41% (9/22) at the 150 mg once-daily dosing schedule. Although no grade 3–4 dermatologic toxicity was reported, 3 patients, including 1 at the 150 mg daily dosing, required termination of treatment because of clinically intolerable rash. Rash and diarrhea were the dose-limiting toxicities at doses higher than 150 mg. Other principal toxicities included mucositis, hyperbilirubinemia, and headache. Pharmacokinetic analysis indicated that a dose of 150 mg daily produced plasma concentrations greater than 0.5 μg/mL in most patients, corresponding to the level expected to result in antitumor activity from the preclinical studies described above.
A phase I study initially presented at the 2004 annual meeting of the American Society of Clinical Oncology (ASCO), since published, evaluated escalating doses of erlotinib in combination with standard-dose gemcitabine (1000 mg/m2 weekly x 7, then weekly x 3 every 4 weeks) in 26 patients, the majority (n = 15) of whom had pancreatic cancer (Porterfield et al 2004; CitationDragovich et al 2007). Two sequential cohorts were enrolled, the first cohort receiving erlotinib at 100 mg daily and the second 150 mg daily. Because 3 of 9 patients at the 100 mg dose level developed grade 3 transaminitis, the study protocol was subsequently amended to increase the stringency of entry criteria, limiting patients to no more than one prior chemotherapy regimen, transaminases less than 1.5× upper limits of normal, and a bilirubin within normal range. The most common non-hematologic toxicity was skin rash, occurring in 18 of 26 patients (69%), none higher than grade 2. Incidence of skin rash was similar at the 100 mg and 150 mg dose levels (67% and 71%, respectively). Other common toxicities included diarrhea in 14 patients (54%), 3 of whom had grade 3; nausea in 11 patients (42%), one of whom had grade 3; and fatigue in 14 patients (54%), 4 of whom had grade 3. Four patients (15%) also developed grade 3 neutropenia. There was 1 fatal episode of pulmonary toxicity in a patient with NSCLC previously treated with chemoradiation. On the whole, the toxicity profile of this combination appeared acceptable, with patients able to tolerate gemcitabine plus erlotinib at 150 mg daily with manageable side effects. Moreover, of the 12 patients with pancreatic cancer evaluable for response, 1 had a partial response (8%), and 9 had stable disease for more than 3 months (75%).
The pivotal study upon which the FDA approval of erlotinib for use in pancreatic cancer was based was PA.3, a phase III trial conducted by the National Cancer Institute of Canada Clinical Trials Group (NCIC-CTG). Results of this study were first presented at the ASCO Gastrointestinal Cancers Symposium in 2005 and have since been recently published (CitationMoore et al 2007b). PA.3 compared gemcitabine plus erlotinib with gemcitabine plus placebo as first-line therapy in patients with advanced pancreatic cancer. The study was a randomized, double-blind, placebo-controlled international trial with 176 centers in 17 countries. Enrollment consisted of 569 patients with locally advanced or metastatic adenocarcinoma of the pancreas, measurable disease, and an ECOG performance status (PS) between 0 and 2. Prior radiotherapy for local disease was allowed with fluorouracil or gemcitabine given concurrently as a radiosensitizer; patients could not have been previously treated with chemotherapy for advanced disease. Appropriate stratifications were made for both PS and disease stage; approximately 80% of patients on each arm had an ECOG PS of 0 or 1, and 75% had metastatic disease. Of note, EGFR overexpression was not required for enrollment.
All patients received gemcitabine 1000 mg/m2 by 30-minute infusion weekly for 7 weeks followed by 1 week rest in cycle 1. Subsequent cycles were administered weekly for 3 weeks followed by 1 week rest. Patients were randomized to receive erlotinib (285 patients) or placebo (284 patients). The vast majority of patients on the study arm received erlotinib at a dose of 100 mg daily until disease progression or unmanageable toxicity. During the latter portion of the study, a small Canadian cohort of 48 patients received erlotinib at 150 mg daily to assess the tolerability of this higher dose.
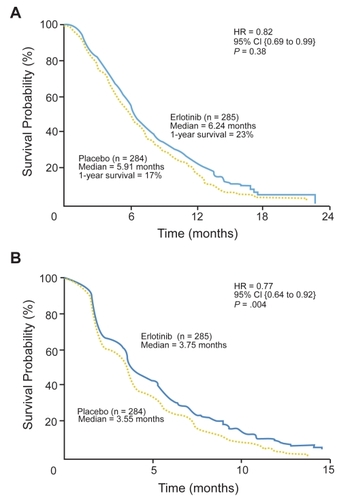
The PA.3 study was powered for overall survival as the primary endpoint. Secondary endpoints included PFS, response rate, response duration, toxicity, quality of life (QOL), and correlation of tumor EGFR expression with clinical outcome. Analysis of results showed median survival in patients randomized to gemcitabine plus erlotinib was 6.24 months as opposed to 5.91 months in the gemcitabine plus placebo group; one year survival was 23% versus 17%, respectively (p = 0.23). The hazard ratio (HR) for overall survival was 0.82 (p = 0.038). PFS was also significantly improved in the erlotinib arm (3.75 months) as compared with the placebo arm (3.55 months) with HR of 0.77 for progression (p = 0.004). Response rates were not significantly different between treatment groups (8.6% versus 8.0%), although a trend towards improved rate of disease control was observed for patients in the erlotinib arm (57.5% versus 49.2%, p = 0.07). There was no difference in response duration between groups. depicts Kaplan-Meier curves for overall survival and progression-free survival in the PA.3 study.
