Abstract
Lamotrigine (LTG), an antiepileptic drug, has been shown to be able to improve cerebral ischemic damage by limiting the presynaptic release of glutamate. The present study investigated further the neuroprotective effect of LTG on hypoxic-ischemic brain damage (HIBD) in neonatal rats and its relations to administration time and doses. The HIBD model was produced in 7-days old SD rats by left common carotid artery ligation followed by 2 h hypoxic exposure (8% oxygen). LTG was administered intraperitoneally with the doses of 5, 10, 20, and 40 mg/kg 3 h after operation and the dose of 20 mg/kg 1 h before and 3 h, 6 h after operation. Blood and brain were sampled 24 h after operation. Nissl staining, terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL), and neuron-specific enolase (NSE) immunohistochemical staining were used for morphological studies. Water content in left cortex and NSE concentration in serum were determined. LTG significantly reduced water content in the cerebral cortex, as well as the number of TUNEL staining neurons in the dentate gyrus and cortex in hypoxic-ischemia (HI) model. Furthermore, LTG significantly decreased the NSE level in serum and increased the number of NSE staining neurons in the cortex. These effects, except that on water content, were dose-dependent and were more remarkable in the pre-treated group than in the post-treated groups. These results demonstrate that LTG may have a neuroprotective effect on acute HIBD in neonates. The effect is more prominent when administrated with higher doses and before HI.
Introduction
Hypoxic ischemia (HI) is a common cause of brain damage in fetus and neonates. It is believed that HI may cause brain damages by activating a cascade of biochemical events, in which glutamate receptor plays an important role in triggering the toxic events (CitationHattori and Wasterlain 1990; CitationStrijbos et al 1996). Previous studies have shown that the antagonists of N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor have a neuroprotective effect on hypoxic-ischemic brain damage (HIBD) in stroke models (CitationNellgard and Wieloch 1992; CitationScatton et al 1994). However, the adverse profile of glutamate receptor antagonists limits their application in clinical practice (CitationScatton et al 1994). An alternative approach to ameliorate ischemic damage is to inhibit the presynaptic release of glutamate. Lamotrigine (LTG), a new kind of anticonvulsant drug with a action of inhibiting excessive presynaptic release of glutamate (CitationLeach et al 1986) by blocking voltage-sensitive sodium channels (CitationCheung et al 1992; CitationLees and Leach 1993), has been demonstrated to be effective for HIBD in focal and global cerebral ischemia model (CitationGraham et al 1993; CitationSmith and Meldumr 1995; CitationWiard et al 1995; CitationCrumrine et al 1997), as well as in the in vitro experiments (CitationCalabresi et al 2003). Although the neuroprotective effects of LTG have been well demonstrated in ischemic models with adult rats and LTG has even been used as combination therapy for the patients with ischemic stroke (CitationChen et al 2002), little is known about its effects on neonatal HIBD (CitationPapazisis et al 2007). Since the immature brain is highly sensitive to glutamate excitotoxicity (CitationBarks and Silverstein 1992; CitationScatton et al 1994; CitationPuka-Sundvall et al 1997), it is conceivable that LTG might be useful for HIBD in neonates. Recent reports have shown that LTG has the ability of extensive placental transfer and also a good tolerability in infants (CitationOhman et al 2000; CitationMikati et al 2002), provided a basis for the clinical application in neonates or infants. The aim of the present study is to investigate the possible neuroprotective effects of LTG on HIBD in the perinatal period, and the relationships between the effect and administration time and doses. HI was induced according to the protocol in a widely used animal model of perinatal asphyxia (CitationRice and Vannucci 1981). Histological and biochemical changes were examined to evaluate the damage of HI. Neuron-specific enolase (NSE), an isoenzyme of the glycolytic enzyme enolase (2-phospho-D-glycerate hydrolase), was adopted as one of important quantitative index in our study, because it was reported that NSE is released into the cerebral spinal fluid (CSF) and systemic circulation when neuronal damage has occurred (Schoerkhuber et al 1990; CitationMartens et al 1998; CitationWang et al 1999; CitationVerdu et al 2001), and NSE is highly specific to the neuron.
