Abstract
C-peptide has long been thought to be an inert byproduct of insulin production, but it has become apparent, and accepted, that C-peptide has important biological properties. C-peptide displays beneficial effects in many tissues affected by diabetic complications, such as increased peripheral blood flow and protection from renal damage. However, the mechanisms mediating these effects remain unclear. C-peptide interacts with cellular membranes at unidentified sites distinctive of the insulin family of receptors, and signals to multiple targets known to play a role in diabetes and diabetic complications, such as Na+/K+-ATPase and NOS. In general, the physiological and molecular effects of C-peptide resemble insulin, but C-peptide also possesses traits separate from those of insulin. These basic studies have been confirmed in human studies, suggesting that C-peptide may lend itself to clinical applications. However, the molecular and physiological properties of C-peptide are not completely elucidated, and large clinical studies have not begun. In order to further these goals, we critically summarize the current state of knowledge regarding C-peptide’s renal and vascular effects and the molecular signaling of C-peptide.
Introduction
Diabetes mellitus (shortened as diabetes) is a class of diseases where an individual cannot properly maintain plasma glucose levels. There are two main types of diabetes, type 1 and type 2. Type 1 diabetes is characterized by a lack of insulin due to loss of beta cells in the pancreatic islets of Langerhans, whereas type 2 diabetes is typified by a loss of response to insulin. Type 2 diabetes progresses through two stages. First there is a loss of target tissue insulin action (termed insulin resistance) that then results in increased insulin production (hyperinsulinemia). Eventually the second stage is reached when the islets of Langerhans cannot maintain such a high level of insulin production, and “pancreatic exhaustion” sets in causing insulin production to cease. Although the diseases have unique etiologies, several long-term complications are associated with both types of diabetes. Intensive treatment of hyperglycemia significantly prevents diabetic complications (CitationDCCT 1993), but glycemic control does not offer sufficient protection from the development of complications.
Besides hyperglycemia, other causal factors appear to contribute to the development of diabetic complications. One such factor is the proinsulin connecting peptide, C-peptide. C-peptide is a cleavage product of insulin synthesis created in the pancreas as part of insulin production, and is released into the circulation with insulin. The average physiological blood concentrations of C-peptide are in the low nM levels in healthy individuals (CitationSamnegard and Brundin 2001). When, as in type 1 diabetes and late type 2 diabetes, insulin synthesis is impaired, synthesis of C-peptide is similarly impaired. Exogenous C-peptide has been shown to improve the function of many tissues commonly affected by diabetes complications (CitationLindstrom et al 1996; CitationJohansson et al 2000; CitationChakrabarti et al 2004; Sima and Li 2006), a development supported by the fact that pancreatic and islet of Langerhans transplantation, restoring not only insulin secretion, but also secretion of C-peptide, will prevent and even reverse diabetic complications (CitationFiorina et al 2003a, Citation2003b, Citation2005a, Citation2005b; CitationLee et al 2006; CitationVenturini et al 2006). Hence, C-peptide deficiency has successively emerged as a possible mechanism for the development of the disproportionate burden of complications affecting insulinopenic diabetes patients. In this paper, some of the beneficial renal and vascular effects of C-peptide are reviewed, as well as the current knowledge regarding C-peptide-mediated molecular mechanisms.
Beneficial effects of C-peptide
Circulatory benefits
It has long been known that diabetes mellitus patients suffer an increased risk of developing, as well as accelerating atherosclerosis – the principal cause of heart attack, stroke and gangrene of the extremities (CitationBrownlee 2001). Microangiopathy, atherosclerosis, and abnormalities in small vessel function significantly contribute to the development of diabetes-induced morbidity (CitationJensen et al 1989). During the development of atherosclerosis, pathological proliferation and migration of vascular smooth muscle cells are necessary to form atherosclerotic plaques. C-peptide has been shown to prevent vascular dysfunction in diabetic rats (CitationIdo et al 1997), and to reduce proliferation of smooth muscle cells exposed to high glucose (CitationKobayashi et al 2005; CitationCifarelli et al 2008), a reduction that may suppress diabetes-induced atherosclerosis. It should be noted that under normoglycemic conditions C-peptide stimulates cultured smooth muscle cell proliferation (CitationWalcher et al 2006).
