Abstract
Glomerulonephritis is a common cause of chronic kidney disease and end stage renal failure. Current therapy relies on variably effective, nonspecific and toxic immunosuppression. Recent insights into underlying biology and disease pathogenesis in human glomerulonephritis combined with advances in the fields of inflammation and autoimmunity promise a cadre of novel targeted interventions. This review highlights the therapeutic potential of two antigens, alpha3 (IV)NC1 collagen and podocyte neutral endopeptidase, and two cell signaling and effector molecules, IgG Fc receptors and complement, judged to be particularly amenable to therapeutic manipulation in man. It is anticipated that continued dissection of pathogenesis in the diverse disorders that comprise the glomerulonephritides will provide the basis for individualized disease-specific therapy.
Introduction
Glomerulonephritis encompasses a cluster of diseases that have been described as the most common single cause of end stage renal disease (ESRD) in the world (CitationTimoshanko and Tipping, 2005). GN was listed as the cause of ESRD in over 70,000 US patients in 2004 (US CitationRenal Data System 2007). Glomerulonephritis and hypertension together are also the most common causes of chronic kidney diseases (CKD) in developing countries, and GN trails only hypertension and diabetes as a leading cause of CKD in the US, Europe, and Japan. Notably, for every patient with clinical GN, an estimated 5–10 patients have undiagnosed subclinical disease (CitationCouser 1999). It is thus striking that an estimated 26 million Americans, or 13% of the US adult population, and over 50 million individuals worldwide have CKD (CitationDirks et al 2005; CitationCoresh et al 2007). Chronic GN may underlie pathogenesis of CKD in a significant proportion of this population. CKD not only places patients at risk for the various complications of renal damage, including increased cardiovascular disease and mortality, but it is estimated that each year more than one million CKD patients develop ESRD. Survival with ESRD requires renal replacement therapy with dialysis or transplantation, costly medical interventions not available in many developing countries. Clearly, effective therapy for GN would have significant impact globally on human health and health care financing.
GN is particularly prevalent among renal allograft recipients (CitationBriganti et al 2002; CitationCouser 2005). Glomerular diseases, including diabetes and glomerulonephritis, account for over 70% of patients receiving renal allografts, and in some countries GN alone accounts for up to 50% of patients (CitationBriganti et al 2002). In a recent review of the Australian registry, among 1505 pts with biopsy-proven GN receiving a primary renal transplant between 1988 and 1997, recurrent or de novo GN occurred in 6%–20% of patients. Recurrence was the third most frequent cause of allograft loss at 10 years, after chronic rejection and death with a functioning transplant. Among patients transplanted due to GN, 8.4% lost their allograft to recurrent GN by 10 years (CitationBriganti et al 2002). These patients may particularly benefit from novel targeted therapies, since disease onset can be readily identified and intervention initiated promptly, if not pre-emptively.
Definition of glomerulonephritis
The glomerulonephritides comprise a group of immunologic diseases that damage the renal glomerulus, the filtering unit of the kidney. This network of capillaries, fed and drained by the afferent and efferent arterioles, respectively, filters plasma across the fenestrated endothelium, glomerular basement membrane (GBM) and podocyte slit diaphragms to collect in Bowman’s space. Filtrate is dramatically modified as it passes through a series of tubules before delivery to the renal pelvis for excretion. The approximately 2 million glomeruli in normal adult kidneys filter over 150 liters of plasma per day. With this constant exposure and high filtering capacity, it is perhaps not surprising that the glomerulus is susceptible to injury from immune cells and their soluble products.
GN is defined as inflammation and cell proliferation in the glomerulus, although injury typically extends to the renal tubules, interstitium and vasculature. Common clinical findings include hematuria, proteinuria, and/or decreased glomerular filtration rate, detected as elevation in creatinine, often accompanied by hypertension, edema, and disease-specific findings. Immunopathogenesis is generally attributed to autoimmunity (see below), exacerbated by a local renal immune and inflammatory response. Common entities categorized as GN are indicated in . These include both renal-limited diseases and nephritides that develop as part of a systemic disorder, as well as those that have both renal-limited and systemic forms, such as anti-GBM nephritis and its systemic counterpart, Goodpasture syndrome. also includes several additional glomerulopathies with presumed immunologic origins but that are considered noninflammatory because they lack prominent cellular proliferation and infiltrates on renal histopathologic examination. These entities are often treated with immunosuppression regimens similar to those used to treat GN and thus are included in this review.
Table 1 Common glomerulonephritides and glomerulopathies with immune origins
Current therapies for GN comprise a modest list of broadly immunosuppressive agents (). Efficacy can be striking, as observed with the combination of steroids and cyclophosphamide, and more recently mycophenolate mofetil, for induction and maintenance therapy in lupus nephritis and the ANCA vasculitides. Newer drugs developed initially to abrogate alloimmunity in transplantation have also proven effective in subsets of GN patients refractory to or intolerant of older established drugs. Nonetheless, these interventions support broad, non-specific blockade of the immune and inflammatory networks, demonstrate variable and often unpredictable efficacy, and carry significant toxicities, including infection, cancer, osteoporosis, avascular necrosis, amenorrhea, sterility, and alopecia. Clearly, more specific safer interventions are urgently needed for this diverse group of diseases. It is therefore notable that a variety of promising new approaches to restore specific immune tolerance to autoantigens, using protein-, cellular-, or gene-based therapies, have entered the clinical arena for multiple autoimmune diseases (CitationWolfraim 2006). Therapy for GN has lagged in this area, in part because the target antigens in most human immune nephritides remain elusive, unlike their experimental counterparts, and in part because little is known about immune regulatory mechanisms controlling nephritogenic lymphocytes. Our goal in this review is to examine in detail two antigens, alpha3 (IV)NC1 collagen and neutral endopeptidase, and two signaling and effector molecules, FcγR and complement, that are judged to be particularly amenable for novel therapeutic intervention in GN in man. The reader is referred to other recent reviews (CitationJavaid and Quigg 2005; CitationFoster 2007) and for a more comprehensive cataloguing of potential biologic interventions in GN.
Table 2 Current common nonspecific therapies for glomerulo-nephritis
Table 3 Potential immunotherapy in autoimmunity and glomeru-lonephritis (after CitationFoster 2007)
Glomerulonephritis: pathogenesis and potential therapeutic targets
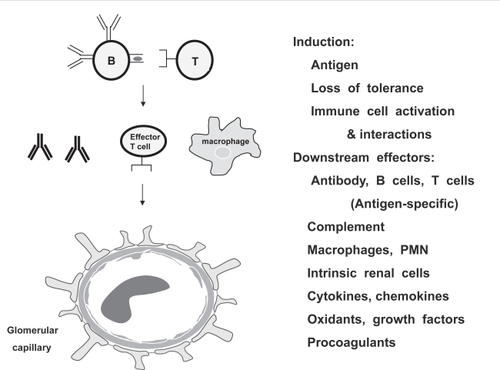
Considerable research in this field is focused on understanding the basic biology and pathogenesis of GN. Animal models have been particularly informative, both in highlighting the role of autoimmunity in the development of most GN and in providing in vivo tools with which to dissect the complex cell interactions that perpetuate disease and specific effector mechanisms that mediate tissue injury and inflammation. These models have revealed multiple targets for intervention, with disease specificity determined by the initiating antigen, the specific receptors on responding B and T lymphocytes, the glomerular antigen targeted locally, and the subset of downstream effectors recruited in a particular disease or individual ().
Figure 1 Pathogenesis of glomerulonephritis. Animal models suggest that autoimmunity underlies pathogenesis of most GN. Disease initiation requires loss of tolerance and activation of self-reactive lymphocytes. Activated B and T cells interact to promote autoantibody and cytokine production, immune complex formation, antibody deposition, macrophage and neutrophil recruitment, renal inflammation, and activation of renal endothelial, mesangial, epithelial and tubular cells. Glomerular and tubulointerstitial antigens may be specifically targeted by autoantibodies and self-reactive CD4 and CD8 TCR alpha/beta effector T cells, gamma/delta T cells and NKT cells. Numerous soluble and cellular mediators participate in subsequent tissue inflammation, injury and repair, and modulate local renal immune responses. The predominant cells and molecules engaged vary depending on the etiology and site of glomerular injury.