Figure 1 Kaplan-Meier curves for (A) overall survival; and (B) progression-free survival. HR = hazard ratio. Reproduced with permission from CitationMoore MJ, Goldstein D, Hamm J, et al 2007b. Erlotinib plus gemictabine compared to gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol, 25:1960–6. Copyright © 2007. American Society of Clinical Oncology.
Toxicity analysis showed treatment to be well-tolerated in both treatment groups. Patients in the erlotinib arm had higher incidence of rash, diarrhea, infection, and stomatitis. Skin rash was the most common toxicity, occurring in 203 of 282 of patients (72%) on the erlotinib-containing arm, fully half (36%) of whom were categorized as having grade 2 or higher. The incidence of grade 3–4 rash was 6% versus 1% in the erlotinib versus placebo arms, respectively.
As for non-dermatologic toxicity, the incidence of grade 3–4 diarrhea was 6% versus 2%. No differences in grade 3–4 hematologic toxicity were observed between the two arms. Of note, the incidence of interstitial lung disease (ILD) syndromes was 2.1% in the erlotinib arm versus 0.4% in the placebo arm. For comparison, the incidence of ILD was reported to be less than 1% in the Tarceva Lung Cancer Evaluation Trial (TALENT) which evaluated gemcitabine plus cisplatin with or without erlotinib in patients with advanced lung cancer (CitationGatzemeier et al 2007). In the PA.3 study, there were 6 protocol-related deaths overall, all of which occurred in the erlotinib arm. Two deaths were attributed to treatment complications (ILD and sepsis), while 4 were attributed to cancer complications combined with possible treatment-related effects.
Dose reductions for toxicity were required in 16% of patients in the erlotinib arm as opposed to 5% in the placebo arm. Dose reductions were more common in the small cohort of patients receiving the higher 150 mg dose of erlotinib (48% of patients at this level required a dose reduction) compared to those receiving a dose of 100 mg daily (dose reduction required in 13%).
Despite these differences in toxicity profiles, there was no difference in global QOL scores using the European Organisation for Research and Treatment of Cancer Core Quality of Life Questionaire C30 (EORTC QLQ-C30) instrument. Individual domain scores were also equivalent with the exception of diarrhea which was reported as worse in the erlotinib arm (p < 0.001). Of note, the EORTC QLQ-C30 instrument does not specifically address quality of life issues relating to skin toxicity. Given that even mild-to-moderate rash can confer impairment and distress, its effects on quality of life may be significant (CitationGridelli et al 2007).
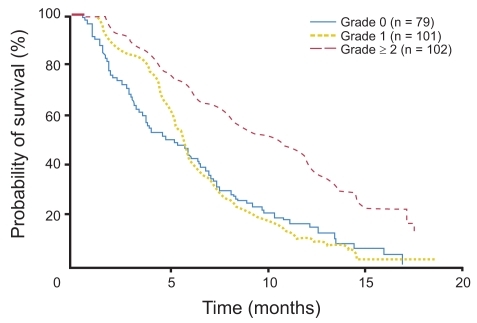
The PA.3 study also assessed EGFR expression in 162 tumor samples adequate for immunohistochemical EGFR analysis. Of these tumors, 86 (53%) were EGFR positive (as defined by at least 10% of cells staining positive) and 76 (47%) were EGFR negative. EGFR status was not significantly associated with response to erlotinib. Interestingly, subgroup analysis was notable for skin rash being associated with disease control with erlotinib (p = 0.05). Median survival for patients with grade 0, 1, and 2+ rash were 5.3, 5.8, and 10.5 months, respectively, with 1-year survival rates of 16%, 9%, and 43% (p < 0.001) ().
Figure 2 Overall survival by grade of rash in patients treated with erlotinib in PA.3 study. Reproduced with permission from CitationMoore MJ, Goldstein D, Hamm J, et al 2007b. Erlotinib plus gemictabine compared to gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol, 25:1960–6. Copyright © 2007. American Society of Clinical Oncology.
In conclusion, the PA.3 study showed that erlotinib in combination with gemcitabine results in a statistically significant improvement in overall survival in patients with advanced pancreatic cancer in the first-line setting. Although the absolute benefit in overall survival was modest with a median survival difference between the two arms of only 2 weeks, the hazard ratio of 0.82 corresponds to a 22% improvement in survival overall (CitationMoore et al 2007b). The hazard ratio may represent a more clinically meaningful parameter in a rapidly-progressive disease like pancreatic cancer in that it measures risk over a time continuum and may better reflect differences in survival than a static time point such as the median. Ultimately, the PA.3 study represents the first phase III study to demonstrate a survival benefit of combination therapy over gemcitabine monotherapy in advanced pancreatic cancer.