Effects on nitric oxide and vasodilation
More than thirty years ago, impaired circulation, relative tissue hypoxia, and an impaired maximal oxygen uptake were reported in insulinopenic diabetes (CitationDitzel and Standl 1975). Shortly after induction of diabetes in lambs, coronary vascular resistance is elevated (CitationLee and Downing 1979), and diabetic animals are prone to develop increased total peripheral resistance and hypertension (CitationBell et al 2006). Administration of C-peptide restores diabetes-impaired skeletal muscle perfusion (CitationJohansson et al 1992a), improves capillary skin red blood cell velocity (CitationForst et al 1998), and myocardial blood perfusion (CitationHansen et al 2002) as well as pancreatic islet arteriole diameter (CitationNordquist et al 2008b). In type 2 diabetes, C-peptide concentrations correlate with the presence of coronary artery disease and peripheral vascular disease (CitationSari and Balci 2005). The C-peptide-induced beneficial effects are, however, not restricted to the diabetic state. Surprisingly, in myocardial ischemia-reperfusion, C-peptide has been shown to exert cardioprotective effects through nitric oxide (NO) release (CitationYoung et al 2000).
Effects on erythrocyte deformability
When erythrocytes are deformed by the narrow space in the capillaries, they release adenosine triphosphate (ATP) (CitationSprague et al 1996). The ATP released from the erythrocyte stimulates NO synthesis and release from vascular endo-thelial cells, an event that will regulate vascular resistance and improve oxygenation (CitationSprague et al 2003). In diabetes, erythrocyte deformability is impaired (CitationBrown et al 2005) and erythrocyte aggregation increases. The impaired deformability results in increased blood viscosity and decreases capillary blood flow and oxygen availability in the tissue. C-peptide levels have been suggested to positively correlate with erythrocyte deformability (CitationDe La Tour et al 1998). Decreased membrane sodium, potassium adenosine triphosphatase (Na+/K+-ATPase) activity in erythrocytes is associated with diabetic complications (CitationRaccah et al 1996; CitationDe La Tour et al 1998), and there are indications that C-peptide concentrations correlate with erythrocyte Na+/K+-ATPase activity in type 1 as well as type 2 diabetic subjects (CitationDe La Tour et al 1998). C-peptide is known to ameliorate the impaired deformability of erythrocytes in blood drawn from insulinopenic diabetic patients, and ouabain-inhibition of this effect suggests that the C-peptide-induced effects on erythrocyte deformability are mediated through restoration of the diabetes-impaired Na+/K+-ATPase activity (CitationKunt et al 1999). Recently, it was shown by CitationMeyer and colleagues (2008) that in the presence of metal ions, C-peptide promotes the release of ATP from erythrocytes, and that this release is mediated via activation of the GLUT1 transporter.
Renoprotection
Early in the development of diabetic nephropathy, the glomerular filtration rate (GFR) increases. Diabetes-induced glomerular hyperfiltration has been proposed as an independent risk factor for the development of renal complications, and it is well known that C-peptide prevents hyperfiltration as well as renal damage in experimental diabetes. C-peptide reduces glomerular hyperfiltration in diabetic patients, and long-term substitution therapy improves renal function (CitationJohansson et al 1992b, Citation2000). In animal models of diabetes mellitus, exogenous C-peptide has been shown to reduce diabetes-induced glomerular hyperfiltration and decrease albuminuria (CitationSamnegard et al 2001, Citation2005; CitationHuang et al 2002; CitationRebsomen and Tsimaratos 2005; CitationMaezawa et al 2006). Structurally, renal glomerular hypertrophy and mesangial matrix expansion is prevented by C-peptide treatment, and it has been shown by CitationMaezawa and colleagues (2006) that podocyte collagen gene expression is normalized by C-peptide treatment in a mouse model of type 1 diabetes (CitationSamnegard et al 2001, Citation2005).
The rat C-peptide carboxy-terminal penta-fragment EVARQ has been shown to abolish diabetes-induced glo-merular hyperfiltration. C-peptide and the penta-fragment both reduce diabetic hyperfiltration (CitationNordquist et al 2007). Hence, it is possible that the five amino acid sequence of the carboxy-terminal of C-peptide is the sequence, or one of the sequences, mediating the normalizing effect on GFR in these diabetic rats. It is common that proteins have defined functional sites. A fragment of gonadotropin-releasing peptide displays increased activity compared to the intact peptide (CitationSandow and Konig 1979; CitationRamasharma et al 1988). Similar to C-peptide gastrin, cholecystokinin and osteogenic growth peptide, have active sites in their C-terminal penta-peptides (CitationPortelli and Renzi 1973; CitationYaijma et al 1977; CitationBab et al 1999; CitationHe et al 2004).