Three of the most extensively studied animal models of nephritis involve antibody interactions with intrinsic glomerular protein. Autoimmune responses to glomerular basement membrane (GBM) alpha3 (IV) collagen and to glomerular epithelial cell (podocyte) membrane antigens can be induced by active immunization of animals with heterologous or autologous kidney extract. Resulting autoantibodies lead to nephritis marked by linear GBM Ig deposits (eg, autoimmune anti-GBM Ig-mediated disease) or disease dominated by granular subepithelial deposits (eg, active Heymann nephritis), respectively. Nonautoimmune nephritis with similar histopathologic characteristics can be induced by passive administration of preformed antigen-reactive antibodies. A third model, mesangioproliferative nephritis, is induced in rats by passive administration of antibodies that recognize a rat mesangial cell phosphatidylinositol-anchored glycoprotein, termed Thy-1 antigen.
These models, as well as the spontaneous murine lupus models, provide considerable insight and are excellent tools with which to study downstream effector pathways. The specific mediators engaged vary by species, strain, and antigen preparation, and collectively effectively model the heterogeneous nephritis phenotypes observed in man (). Conversely, the models that depend on passive transfer of differentiated effectors or Ig provide minimal insight into the events that initiate and regulate renal-restricted autoimmunity. Also of unclear relevance to spontaneous human disease is the use of potent adjuvant in active immunization models. Elucidation of tolerance mechanisms that regulate nephritogenic autoreactivity will require novel autoreactive lymphocyte receptor transgenic models that target the relevant kidney antigens (CitationRudolph et al 2002; CitationZhang et al 2006).
Antigen-based approaches
Kidney antigen is an obvious target for achieving specificity in treating GN. Animal models engage a variety of cell-bound or soluble antigens either native to the glomerulus or that deposit on glomerular structures (). It is likely that similar antigens are involved in human GN, although to date only two classes of antigens have been confirmed in human disease: the noncollagenous (NC1) domains of the alpha 3 and 5 chains of type IV collagen in anti-GBM nephritis, and neutral endopeptidase in the rare but highly informative antenatal membranous nephropathy. These antigens will be considered in turn below, with regard to their therapeutic exploitation.
Table 4 Antigen targets in immune-mediated glomerular injury
Anti-GBM GN and Goodpasture syndrome
The best characterized and until recently only confirmed kidney target in human GN is alpha3 (IV)NC1 collagen. A pathologic autoimmune response to this antigen leads to focal segmental proliferative and necrotizing GN, frequently with crescent formation. This is associated with linear IgG deposition along the GBM, a pathologic hallmark of the disease. Co-involvement of the alveolar basement membrane with resultant pulmonary hemorrhage is termed Goodpasture syndrome (GPS). This restricted organ involvement is directly related to the restricted tissue expression of the target antigen.
Over the past two decades the molecular nature of the specific epitopes targeted by patient autoantibodies has been carefully dissected. The GBM collagen network is composed of heterotrimers of alpha3, alpha4 and alpha5 chains of type IV collagen. Trimers self associate at their NC1 carboxyl termini, forming NC1 hexamers, and at their amino termini, forming a collagen IV network. The epitopes recognized by the high affinity IgG autoantibodies isolated from serum and kidney eluates of GPS patients have been localized to the NC1 domain of the alpha3 (IV) chain (CitationWieslander et al 1984; CitationSaus et al 1988). These pathogenic epitopes are conformational and partially sequestered within the NC1 hexamer, minimally accessible for autoantibody binding unless hexamer dissociation into dimers or monomers exposes critical hydrophobic residues (CitationDavid et al 2001; CitationBorza et al 2002; CitationBorza and Hudson 2003; CitationBorza et al 2005). Environmental or genetic influences that facilitate hexamer dissociation may underlie disease susceptibility (CitationWilson 1997; CitationBorza et al 2005; CitationKalluri et al 2000a). Binding to accessible hydrophilic residues by low affinity autoantibodies may also promote pathogenic epitope exposure by inducing conformational changes and stabilizing monomers (CitationBorza et al 2000). Two immunodominant antibody epitopes recognized by the majority of GPS patient sera have been mapped using chimeric proteins bearing segments of the alpha3 (IV) chain superimposed on a nonimmunoreactive alpha1(IV)NC1 collagen framework (CitationHellmark et al 1999; CitationNetzer et al 1999; CitationBorza et al 2000). A restricted number of target epitopes is supported by demonstration of a shared idiotype, and thus shared structural determinant, among GPS serum autoantibodies (CitationMeyers et al 1998).
Epitope and antibody characteristics suggest several targets for specific therapy in anti-GBM nephritis. Selective immunoadsorption using affinity columns coated with antigen can rapidly reduce circulating autoantibody titers in active disease. As proof of concept, affinity chromatography using either purified bovine or recombinant human alpha3 (IV)NC1 collagen completely and selectively depletes pathogenic autoantibodies from patient plasma (CitationBoutaud et al 1996). Feasibility of this approach in the clinic is further suggested by the success of repetitive immunoadsorption in removing plasma anti-A or anti-B isotype agglutinins in man; when combined with standard anti-rejection immunosuppression, immunoadsorption permits long-term survival of ABO-incompatible kidney allografts (CitationDonauer et al 2006). This approach has an advantage over the current practice of initiating plasmapheresis in acute GPS, in that it does not deplete protective anti-microbial IgG, a major concern in a patient receiving concurrent cyclophosphamide.
Administration of oral or nasal GBM, bovine alpha3 (IV)NC1 dimers or recombinant rat alpha3 (IV)NC1 collagen can prevent induction of anti-GBM nephritis in antigen-immunized rodents (CitationKalluri et al 1997; CitationReynolds and Pusey 2001; CitationReynolds et al 2005). The tolerance mechanisms remain unclear, although downregulation of Th1 T cell responses is implicated. This approach may have particular utility as pre-emptive therapy in a subset of patients in whom onset of antibody production can be anticipated, such as Alport patients undergoing renal transplantation (see below). The consequences of antigen administration in active disease are less clear and require further study to understand the potential for triggering allergic or pathogenic stimulatory signals.
Antigen-based interventions in lupus nephritis, which has been most extensively studied, are instructive. Proposed approaches include neutralization or depletion of antigen-reactive autoantibodies, B cells, or T cells, using infusion of soluble antigen, peptide epitopes, antigen multimers or anti-idiotypic antibodies, either alone or linked to cytotoxins. It is reasoned that autoantigen epitopes recognized by pathogenic B and T cells can delay or reverse disease if administered in a dose and form that can anergize or delete pathogenic cells or induce inhibitory regulatory cells. In this regard, in lupus mice onset of nephritis can be delayed by biweekly subcutaneous administration of sub-nanomolar quantities of nucleosomal histone peptide (CitationKang et al 2005) or by intravenous injection of high doses of a synthetic consensus peptide based on unique T-cell stimulatory epitopes expressed within anti-DNA IgG variable regions (CitationHahn et al 2001). Immunization with peptide epitopes from anti-nucleosomal IgG variable regions has a similar effect (CitationStoll et al 2007). Tolerance depends in part on induction of antigen-specific regulatory CD4+CD25+ T cells and CD8+ inhibitory T cells that secrete TGF-beta (CitationLa Cava et al 2004, Citation2005; CitationHahn et al 2005; CitationKang et al 2005). IL-10+ regulatory T cells induced by nasal histone peptide (CitationWu et al 2002) or high dose intravenous ribonucleoprotein peptide (CitationRiemekasten et al 2004) are reported to provide protection in the (SWRxNZB)F1 and (NZBxNZW)F1 models of lupus nephritis, respectively. Murine lupus nephritis is also delayed by injection of a 21-mer laminin peptide recognized by a subset of pathogenic murine lupus autoantibodies (CitationAmital et al 2005). The antigen peptide is located within the laminin alpha1 chain expressed in normal adult kidney mesangium and diseased GBM (CitationAbrahamson et al 2003). In lupus patients, levels of anti-laminin autoantibodies correlate with disease activity and can be effectively removed by peptide-specific immunoadsorption (CitationAmital et al 2007). In early human trials, lupus nephritic flares were prevented by administration of double stranded DNA tetramers that putatively adsorb and remove anti-DNA IgG and anti-DNA B cells (CitationAlarcon-Segovia et al 2003); phase III clinical trials testing this reagent are ongoing (CitationFurie 2006).