A number of questions arise in evaluating the results of this trial. The high incidence of ILD in the erlotinib arm is noteworthy, particularly by comparison to the lower incidence seen in TALENT which also used erlotinib in combination with gemcitabine. This finding raises the possibility of an additive toxicity with these agents in combination. Also worth pointing out are the subgroup analysis findings identifying female gender as being significantly associated with longer overall survival. This is particularly relevant given the gender imbalance between treatment arms (52.3% females in the erlotinib arm compared to 43.0% in the placebo arm). The authors report that treatment effect remained significant when adjusted for sex, but these data were not presented. The optimal dose of erlotinib in this study also remains in question; the authors recommend a starting dose of 100 mg daily with consideration of dose escalation to 150 mg daily as tolerated. Dose reductions for toxicity in the 150 mg group greatly exceeded those in the 100 mg group, however, suggesting that dose escalations must be undertaken with caution.
Most importantly, the question remains how to identify patients who will benefit from the addition of erlotinib. Although there was no relationship between clinical outcome and EGFR expression by immunohistochemistry, subgroup analysis in the PA.3 study did identify the development of rash as a significant predictor of response, a finding that has been observed in studies evaluating other EGFR inhibitors in other tumor types. This subject will be discussed in further detail in a subsequent section.
In addition to PA.3, smaller studies have evaluated erlotinib in other settings and combinations for advanced pancreatic cancer. A phase II study presented at the annual meeting of ASCO in 2005 evaluated the combination of capecitabine and erlotinib as a second-line regimen in 28 patients with metastatic pancreatic cancer after failure of front line therapy with gemcitabine (CitationBlaszkowsky et al 2005). This study reported a well-tolerated toxicity profile as well as a promising median survival of 6.7 months. Three patients (11%) were assessed as having a partial response and an additional 16 (57%) demonstrated stable disease. Another phase II study presented at the annual meeting of ASCO in 2006 randomized 58 previously untreated patients with advanced pancreatic cancer to treatment arms consisting of bevacizumab (B) and gemcitabine (C) plus either erlotinib (E) or cetuximab (C) (CitationKindler et al 2006). Preliminary analysis suggested activity of both regimens (GBE and GBC) with overall responses of 21% and 19%, respectively; similar six-month survival rates (38% and 41%); and minimal differences in toxicity profiles. A separate phase I study of erlotinib and gemcitabine in combination with bevacizumab is now underway (CitationGomez-Martin et al 2007). Erlotinib may also have very modest activity as a single agent or in combination with bevacizumab for patients who have progressed on prior chemotherapy (CitationEpelbaum et al 2007; CitationKo et al 2007). Clinical trials of erlotinib in advanced pancreatic cancer are reviewed in .
Table 1 Clinical trials of erlotinib in advanced pancreatic cancer
Predicting response to EGFR inhibition
Studies in a variety of solid tumor types have evaluated EGFR expression by immunohistochemical staining and have found equivocal association with clinical outcome in response to EGFR inhibition. A potential limitation of immunohistochemistry (IHC) in the assessment of EGFR expression is the uncertain recognition of truncated or mutated receptors, or receptors that have been internalized to the cytoplasm. Furthermore, EGFR expression may be differentially predictive in particular subsets of cases, leading to confounding results in population studies. In colorectal cancer, EGFR staining does not appear to correlate with clinical response to cetuximab; moreover, patients with negative immunohistochemical staining for EGFR have demonstrated responses to cetuximab (Cunningham et al 2004; CitationChung et al 2005). In NSCLC patients, EGFR expression by IHC was not associated with response to gefitinib in the Iressa Dose Evaluation in Advanced Lung Cancer (IDEAL) trials (CitationBell et al 2005; CitationMarshall 2006). Conversely, pooled data from two randomized, placebo-controlled trials in NSCLC, SWOG 0126 and an Italian study by Cappuzzo et al showed EGFR IHC to be an independent predictive marker of response to gefitinib (CitationCappuzzo et al 2005; CitationHirsch et al 2007). Limited data from a phase II study of erlotinib in head and neck cancers did not find EGFR staining by IHC to be predictive of response (CitationSoulieres et al 2004). In pancreatic cancer, EGFR expression by immunohistochemistry likewise has not been shown to correlate with clinical response. In the PA.3 study discussed above, there was no differential response to therapy in the EGFR positive and EGFR negative subgroups with hazard ratios of 0.80 and 0.83, respectively (CitationMoore et al 2007b). At this time, there is no data to support the decision to use EGFR inhibitor therapy based on EGFR expression by IHC alone.