Afferent arteriolar constriction
The glomerular microcirculation is regulated to a great extent through alterations in the vascular resistance of the afferent and efferent arterioles. In insulinopenic diabetes, afferent diameter is increased (CitationIkenaga et al 2000). C-peptide constricts isolated renal afferent arterioles from diabetic mice, but not from normoglycemic animals (CitationNordquist et al 2008a). Constriction of the afferent arteriole lowers glomerular filtration pressure and thus decreases GFR. However, C-peptide does not appear to diminish renal blood flow (CitationHuang et al 2002, CitationSamnegard et al 2004). Taken together, these results suggest that C-peptide reduces diabetes-induced glomerular hyperfiltration at least partly via constriction of the afferent glomerular arteriole, but with a simultaneous dilation of the efferent arteriole that counterbalances the effect on renal blood flow. It should be noted that efferent dilation and increased afferent vascular tone have also been reported for insulin (CitationJuncos and Itos 1993).
Tubuloglomerular feedback
It has previously been postulated that diabetic hyperfiltration occurs due to alterations in tubuloglomerular feedback (TGF) (CitationVallon et al 1995; CitationThomson et al 2001). TGF is an intrarenal mechanism that stabilizes GFR, and thus the tubular Na+-load to match the tubular Na+ handling capacity. The anatomical prerequisite for TGF is the return of the tubule to its own glomerulus. These, together with the macula densa (MD) make up the juxtaglomerular apparatus. The MD consists of specialized epithelial cells localized where the returning tubule passes the glomerulus, and constitutes a sensor mechanism for Na+ by sensing Cl–, which relates to Na+ levels. Increased tubular flow rate will increase the tubular NaCl load, which is sensed by the MD, and results in a constriction of the afferent arteriole. However, the afferent constriction caused by C-peptide was demonstrated in isolated arterioles, where the arteriole is not set up with an intact tubulus and thus is independent of TGF. In addition, it was recently shown by Sallstrom’ et al that TGF does not mediate diabetes-induced hyperfiltration, since diabetes-induced glomerular hyperfiltration occurs in adenosine A1-receptor-deficient mice known to lack a functional TGF mechanism (CitationSallstrom et al 2007). If TGF is not the mediator of diabetic hyperfiltration, it is unlikely that C-peptide would exert its effect on filtration via a TGF-dependent mechanism. Therefore, a possible conclusion is that C-peptide exerts its effect directly on the glomerular afferent arterioles.
Many aspects remain to be investigated
Blood flow is differentially affected in the kidney vasculature versus peripheral vasculature in the diabetic state. Multiple studies show normalizing effects of C-peptide on diabetes-impaired blood perfusion and blood cell velocity (CitationForst et al 1998; CitationJohansson et al 2003). However, in these studies, C-peptide reverses diabetes-induced decreases in peripheral blood perfusion, whereas in renal vessels C-peptide reverses diabetes-induced increases in blood flow (CitationBank and Aynedjian 1993; CitationWang et al 1993; CitationKomers et al 1994). It remains to be determined whether these beneficial, but seemingly opposing effects of C-peptide reflect tissue-specific mechanisms.
Another aspect of C-peptide as a vasoactive substance is that the vasoconstrictive effect of C-peptide takes considerably longer time to develop compared to well known constrictors such as angiotensin II or norepinephrine (CitationNordquist et al 2008a). An explanation of this could be a sequential cascade of intracellular events leading to vasoconstriction. Although C-peptide clearly possesses a reducing effect on diabetic hyperfiltration, there is still no effector, no receptor, and no downstream signalling cascade reported for this phenomenon. So what are possible causes of the effect of C-peptide on hyperfiltration? In the next section we review likely candidates mediating C-peptide’s physiological properties.
C-peptide cellular signaling
A putative G protein-coupled receptor
The first report of a possible receptor was published in 1986 when rat C-peptide I was found to bind specifically to cultured rat islet tumor B-cells (CitationFlatt et al 1986). Early studies suggested that C-peptide mimics insulin action, but that its action on the renal Na+/K+-ATPase is completely abolished by pretreatment with pertussis toxin (PTX) (CitationOhtomo et al 1996). PTX is an inhibitor of Gαi/o family of heterotrimeric G proteins (CitationKatada and Ui 1982; CitationWest et al 1985), and has been shown to eliminate nearly all C-peptide signaling activities (CitationRigler et al 1999; CitationKitamura et al 2001; CitationZhong et al 2005; CitationAl-Rasheed et al 2006; CitationLindahl et al 2007), something that strongly suggests that the putative C-peptide receptor is a seven transmembrane G protein-coupled receptor (GPCR).