Alport syndrome, alloantibodies and post-transplant anti-GBM GN
Collagen epitopes also may be targeted therapeutically in patients with Alport syndrome who develop ESRD and undergo kidney transplantation. Alport patients develop a hereditary nephritis due to mutations in one of the genes encoding the alpha chains (alpha3, alpha4, or alpha5) of type IV collagen of the GBM (CitationHudson et al 2003). Ultrastructurally this is manifest as splitting, thickening and thinning of the involved basement membranes. Clinical consequences are related to the tissue distribution of these alpha(IV) chains and include progressive renal failure, sensorineural deafness and/or ocular abnormalities. Because Alport patients lack the corresponding collagen proteins and thus lack the Goodpasture epitopes in their native basement membranes (CitationOlson et al 1980; CitationMcCoy et al 1982), those who undergo renal transplantation typically develop alloantibodies that bind GBM in the allograft but not the native kidneys. Of these, 3%–5% of patients will also develop anti-GBM GN, despite concurrent post-transplant immunosuppression. Although not all alloantibody target epitopes are known, alloantibody reactivity to NC1 domains of all three alpha chains (alpha3, alpha4, or alpha5) is reported (CitationKalluri et al 2000b). Anti-alpha3 (IV)NC1 alloantibodies eluted from the failed renal allograft of one patient with autosomal recessive Alport syndrome and post-transplant anti-GBM GN recognized alloepitopes distinct from Goodpasture autoepitopes (CitationWang et al 2005). Thus, Alport patients in particular may benefit from targeted therapies, since disease onset can be readily identified and intervention initiated promptly, if not pre-emptively.
Membranous nephropathy
Membranous nephropathy (MN) is a common cause of CKD and accounts for approximately 20% of nephrotic syndrome in adult Caucasians (CitationPonticelli, 2007). By 10 years, 20%–40% of patients progress to ESRD. Given this considerable disease burden, novel effective disease-specific therapies are urgently needed. Important insight into disease pathogenesis was gleaned from study of the rat Heymann nephritis models (CitationCouser and Nangaku 2006). Active HN is induced experimentally by immunization with kidney antigen preparations. Immunized rats develop marked proteinuria and glomerular pathology almost indistinguishable from human MN: thickened glomerular capillary basement membranes due to immune deposition exclusively in the subepithelial space. While it was initially believed that the granular deposits resulted from passive deposition of circulating immune complexes, key experiments subsequently revealed that antibody perfusion of isolated rat kidney led to immediate in situ formation of glomerular deposits (CitationCouser et al 1978; CitationVan Damme et al 1978). The target antigens were subsequently identified as megalin, a large membrane glycoprotein, multiligand endocytic receptor and member of the LDL-receptor-superfamily, and the receptor-associated protein, RAP, constitutively expressed on the rat glomerular podocyte foot processes (CitationKerjaschki and Farquhar 1982; CitationSaito et al 1994; CitationFarquhar et al 1995). Circulating pathogenic antibodies traverse the GBM, bind the megalin-RAP protein complex within the podocyte clathrin-coated pits, and shed as immune complexes that attach to the GBM. Additional autoantibodies in serum of rats with Heymann nephritis contribute to disease by binding distinct epitopes in the podocyte membrane including glycolipid and integrins to activate complement and induce proteinuria (CitationSusani et al 1994).
Elucidation of pathogenesis in Heymann nephritis spurred a search for podocyte targets in human idiopathic MN. Megalin is not a candidate; although it is found in human renal proximal tubules, it has not been detected in human glomeruli or in immune deposits in patients with MN (CitationKerjaschki et al 1987). Alternative candidates include dipeptidyl-peptidase IV and neutral endopeptidase (NEP), both of which are expressed on human podocytes and implicated in glomerular immune deposition in experimental models (CitationRonco et al 1989; CitationRonco et al 1994), as well as unknown podocyte targets. NEP, an approximately 90 Kd membrane-bound ectoenzyme, was recently identified as target antigen in a rare form of antenatal membranous nephropathy (CitationDebiec et al 2002). Affected infants have subepithelial immune deposits and proteinuria at birth. Their mothers are phenotypically normal but genetically deficient in NEP due to truncating mutations in the Membrane MetalloEndopeptidase (MME) gene (CitationDebiec et al 2004). During pregnancy, the fetus expresses normal NEP protein, the gene for which is inherited from the father. NEP-deficient mothers become alloimmunized to NEP in the placental syncytiotrophoblast and fetal tissues. Maternal anti-NEP IgG are transported across the placenta to the fetal circulation during the third trimester, cross the fetal GBM to bind NEP-expressing podocytes in developing glomeruli, and form immune complexes similar to those observed in experimental HN.
Therapeutic and diagnostic implications of NEP-induced MN
Two immunodominant linear peptide NEP epitopes recognized by serum of alloimmunized mothers have been identified to date (CitationDebiec and Ronco 2007). These epitopes may provide a basis for antigen-based therapies to prevent kidney injury in fetuses at risk. It is proposed that mothers of affected infants be monitored for circulating anti-NEP antibodies during subsequent pregnancies (CitationRonco and Debiec 2007). Rising titers can be managed either nonspecifically by plasma exchange, or in an antigen-specific manner using repetitive antigen adsorption. It is further proposed that undetected NEP-deficiency in transplant recipients with subsequent NEP alloimmunization may account for some cases of de novo MN in kidney allografts. Alloimmunization may similarly play a role in MN that develops after bone marrow or hematopoietic stem cell transplantation (CitationRonco and Debiec 2007). The degree to which maternal NEP-deficiency contributes to CKD in the general population is unknown, but likely to be low. However, observations from antenatal MN have rekindled hopes that nephritogenic podocyte antigens responsible for adult-onset MN will be identified, and ultimately provide novel targets for antigen–specific therapy.
IgG Fc gamma receptors (FcγR) and complement as therapeutic targets in GN
A large number of cellular and soluble mediators and modulators of immunity and inflammation and cell activation are implicated in immune activation, tissue destruction, remodeling and repair in GN (). Each of these is a potential target for therapeutic intervention (). The discussion below focuses on two modulators, IgG Fc gamma receptors (FcγR) and complement, that are particularly amenable to therapeutic manipulation and for which a variety of therapeutic modulators are already in preclinical or early clinical trials.
Notably, whereas the discussion below focuses primarily on the role of FcγR and complement as effectors of renal inflammation, both molecules also modulate activation of B cells and follicular dendritic cells and thus have potent effects on humoral autoimmunity. Both cell types express inhibitory FcγRIIB that engages IgG as well as complement receptors type 1 (CR1, or CD35) and type 2 (CR2, CD21) that recognize activated products of C3 and C4 (reviewed in CitationNimmerjahn and Ravetch 2006, CitationRoozendaal and Carroll, 2007). Thus, manipulation of these receptor-ligand interactions can alter initial lymphocyte tolerance and activation as well as modulate local renal effector mechanisms.
FcγR
IgG receptors link IgG or IgG immune complexes to effector cells by binding the CH2 domain of the Ig heavy chain constant, or Fc, region (CitationRavetch and Bolland 2001). FcγR are constitutively expressed on several cell types that contribute to pathogenesis in nephritis, including macrophages, dendritic cells, neutrophils, mast cells, endothelial cells and B cells, but not T lymphocytes (CitationNimmerjahn and Ravetch 2007). There is also evidence that FcγR may be cytokine-inducible on parenchymal cells, including human mesangial cells (CitationRadeke et al 1994). Many FcγR-bearing cells co-express two functional classes of FcγR: activating and inhibitory. Most activating receptors, FcγRI, FcγRIII, FcγRIIA and mouse FcγRIV, transmit signals via an adaptor protein, typically the common gamma chain coreceptor, that contains an ITAM (Immunoreceptor Tyrosine-based Activating Motif). FcγRIIIB is unique in being glycosylphosphatidyl inositol-anchored with selective expression on neutrophils and eosinophils. The one inhibitory receptor, FcγRIIB, is a single chain receptor that transmits signals through an ITIM, or inhibitory motif. The balance of signaling from the two types of receptors sets the threshold for cell activation after stimulation by IgG or IgG-immune complexes. B cells express only inhibitory FcγRIIB, which negatively regulates activating signals from the B cell receptor.
Individual FcγR and allelic variants have restricted IgG isotype specificity and different affinities for the IgG isotypes (CitationNimmerjahn and Ravetch 2005; CitationNimmerjahn et al 2005). IgG1 binds the inhibitory FcγRIIB and the activating FcγRIII with low affinity, thus favoring inhibition under basal conditions, whereas IgG2a binds three activating FcγR, FcγRI, FcγRIV, and FcγRIII, with varying affinities and binds inhibitory FcγRIIB with low affinity, thus promoting cell activation. Notably, FcγR expression and IgG isotype are regulated by cytokines, and thus both can vary considerably during the course of disease and during therapy. Transforming growth factor-beta, IL-4 and IL-10 downregulate activating FcγR and upregulate inhibitory FcγRIIB on most effector cells, and induce IgG1 and IgG3 isotypes. In contrast, interferon shifts the balance toward activating FcγR and production of IgG2a and IgG2b isotypes. Thus multiple factors determine the outcome of interactions between FcγR and IgG/immune complexes.