FISH and PCR analysis have also been used to evaluate tumor specimens for EGFR gene amplification and presumed resulting overexpression. In NSCLC, increased EGFR copy number appears to correlate with response to the TKI gefitinib (CitationBell et al 2005; CitationHirsch et al 2005; CitationPao and Miller 2005; CitationTakano et al 2005; CitationShepherd and Tsao 2006; CitationCappuzzo et al 2007; CitationHirsch et al 2007). It is not known, however, whether gene amplification alone or in concert with an accompanying EGFR mutation confers increased response to TKI therapy. In a study of 66 pancreatic tumor specimens, increased EGFR copy numbers were detected by PCR in 41% of tumors (26/66); no association was found between copy number and survival (CitationLee et al 2007). In another study of 55 pancreatic tumor specimens, no amplification of EGFR by PCR was found in five cases that had demonstrated response to therapy with capecitabine plus erlotinib (CitationKwak et al 2006). In the PA.3 study, relationship of EGFR gene copy number to outcome was analyzed in a subset of 107 patients in whom tumor tissue was available. FISH-positivity for EGFR (46.7% of samples) was found to be a negative prognostic factor for progression-free survival irrespective of treatment (HR 1.85, p = 0.004) (CitationMoore et al 2007a). Moreover, the hazard ratio showed a trend toward greater benefit of erlotinib in patients who were EGFR FISH negative (HR 0.60, p = 0.08), although the authors concede that the sample size was too small to draw any firm conclusions.
Recent studies in NSCLC have also identified somatic mutations within the EGFR gene that are strongly associated with response to gefitinib or erlotinib. Retrospective studies collectively show that over 80% of NSCLC patients who demonstrated clinical response to either gefitinib or erlotinib harbored a mutation within the EGFR tyrosine kinase domain (CitationPaez et al 2004; CitationLynch et al 2004; CitationPao and Miller 2005, CitationTakano et al 2005). By comparison, the IDEAL trials identified EGFR gene mutations in only 50% of responders, but nonetheless corroborated a significant difference in response to gefitinib therapy in those patients with EGFR mutations (46%) compared to those without (10%) (p = 0.005) (CitationBell et al 2005). In the phase III INTACT trial, however, there was no survival benefit identified with the addition of gefitinib to chemotherapy in NSCLC patients with EGFR gene mutations (CitationBell et al 2005). Interpretation of these findings is complicated by the recognition that EGFR gene mutation and amplification are prognostic as well as predictive factors in NSCLC (CitationShepherd and Tsao 2006).
In pancreatic cancer, preclinical studies suggest EGFR mutations are infrequent, making it difficult to assess their association with responsiveness to anti-EGFR therapy (CitationKwak et al 2006; CitationLee et al 2007; CitationTzeng et al 2007a). As noted previously, an intron 1 polymorphism with variable length of CA repeats was recently characterized in a study of 30 microdissected pancreatic surgical specimens and 9 cell lines (CitationTzeng et al 2007b). A shorter sum of CA repeats (less than 36) was associated with shorter median survival than in patients with greater than or equal to 36 repeats. Additionally, cell lines with a shorter intron 1 length demonstrated an in vitro response to erlotinib (p = 0.02). To date, EGFR intron 1 CA repeat length has yet to be confirmed as a prognostic or predictive factor in clinical trials.
Another potential molecular predictor of response to therapeutic EGFR inhibition is the presence or absence of mutant K-ras. RAS/MAP kinase represents one of the downstream effector pathways of EGFR signaling, and mutations in any of these signaling components could lead to resistance to therapeutic agents such as erlotinib (CitationEberhard et al 2005). Analysis of a subset of NSCLC patients on the TRIBUTE trial of chemotherapy with or without erlotinib demonstrated significantly decreased time to tumor progression and overall survival in those individuals with an activating K-ras mutation (21% of the evaluable cohort) on the erlotinib-containing arm (CitationEberhard et al 2005). Given the very high frequency of K-ras mutations seen in pancreatic cancer, one might similarly ask whether such mutations affect response to erlotinib in this patient population. Moore and colleagues were able to assess the K-ras mutational status in a subset of 117 patients on the PA.3 trial (CitationMoore et al 2007a), 92 (78.6%) of whom had mutated K-ras. While a trend toward greater benefit of erlotinib for overall survival was observed in patients with wild-type K-ras (HR 0.66), this finding was not statistically significant (p = 0.34). A prospective study that is adequately powered to answer this question more precisely is necessary.
In terms of clinical predictors of response, the development of skin rash has been suggested as a surrogate marker of favorable response to EGFR inhibition by both TKI and MAb across a variety of solid tumor types. The characteristic EGFR inhibitor rash is papulopustular, occurring predominantly on the face and upper torso within two weeks of the start of treatment. Although the etiology remains unclear, the rash is postulated to occur secondary to EGFR inhibition in the skin (CitationPerez-Soler and Saltz 2005). The overall incidence of rash with EGFR inhibitor therapy ranges from 50% to 100%, depending on the type of cancer, dose, and the specific EGFR inhibitor used. A much smaller percentage of patients experience grade 3 or 4 rash, characterized by a generalized, symptomatic dermatitis covering greater than 50% of body surface area (CitationSegaert and Van Cutsem 2005). In colorectal cancer, response rates to cetuximab were shown to be significantly higher in patients with adverse skin reactions in several studies (Cunningham et al 2004;Lenz et al 2005). Multiple studies of both TKI and MAb in NSCLC, head and neck cancer, and ovarian cancer also suggest that development of rash is associated with improved outcomes (CitationWacker et al 2007). In pancreatic cancer, the PA.3 study corroborated these findings, showing that the presence of rash correlated with improved outcomes as discussed in the previous section (CitationMoore et al 2007b) ().
