Abstract
Autophagy is an ancient, intracellular degradative system which plays important roles in regulating protein homeostasis and which is essential for survival when cells are faced with metabolic stress. Increasing evidence suggests that autophagy also functions as a tumor suppressor mechanism that harnesses the growth and/or survival of cells as they transition towards a rapidly dividing malignant state. However, the impact of autophagy on cancer progression and on the efficacy of cancer therapeutics is controversial. In particular, although the induction of autophagy has been reported after treatment with a number of therapeutic agents, including imatinib, this response has variously been suggested to either impair or contribute to the effects of anticancer agents. More recent studies support the notion that autophagy compromises the efficacy of anticancer agents, where agents such as chloroquine (CQ) that impair autophagy augment the anticancer activity of histone deacetylase (HDAC) inhibitors and alkylating agents. Inhibition of autophagy is a particularly attractive strategy for the treatment of imatinib-refractory chronic myelogenous leukemia (CML) since a combination of CQ with the HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) compromises the survival of even BCR-ABL-T315I+ imatinib-resistant CML. Additional studies are clearly needed to establish the clinical utility of autophagy inhibitors and to identify patients most likely to benefit from this novel therapeutic approach.
Autophagy and its role in cancer
Autophagy is an evolutionarily conserved bulk protein degradative pathway in which double-membraned autophagosome vesicles engulf target proteins or organelles and deliver them to lysosomes where they are degraded. Accordingly, the autophagic response plays important roles in regulating protein and organelle homeostasis. However, this pathway is also essential to generate alternative energy sources during starvation or other states of significant cellular stress (CitationKlionsky and Emr 2000; CitationYorimitsu and Klionsky 2005). Further, mouse knockout models have demonstrated that autophagy also plays a critical role in mammalian development, where targeted deletion of Beclin-1 leads to mid-gestational embryonic lethality (CitationYue et al 2003) and where loss of either Atg5 or Atg7, two key players in the ubiquitin-like systems of this pathway, results in perinatal lethality (CitationKuma et al 2004; CitationKomatsu et al 2005).
Several recent high-profile investigations have established that autophagy also plays important roles in cancer biology. However, exactly how autophagy intersects with cancer development, disease progression, tumor maintenance, and therapeutic responses is controversial. The most significant studies regarding the role(s) of autophagy in cancer were recently reviewed by White and colleagues (CitationMathew et al 2007). Collectively, studies in this arena indicate that autophagy is a tumor suppressor pathway that holds cancer development in check, by preventing the proliferation of altered cells. For example, mice haploinsufficient for Beclin-1, an established initiator of the autophagy pathway, are tumor prone (CitationLiang et al 1999; CitationQu et al 2003; CitationYue et al 2003). Further, allelic loss of Beclin-1 is a frequent event in certain types of solid tumors and genes encoding other autophagy regulators are localized to “hot spots” for deletions in tumors (CitationAita et al 1999; CitationLiang et al 1999). Thus, impairing the autophagy response may prime cells for oncogenesis and contribute to the intrinsic genetic instability of pre-malignant and cancer cells. Considering this, agents that stimulate autophagy have potential value in cancer chemoprevention.
Paradoxically, it appears that activation of autophagy is detrimental once a cancer has fully developed. In particular, the degradation of cytoplasmic material and organelles by autophagy is essential to prevent bioenergetic failure when tumor cells face nutrient or oxygen deprivation (CitationLum et al 2005a), which are hallmarks of the tumor microenvironment. Thus, here autophagy would be predicted to promote tumor cell survival and consequently, disease progression (CitationLum et al 2005b). Indeed, despite their diverse mechanisms of action, many frontline anticancer agents ultimately shut down conventional metabolic pathways due to the cellular stress that they impose, and they thus can stimulate autophagy. Indeed, the induction of autophagy has been observed in malignant cells after treatment with a number of cancer therapeutics including arsenic trioxide, rapamycin, histone deacetylase (HDAC) inhibitors, tamoxifen, imatinib, and ionizing radiation (CitationBursch et al 1996; CitationPaglin et al 2001; CitationKanzawa et al 2003; CitationShao et al 2004; CitationTakeuchi et al 2005; CitationErtmer et al 2007). Although some of the these studies have suggested that efficacy of these agents correlates with their ability to stimulate autophagy-mediated cell death, such conclusions need rigorous testing, especially since these modalities also trigger apoptosis. In fact, two recent studies have shown that combining inhibitors of autophagy with alkylating agents or HDAC inhibitors significantly enhances their anticancer activity (CitationAmaravadi et al 2007; CitationCarew et al 2007). Collectively, the majority of investigations have shown that apoptosis is a much more efficient mode of cell death than autophagy-mediated cell killing.