FcγR in GN and potential for therapeutic manipulation
In human GN there is evidence that FcγR susceptibility alleles predispose to disease. Perhaps most compelling, Aitman and colleagues demonstrated not only that copy number of the FcγRIIIB gene (FCGR3B) varies between individuals, ranging from none to four copies per cell, but that low FCGR3B copy number and particularly absence of FCGR3B is associated with susceptibility to lupus nephritis (CitationAitman et al 2006; CitationFanciulli et al 2007). Increased risk for lupus nephritis is also described with polymorphisms in FcγRIIB, FcγRIIA and FcγRIIIA in a subset of ethnic groups in some studies (CitationSchwarting 2006).
Multiple observations in rodent experimental nephritis confirm that engagement of activating FcγR promotes nephritis whereas signaling via inhibitory FcγRIIB ameliorates disease. In the nephrotoxic serum nephritis (NSN) model, susceptible animals are administered heterologous antiserum reactive with recipient GBM. Disease onset is accelerated by prior immunization with species-specific IgG. Because antigen (foreign Ig) is planted in the glomerulus in NSN, this model is used primarily to study downstream effector mechanisms that are recruited either by the deposited heterologous IgG or by the autologous anti-foreign-Ig immune response. In the Wistar Kyoto and related rat strains, susceptibility to NSN and macrophage hyperactivity is associated with a polymorphism in the activating FcγR, FcγRIIIB (CitationAitman et al 2006). In murine NSN, disruption of the gene encoding the common FcγR gamma chain (FcγR-KO) renders activating FcγR nonfunctional and markedly improves survival and prevents severe nephritis. Glomerular IgG and C3 deposits are unchanged in these mice, suggesting that protection involves interruption of events downstream of immune deposition (CitationPark et al 1998; CitationWakayama et al 2000). Reciprocal bone marrow transplantation studies in murine accelerated NSN further show that protection depends on loss of FcγR expression on bone marrow-derived cells, presumably leukocytes (CitationTarzi et al 2002). Genetic deficiency of activating FcγR also confers resistance to lupus nephritis in BWF1 mice, despite persistent serum autoantibodies and renal IgG and C3 deposits (CitationClynes et al 1998).
Mice administered monoclonal antibody to block activating receptor FcγRIV were also protected from NSN (CitationKaneko et al 2006a). FcγRIV-expressing macrophages are recruited to glomerular lesions in this model, suggesting that disease is modulated at the site of tissue injury. Protection was not seen with control antibody infusion, by genetic deletion of FcγRIII, or by combined deletion of the FcγRIII and FcγRI activating receptors. At the time of these studies a deletion model of FcγRIV was not yet developed. Administration of high dose intravenous immunoglobulin, or IVIG, the pooled purified IgG fraction, or its Fc portion, also ameliorated nephritis in this model, concurrent with downregulated expression of FcγRIV on kidney-infiltrating leukocytes (CitationKaneko et al 2006a). IVIG also upregulated inhibitory FcγRIIB; however, anti-FcγRIV antibody therapy was effective in NSN in FcγRIIB-deficient mice, confirming a key role for FcγRIV in pathogenesis (CitationKaneko et al 2006a).
It is notable that the dominant activating FcγR engaged in NSN is model specific. C57BL/6 mice immunized with sheep IgG develop a dominant IgG2b mouse anti-sheep response (CitationKaneko et al 2006a). Mouse IgG2b binds both FcγRIV and FcγRIII, but with different affinities. Induction of NSN in mutant C57BL/6 mice rendered genetically deficient in these activating receptors revealed that FcγRIV is the major if not sole activating FcγR involved in pathogenesis in this model (CitationKaneko et al 2006a). In contrast, NSN induced in C57BL/6 mice using rabbit nephrotoxic serum is FcγRIII dependent (CitationFujii et al 2003). Thus, features of the inciting antigen help determine the nature of the subsequent immune response.
In contrast to the proinflammatory role of activating FcγR, the inhibitory receptor FcγRIIB is protective in several models of experimental nephritis. Genetic deletion of FcγRIIB accelerates mortality in murine NSN (CitationKaneko et al 2006a), and leads to severe crescentic GN and massive pulmonary hemorrhage in mice immunized with collagen, an induced model of human spontaneous autoimmune GPS (CitationNakamura et al 2000). Deletion of FcγRIIB also leads to spontaneous autoimmunity and nephritis in a strain-dependent manner in susceptible normal backgrounds (CitationBolland and Ravetch 2000). FcγRIIB-deficient C57BL/6 mice develop proteinuria, immune complex deposition nephritis, activated lymphocytes, anti-chromatin autoantibodies, multiorgan inflammation, and early death. Conversely, deletion of FcγRIIB has no discernable phenotypic effect in the 129/B6 hybrid or BALB/c inbred backgrounds (CitationBolland and Ravetch 2000).
Protection afforded by FcγRIIB appears to include effects at the level of both effector cells and B cell tolerance. Amelioration of NSN by high dose IVIG is accompanied by upregulated expression of FcγRIIB on kidney-infiltrating leukocytes (CitationKaneko et al 2006a), suggesting a direct effect on glomerular inflammation. In contrast, development of autoantibodies and proteinuria in FcγRIIB-deficient C57BL/6 mice requires loss of FcγRIIB on B cells, not myeloid cells, as revealed by reciprocal bone marrow transfer experiments (CitationBolland and Ravetch 2000). This dual action is consistent with the roles of FcγRIIB in setting activation thresholds for both B cells and myeloid cells.
Conversely, enhancing expression of inhibitory FcγRIIB can restore tolerance and improve lupus nephritis. This approach was effective in three lupus strains, BXSB, NZM and B6.FcγRIIB-KO, subjected to isologous bone marrow transplantation (CitationMcGaha et al 2005). Donor bone marrow cells were transduced with vector carrying a DNA construct encoding FcγRIIB. B cell surface expression of FcγRIIB almost doubled in transplanted recipients. This was associated with significantly improved survival, reduced proteinuria, reduced autoantibody levels, reduced B cell activation, and improved nephritis and lung disease in each mouse strain compared to irradiated transplanted control mice that received mock-transduced bone marrow.
FcγRIIB may play a key pathogenic role in systemic lupus erythematosus, and lupus nephritis in particular. Reduced FcγRIIB expression on activated and germinal center B cells is reported in multiple strains of autoimmune mice, including NZB, NOD, BXSB, and MRL. Low FcγRIIB expression has been linked to polymorphisms in the gene promoter (CitationNimmerjahn and Ravetch 2006). In human lupus, reduced FcγRIIB expression on activated B cells and inadequate upregulation of FcγRIIB on memory B cells are reported (CitationMackay et al 2006). These defects in man have been linked to polymorphisms in the FcγRIIB transmembrane domain that impair the ability of FcγRIIB to enter lipid rafts (CitationFloto et al 2005; CitationKono et al 2005,).
Modulation of IgG glycosylation in glomerular inflammatory injury
IgG binding to FcγR requires Ig glycosylation (CitationKaneko et al 2006b). Over 30 different carbohydrate moieties covalently attach to the IgG Fc region via an N-linked glycan at asparagine 297. These glycans can have potent effects on IgG effector functions. The addition of the terminal sialic acid residue decreases IgG binding to selected FcγR by 5- to 10-fold. In an experimental model in which an anti-platelet IgG1 mediates cytotoxicity in vivo, enrichment for IgG sialic acid markedly reduces antibody efficacy and minimizes platelet depletion (CitationKaneko et al 2006b).
IgG glycosylation is also important for the therapeutic efficacy of IVIG. In an experimental arthritis model, enrichment for sialic acid enhances IVIG efficacy, such that a 10-fold lower IVIG dose yields the same marked clinical improvement as does full dose nonenriched IVIG (CitationKaneko et al 2006b). In this arthritis model, the mechanism appears to involve sialic acid-enriched IVIG upregulation of inhibitory FcγRIIB on macrophages. Conversely, enzymatic removal of all carbohydrate residues from IVIG eliminates most of its efficacy (CitationKaneko et al 2006b). These observations may explain the requirement for high doses of IVIG to achieve anti-inflammatory effects clinically; terminally sialylated IgG normally comprises only 5% of total serum IgG, and thus of IVIG.