Identification of GPCRs is not an easy task, and to date the cell surface receptor(s) for C-peptide remain unknown. However, multiple studies have been conducted via manipulating the ligand–C-peptide (CitationIdo et al 1997; CitationOhtomo et al 1998; CitationRigler et al 1999). Initially, rat and human C-peptides, but not pig C-peptide, were shown to act like insulin and return elevated glucose-mediated blood flow to normal in two models (CitationIdo et al 1997). The structure of C-peptide was examined by circular dichroism revealing that human and rat C-peptide begin and end with alpha helical domain linked by a flexible glycine rich domain containing a distinctive proline kink in the middle of the sequence; sadly pig C-peptide was not examined (CitationIdo et al 1997). Additionally, Ido and colleagues indicated that the reverse sequence, all D amino acids, and the middle glycine rich section had similar effects as full length rat and human C-peptide. It should be noted that if there is a receptor involved, then the D amino acids should be inert. The unexpected data lead the authors to conclude that, contrary to previous studies (CitationOhtomo et al 1996), the effects are not receptor dependent and that C-peptide enters cells similar to some bacterial proteins (CitationIdo et al 1997). Shortly thereafter, activity of the glycine rich region was confirmed when examining renal Na+/K+-ATPase activity (CitationOhtomo 1998). The glycine rich region was 20% less effective than full length C-peptide, but interestingly, the last 4 amino acids were only 8% less active than full length C-peptide, and the terminal penta-peptide had slightly greater activity than the full length peptide. Moreover, randomly scrambled C-peptide and the D-amino acid penta-peptide had no effect (CitationOhtomo et al 1998). These data indicate that indeed sequence specificity and chirality, an essential component of ligand-receptor interactions, are essential for C-peptide activity.
Binding studies demonstrated that C-peptide specifically binds to human renal tubular cells, fibroblasts from the upper arm, and saphenous vein endothelial cells, but not umbilical cord endothelial cells (CitationRigler et al 1999). Rigler and colleagues found that the half maximal binding (B50) was 0.3 nM and Bmax = 0.9 nM. Importantly, C-peptide binding was not displaced by all D-amino acid C-peptide, insulin, proinsulin, or insulin like growth factor (IGF) I and II; however, the terminal penta-peptide was as efficient in displacement as full length C-peptide (CitationRigler et al 1999). These data support the theory that the last 5 amino acids mediate the binding of C-peptide to its receptor and that it has a unique receptor independent of the insulin family of receptors. Lastly, CitationRigler and colleagues (1999) confirmed earlier studies that the majority of the receptor is a Gαi/o coupled receptor via use of PTX. However, there appears to be a small pool of receptors that are PTX insensitive, suggesting that C-peptide either has two receptors or a single GPCR that couples to Gαi/o and another Gα subunit (CitationRigler et al 1999). More recently, multiple studies have confirmed that the secondary structure of C-peptide is essential for proper signaling (CitationKitamura et al 2002; CitationLi et al 2003; CitationWalcher et al 2006), and that the penta-peptide mimics C-peptides effects (CitationZhong et al 2005). Therefore, the earlier studies by CitationIdo and colleagues (1997) have not been repeated, and it appears that C-peptide does bind to yet unidentified receptors.
Recently, the coupling of the C-peptide receptor to Gαi has been proven examining C-peptide-mediated activation of Gαi (CitationAl-Rasheed et al 2006). C-peptide, but not scrambled C-peptide, caused the nonhydrolyzable GTP analogue 35S-GTPγS to bind to Gαi, indicating that C-peptide activates Gαi. Unfortunately no other G-protein was examined in this assay, so it is unknown if the PTX insensitive receptor pool is acting through a different G-protein or a different class of receptors. However, these data prove that stimulation of cells with C-peptide activates Gαi.
In contrast to the studies suggesting that C-peptide acts through a GPCR, studies using isolated L6 rat myoblast membranes demonstrated that insulin and C-peptide, but not scrambled C-peptide, increased membrane associated tyrosine kinase activity (CitationGrunberger et al 2001). Moreover, C-peptide enhances insulin receptor tyrosine kinase activity, although with very low level of activation. These data allowed for theories to be generated about C-peptide binding to, or merely activating, the insulin receptor. In response to the re-emergent theory that C-peptide binds to the insulin receptor, surface plasma resonance (SPR), a technique that detects protein-protein interactions, was used to show little to no binding of C-peptide to insulin or IGF-I receptors (CitationHenriksson et al 2006). Moreover, Henricksson and colleagues demonstrated that insulin, but not C-peptide, activates the glucokinase and insulin promoter. Therefore, although the receptors signal similarly in physiological conditions, the molecular mechanisms are not identical and C-peptide binds to a unique receptor.