Material and methods
HIBD model
All experiments were performed in accordance with the institutional animal care guidelines. Common carotid artery occlusion combined with hypoxia in postnatal day 7 (P7) rat pups was performed to induce HIBD (CitationRice and Vannucci 1981). Rat pups were placed in an incubator with ambient temperature of 37 °C. Under deep ether anesthesia, the left common carotid artery was isolated, double ligated, and cut between the ligatures. After the surgical procedure, the animal was put to recover for 2 h in the temperature-controlled incubator and was then exposed to hypoxia for 2 h: placed in an enclosed and vented chamber that was partially submerged in water (37 °C). Hypoxia was induced by continuous flow of warmed, humidified gas with 8% oxygen that was balanced with 92% nitrogen at the flow rate of 0.5 L/ml. All HI animals showed symptoms of hemiplegia and anoxia.
Study design
In order to evaluate the effects of different doses, P7 rats were divided into 4 different dose groups, ie, 5, 10, 20, and 40 mg/kg (n = 14 for each dose group). LTG was given intraperitoneally at 3 h after the ischemia. The dose of 20 mg/kg was selected to determine the therapeutic time window, which was administrated intraperitoneally 1 h before (pre-treated), or 3 h, 6 h after the ischemia (post-treated) (n = 14 for each time point group). The dose range was derived from the previous report (CitationSmith and Meldumr 1995; CitationWiard et al 1995; CitationLee et al 2000) and from our preliminary experiments. Since a pharmacokinetic study in rats has shown that LTG was rapidly absorbed following the intraperitoneal injection with a peak concentration achieved at 0.2–1.0 h for 20 mg/kg (CitationWalker et al 2000), LTG was administered 1 h before ischemia to achieve a stable plasma concentration in the pre-treated group. It has been reported that LTG plasma elimination half-life is approximately 28 h in Wistar rat with 10 mg/kg intraperitoneally (CitationCastel-Branco et al 2005), so a single dose administration was adopted in our study. Pure LTG (Glaxo Wellcome Co.) was suspended in 0.5% methylcellulose (1 mg/ml) before the experiment. Sham-operated controls (sham group, n = 14) and hypoxic-ischemic controls (HI group, n = 14) were only given equal volume of methylcellulose 3 h after HI. Six of 14 rats in each group were used for neuropathological examination, including immunohistochemical staining of NSE, terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL) and Nissl staining. And 8 from each treatment group were used for determination of water content and serum NSE level.
Measurement of water content
A rat was anesthetized with chloral hydrate (350 mg/kg) 24 h after HI, and the brain was rapidly excised. The sample of cortex from left cerebral hemisphere (ipsilateral to common carotid artery ligation) was weighed in milligram unit. The specimen was subsequently desiccated at 110 °C for 24 h. Reweighing the dry weight of the tissue, the water content was obtained by subtraction from the wet weight [water content = (wet weight-dry weight)/wet weight × 100%].
Determination of neuron-specific enolase (NSE) in serum
Blood was collected during excision. Blood samples were left to clot at room temperature for 20–30 min and then centrifuged and frozen at −18 °C until assayed. The concentration of NSE in serum was measured by enzyme immunoassay NSE kit (Canag diagnostics AB Company, Denmark).
Neuropathology
24 h after HI, the rat was anesthetized with 10% chloral hydrate (0.35 ml/100g, ip) and perfused transcardially with 10–15 ml of saline (0.9% NaCl), followed by 20–30 ml of 4% paraformaldehyde in 0.01M PBS (pH 7.4). The brain was excised and postfixed for 24 h at 4 °C, then through 10%, 20%, and 30% sucroses for cytoprotection. OCT-embedded tissue was frozen at –80 °C and sectioned on a cryotome at a thickness of 30 μm along the coronal plane. The sections from B-2.2 to B-4.2 were selected. The adjacent sections were processed for either immunohistochemical staining of NSE or TUNEL and Nissl staining. NSE staining neurons in cortex and TUNEL positive cells in the cortex and dentate gyrus were quantified by a single observer who had no knowledge of the treatment received by the animals using a light microscope (400×). The mean was derived from six counts (three sections from each sample and two areas from each section) in each animal for statistical analysis.