FcγR and IgG-based therapeutic strategies in GN
FcγR offer attractive targets for therapeutic modulation in nephritis. Potential approaches include blockade of the various activating FcγR using neutralizing monoclonal antibodies or small molecules, and upregulation or activation of inhibitory receptors, either on effector cells in target tissues or on pathogenic autoreactive B cells. IVIG is currently used therapeutically in nephritis primarily as salvage therapy in refractory disease (CitationToubi et al 2005). Recent advances in our understanding of mechanisms of action of IVIG provide a blueprint for enhancing its efficacy by glyco-engineering antibody preparations and selectively enriching for desired isotypes. Gene therapy combined with isologous bone marrow transplantation, an approach used to enhance FcγRIIB expression in rodents, is feasible in man. Ultimately, a tailored approach in which FcγR-directed therapy takes into account the IgG isotype that dominates a given disease, the type of effector cells engaged, and the disease- or patient-specific FcγR profile has the potential to revolutionize therapy in GN.
Complement and GN
Glomerular complement deposition is common in immune complex mediated glomerular injury. Targeting complement for intervention, however, requires understanding of the complexity and diverse functions of the complement system (CitationGasque 2004). Over 30 proteins are engaged in the three major pathways of complement activation that converge on components C3, C5 and the terminal pathway that generates the C5b-9 membrane attack complex, or MAC: (1) The classical pathway initiated by immune complex recruitment of C1q, C4 and C2; (2) The alternative pathway in which activating surfaces engage C3b and factors B and D; and, (3) The lectin pathway in which activation depends on microbial carbohydrate interaction with mannose-binding ligand. Activation generates: the anaphylatoxins C3a and C5a that engage specific cell membrane receptors, C3aR and C5aR; C3b that associates with immune complexes; and, the C5b-9 membrane attack complex, or MAC, that inserts into cell membranes to induce sublytic or lytic injury. Complement activation is in turn regulated by a cadre of widely distributed soluble (factor H, C4bp) and cell-bound (decay accelerating factor or DAF, membrane cofactor protein or MCP, CR1, CD59, and rodent CR1-related gene/protein y or Crry) complement inhibitors. Local renal production of complement components, constitutive and inducible renal cell expression of complement receptors and regulatory proteins, and accessibility of locally activated complement proteins to circulating or infiltrating receptor-bearing leukocytes influences the nephritis phenotype.
Complement proteins mediate a variety of key functions that promote immunity and inflammation to protect the host from microorganisms. These same functions occasionally induce autoimmunity and organ damage. Complement activation leads to adjacent cell activation, sublytic injury or death, myeloid cell chemotaxis and activation, and enhanced T-dependent humoral responses. The complement system can also dampen inflammation and immunity, by solubilizing, clearing and processing antigen, immune complexes and apoptotic debris, by regulating B cell responses, and by helping to maintain B cell tolerance. Because of these divergent functions, the therapeutic potential of complement inhibitors in nephritis is complex.
This intricacy is reflected in the paradoxical roles of complement in lupus nephritis. A pathogenic role is supported by the presence of C1q and C3 in inflamed glomeruli and correlation of low serum C3 and C4 levels with disease activity. Yet deficiency, not excess, of complement components C1q, C2 or C4 predisposes to SLE. This paradox is due in part to the primarily protective role of the early classical complement pathway. Genetic deficiency of C1q or C4 worsens nephritis and autoimmunity in murine lupus, and leads to spontaneous onset of autoimmune GN in susceptible normal strains (CitationBotto et al 1998; CitationProdeus et al 1998; CitationChen et al 2000; CitationEinav et al 2002; CitationMitchell et al 2002; CitationPaul et al 2002).
Conversely, the alternative pathway may contribute to renal damage. Genetic deficiency of factor B or factor D improves lupus nephritis in the MRL/lpr strain (CitationWatanabe et al 2000; CitationElliott et al 2004), suggesting a predominantly pathogenic role for these components. The alternative pathway is also implicated in a subset of patients with membranoproliferative GN, or MPGN, types I and II. Genetic deficiency of the soluble complement regulatory protein, Factor H, leads to continuous complement activation and C3 turnover in vivo. Pigs and mice with this deficiency develop MPGN that is amenable to therapy with anti-C5 monoclonal antibody (CitationHogasen et al 1995; CitationJansen et al 1998; CitationPickering et al 2002, Citation2006). A similar pathogenesis is suspected in the subset of patients with MPGN who have deficient factor H activity due either to mutations in Factor H, to anti-factor H autoantibodies, or to C3 nephritic factor (C3NeF), an autoantibody that binds and stabilizes the alternative pathway C3bBb convertase (CitationMeri et al 1992; CitationDragon-Durey et al 2004; CitationZipfel et al 2006).
Results in murine MRL/lpr lupus using blockade of C3, a common central component of the complement pathways, are inconsistent. Genetic deficiency of C3 worsens nephritis, suggesting a primarily protective role, similar to that proposed for C1q and C4 (CitationSekine et al 2001). In contrast, administration of the rodent C3 inhibitor Crry (CR1-related gene/protein y), either as a Crry-Ig fusion protein or in the form of transgenic soluble Crry, improves nephritis, consistent with a pathogenic role for C3 (CitationBao et al 2002, Citation2003).
The contribution of complement to pathogenicity in murine anti-GBM disease is equally complex. Deficiency of either C3 or C4 protects against nephritis in the heterologous phase of NSN (CitationSheerin et al 1997). Conversely, in the autologous phase of disease or in accelerated NSN, C3 deficient mice develop worse nephritis (CitationSheerin et al 2001). Deficiency of complement regulators CD59, a terminal pathway inhibitor, CD55 (decay accelerating factor, or DAF), a C3 inhibitor, double deficiency of CD59 and CD55, or double deficiency of CD55 and Crry exacerbates nephritis or proteinuria in murine NSN (CitationTurnberg et al 2003; CitationLin et al 2004; CitationMiwa et al 2007).
These diverse outcomes reflect the diverse roles for complement at multiple checkpoints during the immune response, including modulation of B cell activation, inflammation, and immune complex clearance. Additionally, some of these discrepancies may be the result of gaps in our understanding of complement biology. In this regard, Huber-Lang et al recently proposed a fourth pathway for complement activation that bypasses C3 (CitationHuber-Lang et al 2006). This novel pathway depends on thrombin and tissue factor to independently generate C5 convertase to activate C5 and the terminal pathway. Lung injury induced by airway instillation of IgG immune complexes was equivalent in C3-sufficient and C3-deficient mice. Importantly, severe lung disease was ameliorated in both cases by administering antibody specific for C5a.
Renal injury resulting from the membrane attack complex, MAC or C5b-9, is best characterized in the noninflammatory passive Heymann nephritis model of membranous nephropathy induced in susceptible rat strains by injection of heterologous antiserum. Proteinuria is largely dependent on in situ complement activation triggered by podocyte-bound IgG, leading to insertion of the MAC into the podocyte membrane and subsequent cell activation, sublytic injury and release of soluble mediators (CitationCybulsky et al 1986, Citation2005). By analogy, it is proposed that proteinuria in patients with MN is predominantly MAC-dependent, a view supported by demonstration of MAC in patient urine and renal biopsies (CitationSchulze et al 1991). Interindividual and strain-dependent differences in mechanisms of proteinuria are likely, however, because proteinuria develops in active Heymann nephritis induced in the complement C6-deficient PVG/c rat strain (CitationLeenaerts et al 1995).
Complement therapeutics in GN
Although as yet no complement-directed therapy has proven efficacy in nephritis, a variety of complement inhibitors are under development or in clinical trials in human autoimmune and inflammatory diseases (CitationMorgan and Harris 2003; CitationTurnberg and Cook 2005; CitationRicklin and Lambris, 2007). Inhibitors being tested include recombinant soluble forms of naturally occurring inhibitors, soluble complement receptor 1 (CR1), small molecule inhibitors of C3, blocking antibodies or single chain Ig Fv fragments against complement components such as C5, inhibitors of serine proteases, and anaphylatoxin analogues to antagonize C3aR or C5aR. Realization that early components of the classical complement pathway have major protective roles in preventing autoimmunity has shifted focus in nephritis to inhibition of the alternative pathway or downstream complement components and local enhancement of complement inhibitors.