Inhibition of autophagy as a therapeutic strategy for imatinib-refractory CML
Chronic myelogenous leukemia (CML) is one of the four most common types of adult leukemia. In contrast to most forms of cancer, the molecular basis of disease development and pathogenesis is very well defined for CML. The underlying cause of CML is the formation of the Philadelphia chromosome due to a translocation between chromosomes 9 and 22. This translocation results in the production of the BCR-ABL fusion protein, a tyrosine kinase that controls critical signaling pathways involved in the development and progression of CML. The disease is subdivided into three phases, chronic phase, accelerated phase, and blast crisis, and disease progression can occur over several years.
Treatment of CML was revolutionized by the advent of imatinib mesylate (Gleevec™; Novartis), a targeted agent that inhibits the tyrosine kinase activity of BCR-ABL by competing with ATP for binding to the active site of the enzyme. Imatinib is very effective for many CML patients particularly during the chronic phase, yet drug resistance is an emerging problem. Several factors contribute to clinical imatinib resistance, including BCR-ABL overexpression and gene amplification and/or loss-of-function mutations in p53 (CitationShah et al 2002; CitationWendel et al 2006). However, the most prevalent cause of drug resistance is the development of missense mutations in BCR-ABL that either precludes the adoption of the inactive conformation of the kinase or that directly interferes with drug binding. A large number of specific mutations have been characterized and tremendous effort has been put forth to develop more effective strategies to treat imatinib-resistant CML patients. Two novel kinase inhibitors, dasatinib (Sprycel™; Bristol-Myers Squibb) and nilotinib (Tasigna™; Novartis), have shown promising clinical activity in some imatinib-refractory patients and both have recently earned FDA approval. Unfortunately, patients with the T315I BCR-ABL mutation do not respond to these new drugs (CitationKantarjian et al 2006; CitationTalpaz et al 2006).
Given these considerations, a major focus in the field of CML therapy is the development of novel strategies that can effectively treat T315I+ patients. Indeed, a large number of anticancer agents are under investigation for this specific indication. HDAC inhibitors are one such class of drugs that may have utility in the clinical treatment of imatinib-resistant CML. HDAC inhibitors are a fascinating class of anticancer agents, due to their multifaceted mechanisms of action and preferential toxicity for malignant cells, and they have demonstrated promising anticancer activity in a number of pre-clinical and clinical studies (CitationKelly and Marks 2005). SAHA (suberoylanilide hydroxamic acid, vorinostat, Zolinza®; Merck) is the most clinically advanced HDAC inhibitor and recently received FDA approval for the treatment of cutaneous T-cell lymphoma (CTCL) in an oral formulation. SAHA’s anticancer mechanism of action has been reported to include the induction of apoptosis, autophagy, polyploidy, reactive oxygen species (ROS), and growth inhibition (CitationYu et al 2003; CitationShao et al 2004; CitationXu et al 2005; CitationNawrocki et al 2006).
Due to its ability to provide essential sources of energy, autophagy has been suggested to blunt the efficacy of several pro-apoptotic therapeutic agents. Therefore, drugs that impair autophagy could theoretically potentiate the anticancer efficacy of SAHA and other HDAC inhibitors by disabling this important cell survival mechanism. We recently tested this hypothesis in a cell line model of imatinib resistance and in imatinib-refractory primary CML cells from patients with BCR-ABL mutations, including T315I (CitationCarew et al 2007). Both chloroquine (CQ) and 3-methyladenine (3-MA), two pharmacological inhibitors of autophagy, synergistically increased the killing power of SAHA against imatinib-resistant CML. Furthermore, this therapeutic strategy was also effective in killing T315I-Bcr-Abl-expressing cells lacking p53, and this combination demonstrated a high degree of selectivity, indicating that a therapeutic index can be achieved.