Statistical analysis
Data are expressed as mean ± SE. Data were analysed using the ANOVA test followed by post-hoc multiple comparisons by Dunnett’s test.
Results
The effect of LTG on brain edema
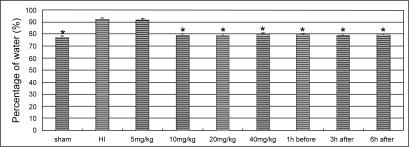
Hypoxic ischemia resulted in a significant increase of the water content. The treatments of LTG ≧10 mg/kg reduced water contents in HI animals significantly (P < 0.01). No significant difference in water content was detected between the each dose groups with LTG higher than 10 mg/kg, nor between the pre-treated and the post-treated groups ().
Figure 1 Changes in the water content in the cortex of ischemic hemisphere. The asterisks indicate statistically significant differences vs HI group value (*P < 0.01, n = 8 for each group). LTG treatment doses and the time points of administration are indicated in horizontal axis.
The effect of LTG on neuron necrosis and apoptosis
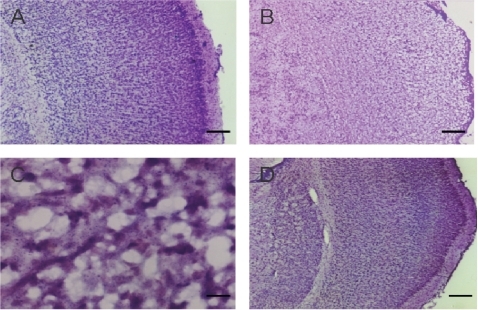
Nissl’s staining exhibited extensive brain damage in the hemisphere that subjected to hypoxic-ischemic injury. The regular architecture was destroyed with a marked loss of Nissl staining. Multiple necroses with shrinkage of cell body, pyknosis of nucleus, loss of Nissl substance, and disappearance of nucleolus, were found in the cortex, hippocampus, striatum, and thalamus. The impairment was reduced in the treated groups with LTG dose higher than 20 mg/kg ().
Figure 2 Photomicrographs of Nissl’s staining of the left cerebral cortex. A. A normal imaging from a sham-operated control. B. The regular architecture was destructed in the HI cortex. C. A high power view of the HI cortex, which shows necrosis of the cells with shrinkage of the cell body, pyknosis of nucleus, loss of Nissl substance, and disappearance of nucleolus. D. An imaging of the cortex from a subject received LTG of 40 mg/kg, 3 h after HI. Scale bar = 250 μm in A, B and D, and 25 μm in C.
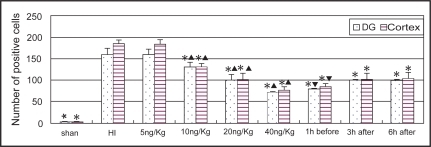
TUNEL positive cells appeared in many regions of the ischemic hemisphere including frontal lobe, parietal lobe, callosal gyrus, striatum and hippocampus, particularly in the pyramidal layer of CA1 region and the granular cell layer of dentate gyrus in the hippocampus (). Most of the TUNEL positive cells had the features of apoptosis with typical demilune and apoptotic bodies. The numbers of TUNEL positive cells in the cortex and dentate gyrus in the ischemic hemisphere were significantly decreased in the LTG-treated groups with doses ≧10 mg/kg (P < 0.01). There were significant differences between each dose groups with LTG of 10, 20, and 40 mg/kg. Significant differences were also found between the pre-treated and the post-treated groups (P < 0.01), but not between the 3 h post-treated and the 6 h post-treated groups ().
Figure 3 Photomicrographs of TUNEL staining of the dentate gyrus. A. Few TUNEL staining in the dentate gyrus from the sham-operated control. B. Marked TUNEL staining in the dentate gyrus in HI group. C. Reduced TUNEL staining in the dentate gyrus from the subject received LTG of 20 mg/kg, 3 h after HI. Scale bar = 250 μm.
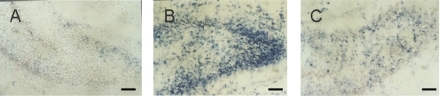
Figure 4 Comparison of the number of TUNEL staining cells in the cortex and dentate gyrus in different groups (n = 6). *Indicates statistically significant differences vs HI group values (P < 0.01). ▴ Indicates statistically significant differences between each dose groups (P < 0.01). ▾ Indicates statistically significant difference between the pre-treated and post-treated groups (P < 0.01).