The mechanism of association between rash and improved outcome in EGFR inhibition remains unclear. The rash may reflect individual differences in drug exposure due to metabolism, differences in the immune system, or EGFR polymorphisms (CitationWacker et al 2007). A potential confounding variable is the duration of time on therapy, which is longer in patients with response to therapy and may result in a higher incidence of skin rash independent of any true surrogacy for outcome. Likewise, nonresponders with poor outcome may become too ill and discontinue therapy prior to development or documentation of rash, leading to underreporting of rash in this subset of patients. To address this confounding variable, a combined analysis was conducted of the phase III NCIC-CTG studies BR.21 of erlotinib monotherapy in advanced NSCLC and PA.3 in pancreatic cancer. This study excluded patients who died within the first month on study to minimize the potential bias created by early deaths. The combined analysis showed that the presence of rash strongly correlated with overall survival in both studies, corroborating previous data (CitationWacker et al 2007). If skin rash is a true surrogate for the degree of EGFR inhibition within the tumor, the question arises whether the dosage of EGFR inhibitors should be escalated to the development of this rash if tolerated without significant other toxicities.
Future directions for EGFR inhibitor therapy in pancreatic cancer
The challenge remains how to identify the subset of advanced pancreatic cancer patients most likely to benefit from EGFR inhibition. In breast cancer, Her2 status by immunohistochemistry and FISH predicts response to trastuzumab, the monoclonal antibody to Her2 which, like EGFR, is a member of the HER family of receptors. In NSCLC, a subset of patients demonstrates a strong, durable response to TKI therapy. Multiple studies suggest that EGFR gene amplification and EGFR mutations are associated with response in these patients, leading to the promise of applying a targeted therapy primarily to those individuals most likely to benefit, especially in a disease in which short survival time renders ineffectual therapy particularly costly. As discussed above, molecular markers predictive of response have not yet been identified among pancreatic cancer patients. Further study is required to evaluate biomarkers that may play a predictive or prognostic role in this aggressive malignancy.
Optimal dosing of erlotinib in advanced pancreatic cancer patients also remains in question. In the PA.3 trial, a dose of 150 mg daily was associated with significantly higher toxicity than 100 mg daily and required dose reductions in 48% of patients (CitationMoore et al 2007b). In vitro studies suggest that drug concentration directly correlates with degree of antitumor effect, however, underscoring the importance of adequate dosing. Furthermore, the incidence of rash is greater with higher doses of EGFR inhibitor (CitationSegaert and Van Cutsem 2005). In concert with data suggesting that rash may be a biomarker for tumor response to EGFR inhibition, this dose-toxicity relationship suggests the need for future study of dose escalation to the development of rash, similar to studies being conducted with cetuximab in colorectal cancer (CitationTejpar et al 2007). It is unknown whether higher doses of erlotinib can induce rash in a greater proportion of patients, and whether this would result in improved clinical outcomes without unacceptable toxicity. Given that even mild-to-moderate skin rash can result in significant patient distress and impairment, the clinical benefit of anti-EGFR therapy must be carefully weighed against the toxicity of this common side effect.
The positive results of the PA.3 trial introduce multiple potential new applications for the TKI erlotinib in pancreatic cancer. In the adjuvant setting, a recent phase III study demonstrated a significant benefit in disease free survival with the use of postoperative gemcitabine in resectable pancreatic cancer (CitationOettle et al 2007). These results together with the results of the PA.3 study raise the possibility of a benefit from erlotinib in combination with cytotoxic chemotherapy in the resectable population as well, although obviously one must apply caution before extrapolating positive results in patients with advanced cancer to those with earlier stages of disease.
Further study may also identify potential additive or synergistic effects of erlotinib in combination with other chemotherapeutic or biologic agents, as inhibition of EGFR signaling by TKI or MAb theoretically may sensitize tumor cells to non-specific cellular stresses and augment the efficacy of other therapies. Another avenue for investigation is the evaluation of erlotinib as a radiosensitizer. It has been previously demonstrated that EGFR activation contributes to radiation resistance (CitationSchmidt-Ullrich et al 1997; CitationDent et al 1999), and in preclinical models, this enhanced tumor-cell survival and radiation resistance may be overcome by inhibition of EGFR (CitationHuang et al 1999; CitationLammering et al 2001; CitationKrishnan et al 2006). A recent phase I study of erlotinib in combination with twice weekly gemcitabine and radiation therapy for locally advanced pancreatic cancer showed this combination to be well-tolerated with promising efficacy, reporting a partial response rate of 35% and stable disease rate of 53% in 17 assessable patients (CitationDuffy et al 2008). Other ongoing studies in resected and locally advanced pancreatic cancer are evaluating erlotinib together with other chemotherapy platforms, such as capecitabine ± gemcitabine and gemcitabine plus oxaliplatin concurrent with radiation.