In support of the GPCR theory, rhodamine-labeled C-peptide binds to Swiss-3T3 and HEK-293 cells and internalizes in a temperature and PTX dependent mechanism (CitationLindahl et al 2007). Additionally, pre-incubation of C-peptide with a C-peptide antibody or unlabeled C-peptide inhibits rhodamine-labeled C-peptide binding. In order to identify a C-peptide receptor, CitationLindahl and colleagues (2007) utilized Biacore sensor chips, SPR, and matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) to identify C-peptide binding partners from HEK-293 cell extracts. Intracellular proteins were found to bind to C-peptide, but although the rhodamine-labeled C-peptide was found to internalize, receptor mediated endocytosis does not expose the ligand to the cytosol. Thus, these data need to be examined further to exclude false positive associations, since the ligand may never be exposed to those proteins unless C-peptide is entering the cells in a nonclassical manner as proposed by CitationIdo and colleagues (1997).
In conclusion, it is clear that C-peptide binds to receptor(s) distinctive of the insulin family of receptors. Additionally, one of the receptors stimulates Gαi. Collectively, the data strongly suggest that the receptor is a GPCR, in fact this amount of evidence was sufficient for classifying receptors before the advent of cloning. However, since the receptor is still unknown this is not a definitive conclusion. To further complicate the potential identity of the C-peptide receptor, activators of G-protein signaling (AGS) family of proteins are known to activate Gαi independently of GPCRs (CitationBlumer et al 2005). Therefore the precise nature of the C-peptide receptor cannot be determined from the literature and requires further investigation.
Receptor-mediated signal transduction
As mentioned previously, the physiological properties mirror, for the most part, insulin-mediated effects. In fact studies conducted in L6 rat myoblasts indicate that C-peptide signals nearly identically to insulin, except in regard to phosphorylation of Akt (CitationGrunberger et al 2001). These data along with the physiological effects of C-peptide were strong contributing factors to the proposal that C-peptide signals through the insulin receptor. In this section we will review the general signaling of C-peptide. However, it must be noted that it has already become evident that there are cell-specific signaling mechanisms. For example, in rat medullary thick ascending limb (mTAL) cells C-peptide causes translocation of protein kinase C (PKC) α, but not PKCδ, ε, or ζ, to the plasma membrane (CitationTsimaratos et al 2003), and in human renal tubular cells C-peptide causes translocation of PKCδ and ε, but not PKCα, γ, ζ, and θ (CitationZhong et al 2005). Similar discrepancies have been shown for Akt (CitationGrunberger et al 2001; CitationZhong et al 2005) and the NOS isoforms (CitationKitamura et al 2003; CitationWallerath et al 2003; CitationLi et al 2004). Therefore, we will include the cell types when describing the signaling pathways, and synthesize a generic signaling paradigm for C-peptide.
Na+/K+-ATPase
Initial studies demonstrated that C-peptide activates the renal Na+/K+-ATPase through a PTX and FK-506-dependent mechanism (CitationOhtomo et al 1996). FK-506 is a specific inhibitor of calcineurin (also known as protein phosphatase 3 and formerly called protein phosphatase 2B), which has been shown to be involved in activation of Na+/K+-ATPase (CitationLea et al 1994). Importantly, calcineurin is activated by binding to calmodulin after increased intracellular Ca2+ levels (CitationKlee et al 1979) indicating that C-peptide must increase intracellular Ca2+ levels. This was confirmed in bovine aortic endothelial cells (CitationWallerath et al 2003). Additionally, extensive studies in human renal tubular cells indicate that the L-type Ca2+ channel blockers nifedipine and verapamil, as well as the phospholipase C (PLC) inhibitor U73122 inhibit C-peptide-mediated signaling, thus indicating that the Ca2+ entry is through L-type Ca2+ channels (CitationZhong et al 2005). The activation of PLC and Ca2+ influx provides a mechanism for C-peptide-mediated activation of classical PKC family members, such as PKCα. In rat mTAL cells C-peptide-mediated Na+/K+-ATPase activity and phosphorylation of the α-subunit of Na+/K+-ATPase is blocked by the general PKC inhibitor GF109203X (CitationTsimaratos et al 2003). Further studies in human renal tubular cells confirm the role of PTX and GF109203X, but also show that phosphorylation of the α-subunit of Na+/K+-ATPase is blocked by PD98059, a MEK inhibitor (CitationZhong et al 2004). Furthermore, the C-peptide-mediated phosphorylation of the α-subunit of Na+/K+-ATPase has been shown to be a threonine residue residing within an ERK consensus site. This suggests that ERK, not PKC, is phosphorylating the Na+/K+-ATPase (CitationZhong et al 2004) and that PKC is involved in activating ERK, as described in the next section.