CQ is a weak base and a known lysosomotropic agent that perturbs lysosomal function. Our data indicated that CQ synergizes with SAHA by disrupting the lysosomal membrane permeability of CML cells, which in turn led to the rapid relocalization and accumulation of the lysosomal aspartic protease cathepsin D to the cytosol. Notably, cathepsin D is a key mediator of a lysosomally triggered apoptotic cascade induced by several anticancer agents (CitationBoya et al 2003; CitationFehrenbacher and Jaattela 2005) and a key substrate of this protease is the antioxidant protein thioredoxin (Trx). Interestingly, the CQ/SAHA combination markedly reduced Trx levels in CML cells, and correspondingly increased ROS (CitationCarew et al 2007), suggesting that a CQ-to-LMP-to-cathepsin D-to-Trx-to-ROS pathway contributes to the demise of imatinib-resistant CML also treated with SAHA.
Collectively, these findings indicate that autophagy promotes the survival of CML cells after metabolic stress that is induced by SAHA. Thus, inhibition of autophagy by CQ interferes with this cytoprotective response and augments SAHA’s anticancer activity. A recent study revealed that imatinib stimulates autophagy in a wide variety of cell types, suggesting that this response contributes to the resistance of several tumor types (CitationErtmer et al 2007). Thus, future investigations are clearly warranted to evaluate the impact of targeting the autophagy pathway on the anticancer activity of imatinib, where one would predict that inhibitors of autophagy such as CQ may also have utility in combination with imatinib (). Indeed, inhibition of autophagy as a novel anticancer strategy may have broad applications for a wide variety of refractory malignancies. This concept is further supported by recent studies demonstrating that inhibition of autophagy by CQ in a Myc-driven mouse model of B cell lymphoma delays tumor onset (CitationMaclean et al 2008) and augments the anticancer activity of the alkylating agent cyclophosphamide in established disease (CitationAmaravadi et al 2007). Indeed, clinical trials testing the efficacy of hydroxychloroquine in combination with bortezomib, temozolomide, temsirolimus, or other agents have been initiated for various malignancies. The knowledge gained from these trials will provide critical insights into the clinical potential of inhibition of autophagy as a novel anticancer strategy.
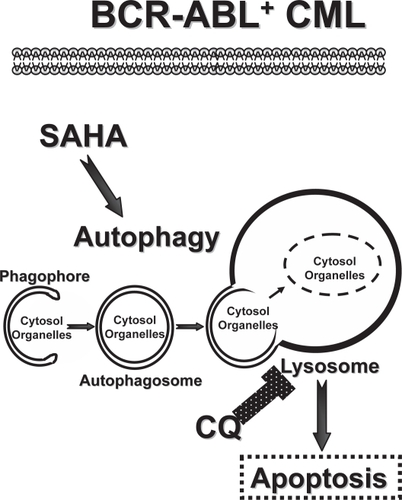
Figure 1 Targeting the autophagy pathway in CML. Imatinib-resistant BCR-ABL+ CML are sensitive to a therapeutic combination of SAHA, a FDA-approved HDAC inhibitor, and CQ, a FDA-approved anti-malarial agent (CitationCarew et al 2007). Here the overall efficacy of SAHA is thought to be compromised by its induction of the autophagy pathway, which is initiated by the formation of double-membraned autophagosomes from vesicular structures coined the phagophore. Autophagosomes deliver their cargo, bulk cytosolic material and organelles, to the lysosome, which degrades the cargo to provide essential building blocks and energy to the CML cell. The lysosomotropic agent CQ compromises lysosomal functions and thus derails the autophagy pathway, augmenting the killing power of SAHA to overcome imatinib resistance provoked by mutations in BCR-ABL and/or p53 (CitationCarew et al 2007).
Concluding remarks
Autophagy is an evolutionarily conserved cell survival pathway that is now rightly receiving attention as a regulator of tumor cell biology. Recent investigations have suggested that autophagy is a promising target for pharmacological inhibition in the context of cancer therapy (CitationCarew et al 2007; CitationAmaravadi et al 2007). On the flip side, others have argued that autophagy represents an alternative cell death pathway that contributes to the killing power of certain anticancer agents (CitationKanzawa et al 2003; CitationTakeuchi et al 2005). Despite these controversies, it is clear that agents such as CQ that inhibit autophagy may have clinical utility in the treatment of imatinib-refractory CML. The observations that imatinib alone induces autophagy, coupled with the evidence that autophagy may play an active role in resistance to anti-cancer agents, mandates the need for additional studies to define whether it represents a novel mechanism of imatinib resistance. It will also be very interesting to assess if imatinib-refractory CML patients have higher basal rates of autophagy than those that respond well to therapy. Hopefully, the knowledge gained from such studies will enable clinicians to use the genetic status of key autophagy regulators to predict therapeutic responses and tailor specific regimens to optimize clinical outcomes for patients with CML, and for those suffering from other resistant malignancies.