In conclusion, the PA.3 study has provided important proof of principle for the role of EGFR tyrosine kinase inhibition in advanced pancreatic cancer. Despite small differences in overall survival, the addition of erlotinib to gemcitabine produced a significant difference in hazard ratio for survival in this rapidly progressive malignancy and now represents an appropriate standard of care option for patients. Given these findings in the context of the preclinical and clinical data presented above, ongoing study is warranted to better characterize the potential benefits of EGFR inhibition as a valuable therapeutic strategy in pancreatic cancer.
Disclosures
AK has received research funding from Genentech for the conduct of investigator-initiated clinical trials in pancreatic cancer, including one that incorporates the use of erlotinib; and has received honoraria from this same company for talks overviewing the treatment landscape for pancreatic cancer.
References
- Abou-AlfaGKLetourneauRHarkerG2006Randomized phase III study of exatecan and gemcitabine compared with gemcitabine alone in untreated advanced pancreatic cancerJ Clin Oncol244441716983112
- ArteagaC2001The epidermal growth factor receptor: from mutant oncogene in nonhuman cancers to therapeutic target in human neoplasiaJ Clin Oncol1918s32s40s11560969
- BellDWLynchTJHaserlatSM2005Epidermal growth factor receptor mutations and gene amplification in non-small-cell lung cancer: molecular analysis of the IDEAL/INTACT gefitinib trialsJ Clin Oncol2380819216204011
- BerlinJDCatalanoPThomasJP2002Phase III study of gemcitabine in combination with fluorouracil versus gemcitabine alone in patients with advanced pancreatic carcinoma: Eastern Cooperative Oncology Group Trial E2297J Clin Oncol203270512149301
- BlaszkowskyLSKulkeKHRyanKP2005A phase II study of erlotinib in combination with capecitabine in previously treated patients with metastatic pancreatic cancerJ Clin Oncol2316S abstract 4099
- BloomstonMBhardwajAEllisonE2006Epidermal growth factor receptor expression in pancreatic carcinoma using tissue microarray techniqueDig Surg2374916717472
- BrunsCJSolorzanoCCHarbisonMT2000Blockade of the epidermal growth factor receptor signaling by a novel tyrosine kinase inhibitor leads to apoptosis of endothelial cells and therapy of human pancreatic carcinomaCancer Res6029263410850439
- BurrisHAMooreMJAndersenJ1997Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trialJ Clin Oncol152403139196156
- CappuzzoFHirschFRRossiE2005Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancerJ Nat Cancer Inst976435515870435
- CappuzzoFLigorioCJannePA2007Prospective study of gefitinib in epidermal growth factor receptor fluorescence in situ hybridization-positive/phospho-Akt-positive or never smoker patients with advanced non-small-cell lung cancer: the ONCOBELL trialJ Clin Oncol2522485517538169
- ChungKYShiaJKemenyN2005Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor by immunohistochemistryJ Clin Oncol2318031015677699
- CohenuramMSaifM2007Epidermal growth factor receptor inhibition strategies in pancreatic cancer: past, present, and the futureJ Pancreas8415
- CunninghamDChauIStockenD2005Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancerECCO 13 European Cancer Conference abstract PS11
- DentPReardonDBParkJS1999Radiation-induced release of transforming growth factor alpha activates the epidermal growth factor receptor and mitogen-activated protein kinase pathway in carcinoma cells, leading to increased proliferation and protection from radiation-induced cell deathMol Biol Cell10249350610436007
- DragovichTHubermanMvon HoffDD2007Erlotinib plus gemcitabine in patients with unresectable pancreatic cancer and other solid tumors: phase IB trialCancer Chemother Pharmacol6029530317149608
- DuffyAKortmanskyJSchwartzGK2008A phase I study of erlotinib in combination with gemcitabine and radiation in locally advanced, non-operable pancreatic adenocarcinomaAnn Oncol19869117878176
- DurkinAJOsborneDAYeatmanTJ2006EGF receptor antagonism improves survival in a murine model of pancreatic carcinomaJ Surg Res13519520116678855
- EberhardDAJohnsonBEAmlerLC2005Predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinibJ Clin Oncol235900916043828
- EpelbaumRSchnaiderJGluzmanA2007Erlotinib as a single-agent therapy in patients with advanced pancreatic cancerASCO Gastrointestinal Cancers Symposium abstract 202
- GatzemeierUPluzanskaASzczesnaA2007Phase III study of erlotinib in combination with cisplatin and gemcitabine in advanced non-small-cell lung cancer: the Tarceva lung cancer investigation trialJ Clin Oncol2515455217442998
- Gomez-MartinCCamaraJCCortesH2007A phase I study of erlotinib, bevacizumab, and gemcitabine in patients with advanced pancreatic cancerJ Clin Oncol2518s abstract 4611