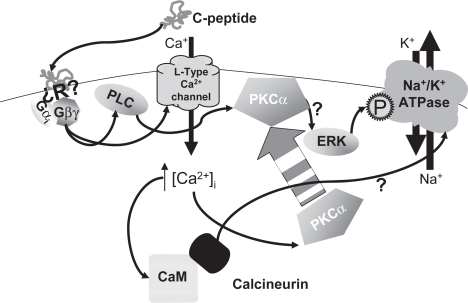
These data collectively suggest that C-peptide activates PLCβ2 through Gβγ subunits associated with Gαi (CitationKatz et al 1992; CitationWu et al 1993), and that C-peptide activates L-type Ca2+ channels through Gβγ subunits (CitationViard et al 1999). The activation of the L-type Ca2+ channel by Gβγ alone is rather minimal and requires phosphatidylinositol 3-kinase (PI3K), which is activated by C-peptide (CitationGrunberger et al 2001; CitationKitamura et al 2001; CitationLi et al 2003; CitationAl-Rasheed et al 2006; CitationWalcher et al 2006), to allow for a full Ca2+, transient (CitationViard et al 1999). Additionally there is a role for PKC in the Gβγ-mediated activation of L-type Ca2+ channels (CitationViard et al 1999). Since PKCδ and ε do not require Ca2+ for activation they may play a role in enhancing the Ca2+ transient since they translocate to the membrane after stimulation with C-peptide (CitationZhong et al 2005). However, the level of C-peptide-mediated activation of the L-type Ca2+ channel was not compared to a positive control, and no experiments examining the mechanism of C-peptide-mediated Ca2+ entry into cells has been conducted. Thus it is unknown if PKC and PI3K are needed for C-peptide-mediated activation of L-type Ca2+ channels and subsequent PKCα activation. As described in the next section, PI3K is most likely needed for activation of ERK, which in humans is a necessary component of C-peptide-mediated phosphorylation of the α-subunit of Na+/K+-ATPase (CitationZhong et al 2004). Further experiments are required to definitively describe the signaling cascade from the C-peptide receptor(s) to Na+/K+ ATPase; the current model is shown in .
Figure 1 Molecular mechanisms of C-peptide-mediated activation of Na+/K+ ATPase. This cartoon simplifies the signaling cascade resulting in activation of the Na+/K+ ATPase. Arrows represented signaling cascades. The activation of ERK is not completely understood. For simplicity it drawn with a single arrow from PKCα; however, it is known not to be a direct mechanism. Similarly, the precise role of calcineurin in regulation of the Na+/K+-ATPase is unknown, and is represented here by a simple arrow. The question mark (?) indicates simplified or unknown pathways.
MAPKs
Activation of mitogen-activated protein kinases (MAPKs) is common to many signaling molecules; there are three major MAPK proteins: ERK, p38, and JNK. However, the mechanism of receptor-mediated, especially GPCR-mediated, activation of MAPKs is not uniform and is still contested. C-peptide activates ERK, p38, and JNK, but as detailed below only ERK is consistently shown to be activated by C-peptide.
C-peptide signaling to ERK was first reported in 2001 (CitationGrunberger et al 2001; CitationKitamura et al 2001). In Swiss 3T3 fibroblasts CitationKitamura and colleagues (2001) demonstrated that C-peptide-mediated phosphorylation of ERK has an EC50 = 0.25 ± 0.05 nM, which is in accordance with the B50 = 0.3 nM (CitationRigler et al 1999), and ERK is phosphorylated within 10 min. Additionally, signaling to ERK is completely inhibited by PTX, nearly abolished by GF109203X, and inhibited by 50% by the PI3K inhibitor wortmannin. Thus, indicating that Gαi and PKC mediate C-peptide-dependent phosphorylation of ERK, and that PI3K also has a significant role. CitationGrunberger and colleagues (2001) demonstrated in L6 rat myoblasts a similar concentration response curve that is bell-shaped and showed no response at 10 nM and higher concentrations of C-peptide. This bell-shaped concentration-response curve is unusual; however, the bell-shaped curve is reported throughout the C-peptide literature. Not all studies with C-peptide result in a bell-shaped curve. For instance, C-peptide stimulation of mouse lung capillary endothelial cells results in a standard sigmoidal activation curve for ERK (CitationKitamura et al 2002). The potential physiological consequences of the bell-shaped curve in regards to diabetes treatment will be discussed in the conclusion.