Anti-C5 antibody blocks generation of C5a fragment, C5b and the MAC. Anti-C5 Ig ameliorates lupus nephritis in the BWF1 strain and nephrotoxic serum nephritis in factor H-deficient mice (CitationWang et al 1996, CitationPickering et al 2006). Prevention of spontaneous MPGN type II by disruption of the C5 gene in factor H-deficient mice suggests that anti-C5 antibody therapy may also be effective in the subset of MPGN patients with aberrant factor H activity (CitationPickering et al 2006; CitationSmith et al 2007). The humanized monoclonal antibody to C5, eculizumab (Alexion), was approved by the FDA in early 2007 for use in patients with chronic hemolytic anemia due to paroxysmal nocturnal hemoglobinuria, an acquired genetic deficiency of complement inhibitors (CitationRicklin and Lambris, 2007). Eculizumab was well tolerated in patients with MN but a relatively short four month course failed to significantly reduce proteinuria (CitationAppel et al 2002). The efficacy of anti-C5 antibody therapy in other human glomerular diseases is yet to be established.
Systemically administered inhibitors that bind the C5aR are in preclinical studies. C5aR is expressed on monocytes, neutrophils and cultured human mesangial cells (CitationBraun and Davis, 1998) and increased in lupus nephritis kidneys (CitationAbe et al 2001; CitationBao et al 2005). Pharmacological C5aR blockade using a small molecule antagonist prevents progressive lupus nephritis in MRL/lpr mice (CitationBao et al 2005).
Alternative strategies that target the site of complement activation and tissue injury in vivo are in development. Targeting permits local concentration of inhibitor while avoiding systemic effects that may compromise host defense or important physiological functions of C3 or C5. Efficacy was demonstrated in experimental proteinuria-induced renal tubulointerstitial injury using rat Crry or CD59 linked to a single chain Fv that targeted a rat proximal tubular epithelial cell antigen (CitationHe et al 2005). A fusion protein engineered to link complement receptor 2 (CR2), a binding partner for C3 activation fragments, to a soluble form of the membrane complement inhibitor DAF successfully targeted the inhibitor to inflamed kidney in mouse models of lupus nephritis (CitationSong et al 2003). Ongoing investigation is dissecting the contributions of locally expressed complement components, receptors and regulatory proteins in experimental and human nephritis. It is anticipated that knowledge of the biology of complement in individual nephritides, throughout the course of disease and during concurrent immunosuppressive therapy, will ultimately permit appropriate therapeutic targeting.
Summary and conclusion
GN is a common cause of renal injury and CKD that continues to rely on non specific immunosuppression for therapeutic intervention. Recent insights into underlying biology and disease pathogenesis in human nephritis combined with advances in the fields of inflammation and autoimmunity bring the promise of new therapies. Notable breakthroughs include the discovery that alloimmunity to a glomerular antigen underlies antenatal membranous nephropathy, that podocytes are key regulators of glomerular filtration, that complement inhibitors are pivotal to pathogenesis in MPGN, and that IgG Fc receptors form a crucial link between antibody deposition and tissue injury. These discoveries provide the basis for novel therapies based on specific antigen or antibody responses or dominant effector cells or molecules engaged in individual nephritides. Future additional insights into cellular immunity, immune tolerance and regulatory networks, effector mechanisms, and genetic and environmental disease susceptibility will extend this armamentarium and provide the foundation for tailored individualized therapy.
Abbreviations
| GN | = | glomerulonephritis |
| ESRD | = | end stage renal disease |
| CKD | = | chronic kidney disease |
| GBM | = | glomerular basement membrane |
| NSN | = | nephrotoxic serum nephritis |
Acknowledgements
This work was supported by National Institutes of Health Grant DK47424 and a Department of Veterans Affairs Merit Award.
Disclosures
Dr. Foster has served as a paid consultant for La Jolla Pharmaceutical Company.
References
- AbeKMiyazakiMKojiT2001Enhanced expression of complement C5a receptor mRNA in human diseased kidney assessed by in situ hybridizationKidney Int601374611422745
- AbrahamsonDRPrettymanACRobertB2003Laminin-1 reexpression in Alport mouse glomerular basement membranesKidney Int638263412631063
- AitmanTJDongRVyseTJ2006Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humansNature439851516482158
- Alarcon-SegoviaDTumlinJAFurieRA2003LJP 394 for the prevention of renal flare in patients with systemic lupus erythematosus: results from a randomized, double-blind, placebo-controlled studyArthritis Rheum484425412571854
- AmitalHHeilweilMUlmanskyR2005Treatment with a laminin-derived peptide suppresses lupus nephritisJ Immunol17555162316210660
- AmitalHHeilweil-HarelMUlmanskyR2007Antibodies against the VRT101 laminin epitope correlate with human SLE disease activity and can be removed by extracorporeal immunoadsorptionRheumatology (Oxford)461433717686790
- AppelGNachmanPHoganS2002Eculizumab (C5 complement inhibitor) in the treatment of idiopathic membranous nephropathy (IMN): Preliminary baseline and pharmacokinetic (PK)/Pharmacodynamic (PD) dataJ Am Soc Nephrol13668A11856770
- BaoLHaasMBoackleSA2002Transgenic expression of a soluble complement inhibitor protects against renal disease and promotes survival in MRL/lpr miceJ Immunol1683601711907125
- BaoLHaasMKrausDM2003Administration of a soluble recombinant complement C3 inhibitor protects against renal disease in MRL/lpr miceJ Am Soc Nephrol14670912595503
- BaoLOsaweIPuriT2005C5a promotes development of experimental lupus nephritis which can be blocked with a specific receptor antagonistEur J Immunol35249650616052609
- BollandSRavetchJ2000Spontaneous autoimmune disease in Fcgamma. RIIB-deficient mice results from strain-specific epistasisImmunity132778510981970
- BorzaDBondarOToddP2002Quaternary organization of the goodpasture autoantigen, the alpha 3(IV) collagen chain. Sequestration of two cryptic autoepitopes by intrapromoter interactions with the alpha4 and alpha5 NC1 domainsJ Biol Chem277400758312193605
- BorzaDBBondarOColonS2005Goodpasture autoantibodies unmask cryptic epitopes by selectively dissociating autoantigen complexes lacking structural reinforcement: novel mechanisms for immune privilege and autoimmune pathogenesisJ Biol Chem280271475415917228
- BorzaDBHudsonBG2003Molecular characterization of the target antigens of anti-glomerular basement membrane antibody diseaseSpringer Semin Immunopathol243456112778332
- BorzaDBNetzerKOLeinonenA2000The goodpasture autoantigen. Identification of multiple cryptic epitopes on the NC1 domain of the alpha3 (IV) collagen chainJ Biol Chem2756030710681598
- BottoMDell’AgnolaCBygraveAE1998Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodiesNat Genet195699590289
- BoutaudAAKalluriRKahsaiTZ1996Goodpasture syndrome: selective removal of anti-alpha 3 (IV) collagen autoantibodies. A potential therapeutic alternative to plasmapheresisExp Nephrol4205128864724
- BraunMDavisAE3rd1998Cultured human glomerular mesangial cells express the C5a receptorKidney Int54154299844130
- BrigantiEMRussGRMcNeilJJ2002Risk of renal allograft loss from recurrent glomerulonephritisN Engl J Med347103912110738
- ChenZKoralovSBKelsoeG2000Complement C4 inhibits systemic autoimmunity through a mechanism independent of complement receptors CR1 and CR2J Exp Med19213395211067882
- ClynesRDumitruCRavetchJV1998Uncoupling of immune complex formation and kidney damage in autoimmune glomerulonephritisScience279105249461440
- CoreshJSelvinEStevensLA2007Prevalence of chronic kidney disease in the United StatesJAMA29820384717986697
- CouserWG1999GlomerulonephritisLancet35315091510232333