The effect of LTG on NSE level in serum and the number of NSE staining neurons
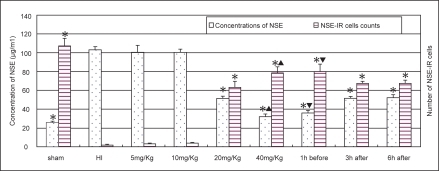
In HI group, NSE staining cells were rarely observed in the cerebral cortex and statistic analysis showed a significant decrease in cell count (). The NSE level in serum was significantly increased in HI group. The treatment of LTG ≧ 20 mg/kg resulted in a significant increase of NSE staining cells in the cortex and a decrease of NSE level in serum (P < 0.01) (). These changes were more significant in the pre-treated group than in the post-treated groups (P < 0.01). There was no significant difference between the 3 h post-treated and the 6 h post-treated groups.
Figure 5 Photomicrographs of NSE immunohistochemical staining of the left cerebral cortex. A. Imaging shows the NSE staining neurons in the cortex from the sham-operated control. B. Reduced NSE staining in HI group. C. Imaging shows the NSE staining neurons in the cortex from the subject received LTG of 20 mg/kg, 3 h after HI. Scale bar = 50 μm.

Figure 6 Alternations of the NSE level in serum (n = 8) and the number of NSE staining cells in the cortex (n = 6) in different groups. *Indicates statistically significant differences vs HI group values (P < 0.01). NSE level in serum is decreased, whereas the number of NSE staining cells is increased, comparing with HI controls. ▴Indicates statistically significant differences between LTG treated-groups of 20 mg/kg and 40 mg/kg (P < 0.01). ▾Indicates a statistically significant difference between the pre-treated and post-treated groups (P < 0.01).
Discussion
The neuroprotective effect of LTG on HIBD in neonatal model has recently been reported (CitationPapazisiset al 2007). Further to our preliminary study (CitationYi et al 2003), the present data demonstrate that LTG has a neuroprotective effect on HIBD in neonatal rats, shown by reducing the brain edema, protecting the neurons from necrosis and apoptosis, and inhabiting the leakage of NSE from the neurons. Results from the present study provided some evidence of neuroprotection of LTG for neonatal HIBD.
The dose-efficacy relationship on the neuroprotection of LGT has not well defined in previous studies, although there is a report from an in vitro experiment (CitationWang et al 2001). In order to study the dose-efficacy relationship and to know the effective dose of LTG for possible clinical application, we tried to quantify HIBD by measuring the water content in the cortex, TUNEL, and NSE staining cells in the brain, and NSE in serum. Brain edema is one of the basic pathological changes during HI. It is shown that LTG of more than 10 mg/kg can significantly decrease the water content in the HI cortex. However, we failed to find a difference between each dose, which might be imputed to the narrow range (77.2%–92.4%) of changes in water content, which is not sensitive enough to show minor alternations.
A good correlation between dose and efficacy is demonstrated in the dose range of 5–40 mg/kg, and the effective dose is from 10 mg/kg, shown by the TUNEL cell number. The correlation dose range is 10–40 mg/kg and the effective dose is from 20 mg/kg, shown by both the NSE staining cell number and the NSE concentrations in serum. The previous report has demonstrated a good relationship between the neurological outcome and serum NSE levels in the patients after cardiac arrest (Schoerkhuber et al 1990), suggesting that serum NSE levels can reflect the degrees of brain injury. The increase level of NSE in serum has been commonly accepted to be as a maker of the neuronal damage. Our results show an elevated NSE level in serum with decreased NSE staining in the neurons in neonatal rats after HI, confirmed that ischemia may cause the leakage of cytosolic brain enzymes into the blood and the elevation of NSE in serum can reflect the degree of brain damage. It is clinical significant because only blood sample can be obtained in practice. The effective dose rang of 10–40 mg/kg in the present study covered the doses of 10–20 mg/kg that was reported previously (CitationSmith and Meldumr 1995; CitationPapazisis1 et al 2007). LTG of 10–40 mg/kg in rats is equivalent to 1.6–6.4 mg/kg in human, lower than the dose of 5–15 mg/kg for monotherapy in patients with epilepsy. Since LTG could be well tolerated within this dose range for partial seizures and infantile spasms in infants (CitationMikati et al 2002), it is reasonable to conceive that LTG could be used as a neuroprotective agent for HIBD in neonates clinically. The fact that LTG of higher doses within the range of 10–40 mg/kg is more effective in protecting neuronal damage, shown in the present study, suggests that a higher dose within tolerated limit could be adopted in practice.