- GrandisJRSokJC2004Signaling through the epidermal growth factor receptor during the development of malignancyPharmacol Ther102374615056497
- GridelliCMaionePAmorosoD2007Clinical significance and treatment of skin rash from erlotinib in non-small cell lung cancer patients: Results of an Experts Panel MeetingCrit Rev OncIn press
- GrunwaldVHidalgoM2003Developing inhibitors of the epidermal growth factor receptor for cancer treatmentJ Nat Cancer Inst958516712813169
- HeinemannVHinkeABöckSGemcitabine-based combinations (gem+x) vs gemcitabine (gem) alone in the treatment of advanced pancreatic cancer: a meta-analysis of sixteen randomized trialsJ Clin Oncol2518s abstract 4515
- HeinemannVLabiancaRHinkeALouvetC2007Increased survival using platinum analog combined with gemcitabine as compared to single-agent gemcitabine in advanced pancreatic cancer: pooled analysis of two randomized trials, the GERCOR/GISCAD intergroup study and a German multicenter studyAnn Oncol18101652917660491
- HerrmannRBodokyGRuhstallerT2007Gemcitabine plus capecitabine compared with gemcitabine alone in advanced pancreatic cancer: a randomized, multicenter, phase III trial of the Swiss Group for Clinical Cancer Research and the Central European Cooperative Oncology GroupJ Clin Oncol252212717538165
- HidalgoMSiuLNemunaitisJ2001Phase I and pharmacologic study of OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in patients with advanced solid malignanciesJ Clin Oncol1932677911432895
- HirschFRVarella-GarciaMCappuzzoF2007Combination of EGFR gene copy number and protein expression predicts outcome for advanced non-small-cell lung cancer patients treated with gefitinibAnnals of Oncology187526017317677
- HirschFRVarella-GarciaMMcCoyJ2005Increased epidermal growth factor recptor gene copy number detected by fluorescence in situ hybridization associates with increased sensitivity to gefitinib in patients with bronchioloalveolar carcinoma subtypes: a Southwest Oncology Group StudyJ Clin Oncol2368384515998906
- HuangSMBockJMHarariPM1999Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of head and neckCancer Res5919354010213503
- JemalASiegelRWardE2007Cancer statistics 2007CA Cancer J Clin57436617237035
- KindlerHLBylowKAHochsterHS2006A randomized phase II study of bevacizumab (B) and gemcitabine (G) plus cetuximab (C) or erlotinib (E) in patients (pts) with advanced pancreatic cancer (PC): a preliminary analysisJ Clin Oncol2418s abstract 4040
- KoAHDitoESchillingerB2007A phase II trial of bevacizumab plus erlotinib for patients with metastatic gemcitabine-refractory pancreatic cancerASCO Gastrointestinal Cancers Symposium abstract 187
- KolbAKeelfJArnoldN2006Expression and differential signaling of heregulins in pancreatic cancer cellsInt J Cancer1205142317096356
- KrishnanSBrownPDBallmanKV2006Phase I trial of erlotinib with radiation therapy in patients with glioblastoma multiforme: results of North Central Cancer Treatment Group protocol N0177Int J Radiat Oncol Biol Phys651192916626884
- KwakELJankowskiJThayerSP2006Epidermal growth factor receptor kinase domain mutations in esophageal and pancreatic adeno-carcinomasClin Cancer Res124283716857803
- LammeringGValerieKLinPS2001Radiosensitization of malignant glioma cells through overexpression of dominant-negative epidermal growth factor receptorClin Cancer Res76829011297265
- LeeJJangKTKiCS2007Impact of epidermal growth factor receptor (EFGR) kinase mutations, EGFR gene amplifications, and KRAS mutations on survival of pancreatic adenocarcinomaCancer1091561917354229
- LenzH-JVan CutsemEKhambata-FordS2006Multicenter phase II and translational study of cetuximab in metastatic colorectal carcinoma refractory to irinotecan, oxaliplatin, and fluoropyrimidinesJ Clin Oncol2449142117050875
- LiJKleefJGieseN2004Gefitinib (Iressa, ZD1839), a selective epidermal growth factor receptor tyrosine kinase inhibitor, inhibits pancreatic cancer cell growth, invasion, and colony formationInt J Oncol252031015202007
- LouvetCLabiancaRHammelP2005Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trialJ Clin Oncol2335091615908661
- LynchTJBellDWSordellaR2004Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinibN Engl J Med35021293915118073
- MarshallJ2006Clinical implications of the mechanism of epidermal growth factor receptor inhibitorsCancer10712071816909423
- MendelsohnJBaselgaJ2003Status of epidermal growth factor receptor antagonists in the biology and treatment of cancerJ Clin Oncol212787279912860957
- MooreMJGoldsteinDHammJ2005Erlotinib plus gemcitabine compared to gemcitabine alone in patients with advanced pancreatic cancer. A phase III trial of the National Cancer Institute of Canada Clinical Trials Group (NCIC-CTG)J Clin Oncol2316s abstract 1
- MooreMJda Cunha SantosGKamel-ReidS2007aThe relationship of K-ras mutations and EGFR gene copy number to outcome in patients treated with erlotinib on National Cancer Institute of Canada Clinical Trials Group trial study PA.3J Clin Oncol2518S abstract 4521