The studies by CitationKitamura and colleagues (2002, Citation2003) indicate that C-peptide increased the phosphorylation of ERK and p38, but not JNK. These studies were reinforced via kinase assays, and thus are the first conclusive proof that C-peptide activates ERK and p38 (CitationKitamura et al 2002). C-peptide-mediated activation of ERK and p38 was confirmed in rat aortic endothelial cells (CitationKitamura et al 2003). In contrast, in human renal tubular cells C-peptide increases phosphorylation of ERK and JNK, but not p38 (CitationZhong et al 2005). These and other data suggest that ERK is the primary MAPK activated by C-peptide. Via use of specific inhibitors, Kitamura and colleagues demonstrated that ERK leads to phosphorylation of RSK1, but not MSK1. C-peptide-mediated phosphorylation and activation of RSK1 also occurs in L6 rat myoblasts (CitationGrunberger et al 2001). Importantly, RSK1 is downstream of ERK and PDK1, and is involved in multiple signaling cascades including inactivation of glycogen synthase kinase (CitationHauge and Frodin 2006). Thus C-peptide-mediated activation of ERK may be important in processing glucose.
The most complete C-peptide-mediated signaling to ERK has been worked out in vascular smooth muscle cells. C-peptide-mediated ERK and JNK phosphorylation absolutely requires PLC, L-type Ca2+ channels, PKC, and MEK activity (CitationWalcher et al 2006). Furthermore, C-peptide signaling to ERK is significantly reduced by treatment with the src family kinase inhibitor PP2 and the PI3K inhibitor LY294002 (CitationWalcher et al 2006). PP2 is a dirty drug (CitationBain et al 2007), however Walcher and colleagues also used siRNA demonstrating that c-src is involved in C-peptide-mediated phos-phorylation of ERK. Additionally, PP2 significantly, but not completely, blocks C-peptide mediated generation of PIP3, indicating that src is involved in the activation of PI3K. Activation of ERK in vascular smooth muscle cells leads to cellular proliferation and increased cyclin D1 expression (CitationWalcher et al 2006).
Collectively the data indicate that C-peptide is a potent stimulant of MAPKs, specifically ERK. Most of the studies have used insulin or PDGF as a positive control and found C-peptide to activate ERK to a much lesser extent; however, how C-peptide acts in comparison to ligands with similarly expressed receptors has not been examined. It is known that the C-peptide receptor(s) are less dense than the insulin receptor (CitationRigler et al 1999). Although there appears to be tissue specificity as to which MAPKs are activated, in all cases ERK is activated. ERK phosphorylation is a common occurrence for Gαi-coupled receptors (CitationL’Allemain et al 1991), and can occur through multiple pathways including through Gα (CitationHedin et al 1999) and through Gβγi (CitationCrespo et al 1994; CitationKoch et al 1994) activation of PI3K (CitationStoyanov et al 1995; CitationHawes et al 1996; CitationLopez-Ilasaca et al 1997). CitationHedin and colleagues (1999) demonstrate that the Gαi-dependent mechanism is independent of Ras and PI3K, but requires PKC, which may explain the 50% inhibition seen with wortmannin (CitationKitamura et al 2001) and allow for more traditional roles for PKC (CitationSchonwasser et al 1998) and c-src (CitationLuttrell et al 1996) as is eluded to by CitationWalcher and colleagues (2006). These accepted pathways partially unite the disparate C-peptide signaling pathways into one mechanism. However, the precise molecular mechanisms of how ERK is activated remain unknown.
In conclusion, the use of PTX, PI3K, and MEK inhibitors suggest that the mechanism involves the classical Ras-Raf/MEK/ERK cascade that is activated by Gβγ-mediated activation of PI3K. However, this does not account for how PKC and c-src are involved in activating this cascade. Thus, although C-peptide clearly activates ERK through a Gαi-mediated mechanism that involves PI3K, the multiple cell types used, lack of standardized experiments across cell types, and lack of a known receptor make it impossible to clearly unite all of the data into a single pathway. Our experience indicates that signaling to ERK is cell type-dependent (CitationEscano et al 2008). Additionally, the disparities between the phosphorylation of p38 and JNK support the idea that cell type dependent mechanisms are involved in the activation of the MAPKs. Thus, we are hesitant to combine all of the data into one pathway.