The therapeutic time window is crucial for a neuroprotective agent, which is not well defined for LTG. Previous studies have selected different time points for observation, from 1 h before HI in histotoxic hypoxia model (CitationSchulz et al 1995) to 5 h after recovery from cardiac arrest in rat (CitationCrumrine et al 1997). The present results show that LTG given either before or after HI, may attenuate HIBD in neonatal rats, with better effect in the pre-treated group and equivalent effect in the groups treated 3 h and 6 h after HI. Extended time points are needed to define the exact time window in further studies.
Clinically, neuroprotective agents may be used either in patient known at the risk of hypoxic-ischemic brain injury or as post-insult “rescue” therapy. HIBD in neonatal occurs most frequently in the antepartum or intrapartum periods during which the drug administration is usually difficult. Therefore, the essential characteristics for a neuroprotective agent in prevention and treatment of neonatal HIBD should include: First of all, a wide therapeutic time window; secondly, the ability to reach peak concentration in the brain rapidly after administration; thirdly, it can pass through the placenta when it is given during antepartum or intrapartum periods. The present study has shown that LTG has a therapeutic time window of at least 6 h after hypoxic-ischemia. It is demonstrated that LTG can be detected in the CSF 10 min after intraperitoneal administration and the penetration half-time into brain extracellular fluid is 0.51 ± 0.11 h (CitationWalker et al 2000). It has been reported that the maternal plasma LTG concentration at delivery is similar to that from the umbilical cord, and the plasma concentration in newborn declined slowly after then (CitationOhman et al 2000; CitationMikati et al 2002). Together, these data suggest that LTG has the potential for use as a neuroprotective agent in acute HI in neonates, as well as in pregnant women known at the risk of perinatal asphyxia.
In summary, the present results demonstrated that LTG of tolerable doses initiated either before or after HI can attenuate the brain damage in P7 rats, suggesting the potential for clinical use in prevention and treatment of HIBD.
Acknowledgements
This work is supported by a Grant for Medical Scientific Research from the health bureau of Guangdong province, R. P. China. (No. A2002298).
Reference
- BarksJDSilversteinFS1992Excitatory amino acids contribute to the pathogenesis of perinatal hypoxic-ischemic brain injuryBrain Pathol2235431343839
- CalabresiPCentonzeDCupiniLM2003Ionotropic glutamate receptors: still a target for neuroprotection in brain ischemia? Insights from in vitro studiesNeurobiol Dis1282812609492
- CalabresiPMartiMPicconiB2003Lamotrigine and remacemide protect striatal neurons against in vitro ischemia: an electrophysiological studyExp Neurol182461912895457
- CalabresiPPicconiBSaulleE2000Is pharmacological neuroprotection dependent on reduced glutamate release?Stroke317667310700517
- Castel-BrancoMMFalcãoACFigueiredoIV2005Lamotrigine pharmacokinetic/pharmacodynamic modelling in ratsFundam Clin Pharmacol196697516313279
- ChenSDLeeJMYangDI2002Combination therapy for ischemic stroke: potential of neuroprotectants plus thrombolyticsAm J Cardiovasc Drugs23031314727960
- CheungHKampDHarrisE1992An in vitro investigation of the action of lamotrigine on neuronal voltage-activated sodium channelsEpilepsy Res13107121334455
- CrumrineRCBergstrandKCooperAT1997Lamotrigine protects hippocampal CA1 neurons from ischemic damage after cardiac arrestStroke28223079368570
- GrahamSHChenJSharpFR1993Limiting ischemic injury by inhibition of excitatory amino acid releaseJ Cereb Blood Flow Metab1388978093250