- MooreMJGoldsteinDHammJ2007bErlotinib plus gemictabine compared to gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials GroupJ Clin Oncol251960617452677
- MoyerJDBarbacciEGIwataKK1997Induction of apoptosis and cell cycle arrest by CP-358,774, an inhibitor of epidermal growth factor receptor tyrosine kinaseCancer Res574838489354447
- MurphyLOCluckMWLovasS2001Pancreatic cancer cells require an EGF receptor-mediated autocrine pathway for proliferation in serum-free conditionsBrit J Cancer8492693511286473
- NgSSTsaoMSNickleeT2002Effects of the epidermal growth factor receptor inhibitor OSI-774, Tarceva, on downstream signaling pathways and apoptosis in human pancreatic adenocarcinomaMol Cancer Ther17778312492110
- OettleHPostSNeuhausPGellertK2007Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancerJAMA2972677717227978
- PaezJGJannePALeeJC2004EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapyScience304149750015118125
- PaoWMillerVA2005Epidermal growth factor receptor mutations, small-molecule kinase inhibitors, and non-small-cell lung cancer: current knowledge and future directionsJ Clin Oncol2325566815767641
- Perez-SolerRSaltzL2005Cutaneous adverse effects with HER1/EGFR-targeted agents: is there a silver lining?J Clin Oncol2352354616051966
- PhilipPABenedettiJFenoglio-PreiserC2007Phase III study of gemcitabine [G] plus cetuximab [C] versus gemcitabine in patients [pts] with locally advanced or metastatic pancreatic adenocarcinoma [PC]: SWOG S0205 studyJ Clin Oncol2518s abstract LBA4509
- PollackVASavageDMBakerDA1999Inhibition of epidermal growth factor receptor-associated tyrosine phosphorylation in human carcinomas with CP-358,774: dynamics of receptor inhibition in situ and antitumor effects in athymic miceJ Pharmacol Exp Ther2917394810525095
- PorterfieldBWDragovichTPatnaikAErlotinib + gemcitabine in patients with unresectable pancreatic carcinoma: results from a phase IB trialJ Clin Oncol2214S abstract 4110
- Rocha LimaCMGreenMRRotcheR2004Irinotecan plus gemcitabine results in no survival advantage compared with gemcitabine monotherapy in patients with locally advanced or metastatic pancreatic cancer despite increased tumor response rateJ Clin Oncol2237768315365074
- Schmidt-UllrichRKMikkelsenRBDentP1997Radiation-induced proliferation of the human A431 squamous carcinoma cells is dependent on EGFR tyrosine phosphorylationOncogene15119179294612
- SegaertSVan CutsemE2005Clinical signs, pathophysiology and management of skin toxicity during therapy with epidermal growth factor receptor inhibitorsAnn Oncol1614253316012181
- ShepherdFATsaoMS2006Unraveling the mystery of prognostic and predictive factors in epidermal growth factor receptor therapyJ Clin Oncol2412192016505443
- SoulieresDSenzerNNVokesEE2004Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrpsine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neckJ Clin Oncol22778514701768
- TakanoTOheYSakamotoH2005Epidermal growth factor receptor gene mutations and increased copy numbers predict gefitinib sensitivity in patients with recurrent non-small-cell lung cancerJ Clin Oncol2368293715998907
- TejparSPeetersMHumbletY2007Phase I/II study of cetuximab dose-escalation in patients with metastatic colorectal cancer (mCRC) with no or slight skin reactions on cetuximab standard dose treatment (EVEREST): Pharmacokinetic (PK), pharmacodynamic (PD), and efficacy dataJ Clin Oncol2518s abstract 4037
- TobitaKKijimaHDowakiS2003Epidermal growth factor receptor expression in human pancreatic cancer: significance for liver metastasesInt J Mol Med11305912579331
- TzengCWFrolovAFrolovaN2007Epidermal growth factor receptor (EGFR) is highly conserved in pancreatic cancerSurgery141464917383523
- TzengCWFrolovAFrolovaN2007Pancreatic cancer epidermal growth factor receptor (EGFR) intron 1 polymorphism influences postoperative patient survival and in vitro erlotinib responseAnn Surg Onc1421508
- UedaSOgataSTsudaH2004The correlation between cytoplasmic overexpression of epidermal growth factor receptor and tumor aggressiveness; poor prognosis in patients with pancreatic ductal adenocarcinomaPancreas29e1815211117
- WackerBNagraniTWeinbergJ2007Correlation between development of rash and efficacy in patients treated with the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib in two large phase III studiesClin Cancer Res1339132117606725
- YardenYSliwkowskiM2001Untangling the ErbB signaling networkNat Rev Mol Cell Biol21273711252954
- YipDKarapetisCStricklandA2006Chemotherapy and radio-therapy for inoperable advanced pancreatic cancerCochrane Database Syst Rev3CD00209316855985
- YamanakaYFriessHKobrinMS1993Coexpression of epidermal growth factor receptor and ligands in human pancreatic cancer is associated with enhanced tumor aggressivenessAnticancer Res1356598317885