- CouserW2005Recurrent glomerulonephritis in the renal allograft: an update of selected areasExp Clin Transplant3283815989671
- CouserWSteinmullerDStilmantM1978Experimental glomerulonephritis in the isolated perfused rat kidneyJ Clin Invest62127587372233
- CouserWGNangakuM2006Cellular and molecular biology of membranous nephropathyJ Nephrol1969970517173240
- CybulskyAVQuiggRJSalantDJ2005Experimental membranous nephropathy reduxAm J Physiol Renal Physiol289F6607116159900
- CybulskyARennkeHFeintzeigI1986Complement-induced glomerular epithelial cell injury. Role of the membrane attack complex in rat membranous nephropathyJ. Clin Invest7710961073514672
- DavidMBorzaDLeinonenA2001Hydrophobic amino acid residues are critical for the immunodominant epitope of the Goodpasture autoantigen. A molecular basis for the cryptic nature of the epitopeJ Biol Chem2766370711098057
- DebiecHGuigonisVMougenotB2002Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodiesN Engl J Med34620536012087141
- DebiecHNautaJCouletF2004Role of truncating mutations in MME gene in fetomaternal alloimmunisation and antenatal glomerulopathiesLancet3641252915464186
- DebiecHRoncoP2007Fetomaternal alloimmunization with antenatal glomerulopathiesAnn N Y Acad Sci11105596617911472
- DirksJHde ZeeuwDAgarwalSK2005Prevention of chronic kidney and vascular disease: toward global health equity – the Bellagio 2004 DeclarationKidney Int SupplS16
- DonauerJWilpertJGeyerM2006ABO-incompatible kidney transplantation using antigen-specific immunoadsorption and rituximab: a single center experienceXenotransplantation131081016623802
- Dragon-DureyMAFremeaux-BacchiVLoiratC2004Heterozygous and homozygous factor H deficiencies associated with hemolytic uremic syndrome or membranoproliferative glomerulonephritis: report and genetic analysis of 16 casesJ Am Soc Nephrol157879514978182
- EinavSPozdnyakovaOOMaM2002Complement C4 is protective for lupus disease independent of C3J Immunol16810364111801636
- ElliottMKJarmiTRuizP2004Effects of complement factor D deficiency on the renal disease of MRL/lpr miceKidney Int651293814675043
- FanciulliMNorsworthyPJPetrettoE2007FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunityNat Genet39721317529978
- FarquharMGSaitoAKerjaschkiD1995The Heymann nephritis antigenic complex: megalin (gp330) and RAPJ Am Soc Nephrol635477579068
- FlotoRAClatworthyMRHeilbronnKR2005Loss of function of a lupus-associated FcgammaRIIb polymorphism through exclusion from lipid raftsNat Med111056816170323
- FosterMH2007T cells and B cells in lupus nephritisSemin Nephrol27475817336688
- FujiiTHamanoYUedaS2003Predominant role of FcgammaRIII in the induction of accelerated nephrotoxic glomerulonephritisKidney Int6414061612969160
- FurieR2006Abetimus sodium (riquent) for the prevention of nephritic flares in patients with systemic lupus erythematosusRheum Dis Clin North Am321495616504827
- GasqueP2004Complement: a unique innate immune sensor for danger signalsMol Immunol4110899815476920
- HahnBSinghRLa CavaA2005Tolerogenic treatment of lupus mice with consensus peptide induces Foxp3-expressing, apoptosis-resistant, TGFbeta-secreting CD8+ T cell suppressorsJ Immunol17577283716301683
- HahnBHSinghRRWongWK2001Treatment with a consensus peptide based on amino acid sequences in autoantibodies prevents T cell activation by autoantigens and delays disease onset in murine lupusArthritis Rheum444324111229475
- HeCImaiMSongH2005Complement inhibitors targeted to the proximal tubule prevent injury in experimental nephrotic syndrome and demonstrate a key role for C5b-9J Immunol1745750715843577
- HellmarkTBurkhardtHWieslanderJ1999Goodpasture disease. Characterization of a single conformational epitope as the target of pathogenic autoantibodiesJ Biol Chem27425862810464328
- HogasenKJansenJHMollnesTE1995Hereditary porcine membranoproliferative glomerulonephritis type II is caused by factor H deficiencyJ Clin Invest951054617883953
- Huber-LangMSarmaJVZetouneFS2006Generation of C5a in the absence of C3: a new complement activation pathwayNat Med12682716715088
- HudsonBGTryggvasonKSundaramoorthyM2003Alport’s syndrome, Goodpasture’s syndrome, and type IV collagenN Engl J Med34825435612815141
- JansenJHHogasenKHarboeM1998In situ complement activation in porcine membranoproliferative glomerulonephritis type IIKidney Int53331499461093
- JavaidBQuiggRJ2005Treatment of glomerulonephritis: will we ever have options other than steroids and cytotoxics?Kidney Int67169270315840015
- KalluriRCantleyLKerjaschkiD2000aReactive oxygen species expose cryptic epitopes associated with autoimmune Goodpasture syndromeJ Biol Chem275200273210748075
- KalluriRDanoffTMOkadaH1997Susceptibility to anti-glomerular basement membrane disease and Goodpasture Syndrome is linked to MHC class II genes and the emergence of T cell-mediated immunity in miceJ Clin Invest1002263759410904
- KalluriRTorreAShieldCF3rd2000bIdentification of alpha3, alpha4, and alpha5 chains of type IV collagen as alloantigens for Alport posttransplant anti-glomerular basement membrane antibodiesTransplantation696798310708133
- KanekoYNimmerjahnFMadaioMP2006aPathology and protection in nephrotoxic nephritis is determined by selective engagement of specific Fc receptorsJ Exp Med2037899716520389
- KanekoYNimmerjahnFRavetchJV2006bAnti-inflammatory activity of immunoglobulin G resulting from Fc sialylationScience313670316888140
- KangHKMichaelsMABernerBR2005Very low-dose tolerance with nucleosomal peptides controls lupus and induces potent regulatory T cell subsetsJ Immunol17432475515749855
- KerjaschkiDFarquharM1982The pathogenic antigen of Heymann nephritis is a membrane glycoprotein of the renal proximal tubule brush borderProc Natl Acad Sci USA795557616752952
- KerjaschkiDHorvatRBinderS1987Identification of a 400-kd protein in the brush borders of human kidney tubules that is similar to gp330, the nephritogenic antigen of rat Heymann nephritisAm J Pathol129183912444109
- KonoHKyogokuCSuzukiT2005FcgammaRIIB Ile232Thr transmembrane polymorphism associated with human systemic lupus erythematosus decreases affinity to lipid rafts and attenuates inhibitory effects on B cell receptor signalingHum Mol Genet1428819216115811
- La CavaAEblingFMHahnBH2004Ig-reactive CD4+CD25+ T cells from tolerized New Zealand Black x New Zealand White.F1 mice suppress in vitro production of antibodies to DNAJ Immunol1733542815322219
- La CavaAFangCJSinghRP2005Manipulation of immune regulation in systemic lupus erythematosusAutoimmun Rev4515916214088
- LeenaertsPLHallBMvan DammeBJ1995Active Heymann nephritis in complement component C6 deficient ratsKidney Int47160416147643529
- LinFSalantDJMeyersonH2004Respective roles of decay-accelerating factor and CD59 in circumventing glomerular injury in acute nephrotoxic serum nephritisJ Immunol17226364214764738
- MackayMStanevskyAWangT2006Selective dysregulation of the FcgammaIIB receptor on memory B cells in SLEJ Exp Med20321576416923849
- McCoyRCJohnsonHKStoneWJ1982Absence of nephritogenic GBM antigen(s) in some patients with hereditary nephritisKidney Int21642527047864
- McGahaTLSorrentinoBRavetchJV2005Restoration of tolerance in lupus by targeted inhibitory receptor expressionScience307590315681388
- MeriSKoistinenVMiettinenA1992Activation of the alternative pathway of complement by monoclonal lambda light chains in membranoproliferative glomerulonephritisJ Exp Med175939501532415
- MeyersKEKinniryPAKalluriR1998Human Goodpasture anti-alpha3 (IV)NC1 autoantibodies share structural determinantsKidney Int5340279461099
- MitchellDAPickeringMCWarrenJ2002C1q deficiency and autoimmunity: the effects of genetic background on disease expressionJ Immunol16825384311859149
- MiwaTZhouLTudoranR2007DAF/Crry double deficiency in mice exacerbates nephrotoxic serum-induced proteinuria despite markedly reduced systemic complement activityMol Immunol441394616887189
- MorganBPHarrisCL2003Complement therapeutics; history and current progressMol Immunol401597012914822