Acknowledgements
The authors’ studies were supported by a grant CA076379 (to JLC) from the National Cancer Institute and by monies awarded by the State of Florida to Scripps-Florida.
Disclosures
None of the authors has any conflicts of interest to disclose.
References
- AitaVMLiangXHMurtyVV1999Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21Genomics59596510395800
- AmaravadiRKYuDLumJJ2007Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphomaJ Clin Invest1173263617235397
- BoyaPAndreauKPoncetD2003Lysosomal membrane permeabilization induces cell death in a mitochondrion-dependent fashionJ Exp Med19713233412756268
- BurschWEllingerAKienzlH1996Active cell death induced by the anti-estrogens tamoxifen and ICI 164 384 in human mammary carcinoma cells MCF-7. in culture: the role of autophagyCarcinogenesis1715956078761415
- CarewJSNawrockiSTKahueCN2007Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistanceBlood1103132217363733
- ErtmerAHuberVGilchS2007The anticancer drug imatinib induces cellular autophagyLeukemia219364217330103
- FehrenbacherNJaattelaM2005Lysosomes as targets for cancer therapyCancer Res652993515833821
- KantarjianHGilesFWunderleL2006Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALLN Engl J Med35425425116775235
- KanzawaTKondoYItoH2003Induction of autophagic cell death in malignant glioma cells by arsenic trioxideCancer Res632103812727826
- KellyWKMarksPA2005Drug insight: Histone deacetylase inhibitors – development of the new targeted anticancer agent suberoylanilide hydroxamic acidNat Clin Pract Oncol2150716264908
- KlionskyDJEmrSD2000Autophagy as a regulated pathway of cellular degradationScience29017172111099404
- KomatsuMWaguriSUenoT2005Impairment of starvation-induced and constitutive autophagy in Atg7-deficient miceJ Cell Biol1694253415866887
- KumaAHatanoMMatsuiM2004The role of autophagy during the early neonatal starvation periodNature4321032615525940
- LiangXHJacksonSSeamanM1999Induction of autophagy and inhibition of tumorigenesis by beclin 1Nature402672610604474
- LumJJBauerDEKongM2005aGrowth factor regulation of autophagy and cell survival in the absence of apoptosisCell1202374815680329
- LumJJDeberardinisRJThompsonCB2005bAutophagy in metazoans: cell survival in the land of plentyNat Rev Mol Cell Biol64394815928708
- MacleanKHDorseyFCClevelandJL2008Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesisJ Clin Invest118798818097482
- MathewRKarantza-WadsworthVWhiteE2007Role of autophagy in cancerNat Rev Cancer7961717972889
- NawrockiSTCarewJSPinoMS2006Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cellsCancer Res6637738116585204
- PaglinSHollisterTDeloheryT2001A novel response of cancer cells to radiation involves autophagy and formation of acidic vesiclesCancer Res614394411212227
- QuXYuJBhagatG2003Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy geneJ Clin Invest11218092014638851
- ShahNPNicollJMNagarB2002Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib STI571. in chronic phase and blast crisis chronic myeloid leukemiaCancer Cell21172512204532
- ShaoYGaoZMarksPA2004Apoptotic and autophagic cell death induced by histone deacetylase inhibitorsProc Natl Acad Sci USA10118030515596714
- TakeuchiHKondoYFujiwaraK2005Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitorsCancer Res6533364615833867
- TalpazMShahNPKantarjianH2006Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemiasN Engl J Med35425314116775234
- WendelHGde StanchinaECeperoE2006Loss of p53 impedes the antileukemic response to BCR-ABL inhibitionProc Natl Acad Sci USA1037444916651519
- XuWSPerezGNgoL2005Induction of polyploidy by histone deacetylase inhibitor: a pathway for antitumor effectsCancer Res657832916140952
- YorimitsuTKlionskyDJ2005Autophagy: molecular machinery for self-eatingCell Death Differ12Suppl 215425216247502
- YuCSublerMRahmaniM2003Induction of apoptosis in BCR/ABL+ cells by histone deacetylase inhibitors involves reciprocal effects on the RAF/MEK/ERK and JNK pathwaysCancer Biol Ther25445114614324
- YueZJinSYangC2003Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressorProc Natl Acad Sci USA100150778214657337