- HattoriHWasterlainCG1990Excitatory amino acids in the developing brain: ontogeny, plasticity, and excitotoxicityPediatr Neurol6219282169749
- LeeWTShenYZChangC2000Neuroprotective effect of lamotrigine and MK-801 on rat brain lesions induced by 3-nitropropionic acid: evaluation by magnetic resonance imaging and in vivo proton magnetic resonance spectroscopyNeuroscience95899510619465
- LeachMJMardenCMMillerAA1986Pharmacological studies on lamotrigine, a novel potential antiepileptic drug: II. Neurochemical studies on the mechanism of actionEpilepsia2749073757936
- LeesGLeachMJ1993Studies on the mechanism of action of the novel anticonvulsant lamotrigine (Lamictal) using primary neurological cultures from rat cortexBrain Res61219097687190
- MartensPRaabeAJohnssonP1998Serum S-100 and neuron-specific enolase for prediction of regaining consciousness after global cerebral ischemiaStroke29236369804649
- MikatiMAFayadMKoleilatM2002Efficacy, tolerability, and kinetics of lamotrigine in infantsJ Pediatr14131512091848
- NellgårdBWielochT1992Postischemic blockade of AMPA but not NMDA receptors mitigates neuronal damage in the rat brain following transient severe cerebral ischemiaJ Cereb Blood Flow Metab122111345757
- OhmanIVitolsSTomsonT2000Lamotrigine in pregnancy: pharmacokinetics during delivery, in the neonate, and during lactationEpilepsia417091310840403
- PapazisisGKallarasKKaiki-AstaraA2007Neuroprotection by lamotrigine in a rat model of neonatal hypoxic-ischaemic encephalopathyInt J Neuropsychopharmacol2619
- Puka-SundvallMSandbergMHagbergH1997Brain injury after hypoxia-ischemia in newborn rats: relationship to extracellular levels of excitatory amino acids and cysteineBrain Res75032589098561
- RiceJE3rdVannucciRCBrierleyJB1981The influence of immaturity on hypoxic-ischemic brain damage in the ratAnn Neurol9131417235629
- ScattonBFrostJGeorgeP1994Present developments in NMDA receptor antagonists against cerebral ischaemiaCurrent Patents Lad0926–2594523545
- SchoerkhuberWKittlerHSterzF1999Time course of serum neuron-specific enolase a predictor of neurological outcome in patients resuscitated from cardiac arrestStroke30159860310436107
- SchulzJBMatthewsRTJenkinsBG1995Improved therapeutic window for treatment of histotoxic hypoxia with a free radical spin trapJ Cereb Blood Flow Metab15948527593355
- SmithSEMeldrumBS1995Cerebroprotective effect of lamotrigine after focal ischemia in ratsStroke26117227839380
- StrijbosPJLeachMJGarthwaiteJ1996Vicious cycle involving Na+ channels, glutamate release, and NMDA receptors mediates delayed neurodegeneration through nitric oxide formationJ Neurosci165004138756431
- Verdú PérezAFaleroMPArroyosA2001Blood neuronal specific enolase in newborns with perinatal asphyxiaRev Neurol327141711391504
- WangSJSihraTSGeanPW2001Lamotrigine inhibition of glutamate release from isolated cerebrocortical nerve terminals (synaptosomes) by suppression of voltage-activated calcium channel activityNeuroreport122255811447345
- WangXHQingMFangSZ1999A study on the changes of S-100, CK-BB, NSE and MBP levels in the blood and the cerebrospinal fluid after hypoxic-ischemic brain damage in ratsChin J Pediatr376702
- WalkerMCTongXPerryH2000Comparison of serum, cerebrospinal fluid and brain extracellular fluid pharmacokinetics of lamotrigineBr J Pharmacol130242810807660
- WiardRPDickersonMCBeekO1995Neuroprotective properties of the novel antiepileptic lamotrigine in a gerbil model of global cerebral ischemiaStroke26466727886726
- YiYHGuoWCLiaoWP2003Neuroprotective effects of lamotrigine on hypoxic-ischemic encephalopathy in neonatal rats: Relationships with administration timeEpilepsia44s8177