- NakamuraAYuasaTUjikeA2000Fcgamma receptor IIB-deficient mice develop Goodpasture’s syndrome upon immunization with type IV collagen: a novel murine model for autoimmune glomerular basement membrane diseaseJ Exp Med19189990610704470
- NetzerKOLeinonenABoutaudA1999The goodpasture auto-antigen. Mapping the major conformational epitope(s) of alpha3 (IV) collagen to residues 17–31 and 127–141 of the NC1 domainJ Biol Chem274112677410196215
- NimmerjahnFBruhnsPHoriuchiK2005FcgammaRIV: a novel FcR with distinct IgG subclass specificityImmunity23415116039578
- NimmerjahnFRavetchJV2005Divergent immunoglobulin g subclass activity through selective Fc receptor bindingScience3101510216322460
- NimmerjahnFRavetchJ2006Fcgamma receptors: old friends and new family membersImmunity24192816413920
- NimmerjahnFRavetchJV2007Fc-receptors as regulators of immunityAdv Immunol9617920417981207
- OlsonDLAnandSKLandingBH1980Diagnosis of hereditary nephritis by failure of glomeruli to bind anti-glomerular basement membrane antibodiesJ Pediatr9669797359277
- ParkSYUedaSHamanoY1998Resistance of Fc receptor-deficient mice to fatal glomerulonephritisJ Clin Invest541229389739057
- PaulEPozdnyakovaOOMitchellE2002Anti-DNA autoreactivity in C4-deficient miceEur J Immunol322672912207352
- PickeringMCCookHTWarrenJ2002Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor HNat Genet31424812091909
- PickeringMCWarrenJRoseKL2006Prevention of C5 activation ameliorates spontaneous and experimental glomerulonephritis in factor H-deficient miceProc Natl Acad Sci USA10396495416769899
- PonticelliC2007Membranous nephropathyJ Nephrol202688717557260
- ProdeusAPGoergSShenLM1998A critical role for complement in maintenance of self-toleranceImmunity9721319846493
- RadekeHHGessnerJEUciechowskiP1994Intrinsic human glomerular mesangial cells can express receptors for IgG complexes (hFc gamma RIII-A) and the associated Fc epsilon RI gamma-chainJ Immunol1531281928027555
- RavetchJVBollandS2001IgG Fc receptorsAnnu Rev Immunol192759011244038
- ReynoldsJProdromidiEIJuggapahJK2005Nasal administration of recombinant rat alpha3 (IV)NC1 prevents the development of experimental autoimmune glomerulonephritis in the WKY ratJ Am Soc Nephrol161350915814836
- ReynoldsJPuseyCD2001Oral administration of glomerular basement membrane prevents the development of experimental autoimmune glomerulonephritis in the WKY ratJ Am Soc Nephrol12617011134251
- RicklinDLambrisJD2007Complement-targeted therapeuticsNat Biotechnol2512657517989689
- RiemekastenGLangnickeLDEnghardP2004Intravenous injection of a D1 protein of the Smith proteins postpones murine lupus and induces type 1 regulatory T cellsJ Immunol17358354215494537
- RoncoPAllegriLBriantiE1989Antigenic targets in epimembranous glomerulonephritis. Experimental data and potential application in human pathologyAppl Pathol785982567175
- RoncoPMArdaillouNVerroustP1994Pathophysiology of the podocyte: a target and a major player in glomerulonephritisAdv Nephrol Necker Hosp23911318154367
- RoncoPDebiecH2007Target antigens and nephritogenic antibodies in membranous nephropathy: of rats and menSemin Immunopathol294455817899086
- RoozendaalRCarrollMC2007Complement receptors CD21 and CD35 in humoral immunityImmunol Rev21915716617850488
- RudolphEHCongdonKLSackeyFN2002Humoral autoimmunity to basement membrane antigens is regulated in C57BL/6 and MRL/MpJ mice transgenic for anti-laminin Ig receptorsJ Immunol16859435312023401
- SaitoAPietromonacoSLooAK1994Complete cloning and sequencing of rat gp330/“megalin”, a distinctive member of the low density lipoprotein receptor gene familyProc Natl Acad Sci, USA91972597937880
- SausJWieslanderJLangeveldJ1988Identification of the Good-pasture antigen as the alpha 3(IV) chain of collagen IVJ Biol Chem26313374803417661
- SchulzeMDonadioJVPruchnoCJ1991Elevated urinary excretion of the C5b-9 complex in membranous nephropathyKidney Int4053381787650
- SchwartingA2006Genetic predisposition – is lupus nephritis a question of copy numbers?Nephrol Dial Transplant212378916864599
- SekineHReillyCMMolanoID2001Complement component C3 is not required for full expression of immune complex glomerulonephritis in MRL/lpr miceJ Immunol16664445111342671
- SheerinNSSpringallTAbeK2001Protection and injury: the differing roles of complement in the development of glomerular injuryEur J Immunol3112556011298352
- SheerinNSSpringallTCarrollMC1997Protection against anti-glomerular basement membrane GBM-mediated nephritis in C3- and C4-deficient miceClin Exp Immunol11040399409643
- SmithRJAlexanderJBarlowPN2007New approaches to the treatment of dense deposit diseaseJ Am Soc Nephrol1824475617675665
- SongHHeCKnaakC2003Complement receptor 2-mediated targeting of complement inhibitors to sites of complement activationJ Clin Invest11118758512813023
- StollMLPriceKDSilvinCJ2007Immunization with peptides derived from the idiotypic region of lupus-associated autoantibodies delays the development of lupus nephritis in the SWR x NZB.F1 murine modelJ Autoimmun2930717459659
- SusaniMSchulzeMExnerM1994Antibodies to glycolipids activate complement and promote proteinuria in passive Heymann nephritisAm J Pathol144807198160779
- TarziRMDaviesKARobsonMG2002Nephrotoxic nephritis is mediated by Fcgamma receptors on circulating leukocytes and not intrinsic renal cellsKidney Int6220879612427132
- TimoshankoJRTippingPG2005Resident kidney cells and their involvement in glomerulonephritisCurr Drug Targets Inflamm Allergy43536216101545
- ToubiEKesselAShoenfeldY2005High-dose intravenous immunoglobulins: an option in the treatment of systemic lupus erythematosusHum Immunol6639540215866703
- TurnbergDBottoMWarrenJ2003CD59a deficiency exacerbates accelerated nephrotoxic nephritis in miceJ Am Soc Nephrol142271912937303
- TurnbergDCookHT2005Complement and glomerulonephritis: new insightsCurr Opin Nephrol Hypertens14223815821414
- US Renal Data System2007ESRD incidence and prevalenceIn USRDS 2007 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United StatesNational Institutes of Health, National Institute of Diabetes and Digestive and Kidney DiseasesBethesda, MD8198
- van DammeBFleurenGBakkerW1978Experimental glomerulonephritis in the rat induced by antibodies directed against tubular antigens. V. Fixed glomerular antigens in the pathogenesis of heterologous immune complex glomerulonephritisLab Invest3850210147961
- WakayamaHHasegawaYKawabeT2000Abolition of anti-glomerular basement membrane antibody-mediated glomerulonephritis in FcRgamma-deficient miceEur J Immunol3011829010760808
- WangXPFogoABColonS2005Distinct epitopes for anti-glomerular basement membrane alport alloantibodies and goodpasture autoantibodies within the noncollagenous domain of alpha3 (IV) collagen: a janus-faced antigenJ Am Soc Nephrol1635637116236801
- WangYHuQMadriJA1996Amelioration of lupus-like auto-immune disease in NZB/WF1 mice after treatment with a blocking monoclonal antibody specific for complement component C5Proc Natl Acad Sci USA93856388710910
- WatanabeHGarnierGCircoloA2000Modulation of renal disease in MRL/lpr mice genetically deficient in the alternative complement pathway factor BJ Immunol157869410623824
- WieslanderJBarrJFButkowskiRJ1984Goodpasture antigen of the glomerular basement membrane: localization to noncollagenous regions of type IV collagenProc Natl Acad Sci USA813838426328527
- WilsonCB1997Immune Models of Glomerular InjuryNeilsonEGCouserWGImmunologic Renal DiseasesPhiladelphiaLippincott-Raven
- WolfraimLA2006Treating autoimmune diseases through restoration of antigen-specific immune toleranceArch Immunol Ther Exp (Warsz)5411316642252
- WuHYWardFJStainesNA2002Histone peptide-induced nasal tolerance: suppression of murine lupusJ Immunol16911263412097422
- ZhangYSCSuDBHecox2006Anti-Goodpasture autoantibodies escape regulation in a novel Ig transgenic modelJ Am Soc Nephrol1786 abst
- ZipfelPFHeinenSJozsiM2006Complement and diseases: defective alternative pathway control results in kidney and eye diseasesMol Immunol439710616026839