NO synthase
There are three isoforms of nitric oxide synthase (NOS): iNOS, nNOS, and eNOS (CitationMashimo and Goyal 1999). C-peptide has been shown to activate eNOS in rat and bovine aortic endothelial cells (CitationKitamura et al 2003; CitationWallerath et al 2003). Wallerath and colleagues demonstrated that C-peptide rapidly generates cGMP in a concentration-dependent manner. cGMP levels peak within 5 min and by 10 min the levels are similar to baseline levels, which can be attributed to phosphodiesterases degrading the cGMP. Furthermore, CitationWallerath and colleagues (2003) indicate that C-peptide-mediated increase in cGMP levels were abrogated by the NOS inhibitor L-NNA and by Ca2+ free media. This suggests that eNOS is involved in C-peptide-mediated NO production. C-peptide did not increase eNOS mRNA. In contrast, CitationKitamura and colleagues (2003) demonstrated that C-peptide transiently increases eNOS, but not iNOS, and that mRNA levels and eNOS protein levels are increased for up to 6 hours. eNOS expression was blocked by inhibiting gene transcription with actinomycin D and inhibition of MEK. Thus, CitationKitamura and colleagues (2003) indicate that the ERK MAPK pathway is important in C-peptide-mediated generation of NO. Additionally, CitationKitamura and colleagues (2003) observe NO production utilizing the NO sensitive dye DAF-2. The procedure followed, 3 hours of treatment followed by washing and then exposure to DAF-2 for 1 hour, cannot discriminate between the increased levels of eNOS generating increased basal NO and C-peptide-mediated release of NO. Given that CitationWallerath and colleagues (2003) show that C-peptide-mediated production of cGMP occurs within 10 min, the DAF-2 results from Kitamura and colleagues are most likely due to the enhanced eNOS protein levels. Supporting, in part, the study by CitationKitamura and colleagues (2003), CitationLi and colleagues (2004) indicate that 24 hour exposure of human smooth muscle cells to C-peptide increases iNOS and eNOS mRNA and protein levels. Though generally reported to be a stimulator of NOS, C-peptide counteracts diabetes-induced increases in renal eNOS levels (CitationKamikawa et al 2008). The seeming contradictions in the data may be due to altered mechanisms in the diabetic state, cell type, or methodology, which is not standard between the studies. In summary, C-peptide, like insulin, is involved in generation of NO through activation of eNOS, but the mechanism is largely unknown.
NF-κB activation and protection of diabetic nephropathy
There are a few reports indicating that C-peptide activates nuclear factor κB (NF-κB) (CitationAl-Rasheed et al 2006; CitationKitazawa et al 2006). In Swiss 3T3 cells inhibition of PKC inhibits NF-κB activation (CitationKitazawa et al 2006). In OK cells C-peptide protects against tumor necrosis factor α (TNF-α)-mediated apoptosis through an NF-κB mediated mechanism (CitationAl-Rasheed et al 2006). C-peptide-mediated NF-κB activity was ablated by PTX and wortmannin, but not PD98059. CitationAl-Rasheed and colleagues (2006) also demonstrate that C-peptide increases TRAF2, an NF-κB driven survival gene, and blocks high concentration TNF-α-mediated decreased expression of TRAF2. Together the data indicate that C-peptide may activate NF-κB through a PKC- and PI3K-dependent phosphorylation of IκB that then leads to increased translation of NF-κB driven genes that can inhibit TNF-α-mediated signaling. It should be noted that recent data suggest C-peptide rather reduces NF-κB activation (CitationCifarelli et al 2008).
Importantly, TNF-α has been suggested in the pathogenesis of diabetic nephropathy (CitationMoriwaki et al 2007). Additionally, TNF-α is increased in diabetic nephropathy (CitationKalantarinia et al 2003) and correlates with urinary protein excretion (CitationMoriwaki et al 2007). Injection of TNF-α decreases renal function (CitationSchmidt et al 2007), and CitationMoriwaki and colleagues (2007) demonstrated that inhibiting TNF-α reduces albuminuria in experimental diabetic rats. Therefore, C-peptide-mediated inhibition of TNF-α-mediated effects in renal cells may have clinical benefits.
Activation of RhoA and physiological consequences
C-peptide activates the small guanosine triphosphatase (GTPase) RhoA through an unknown mechanism in human renal tubular cells (CitationZhong et al 2005). Physiologically, Rho-kinase, an effector of RhoA, is known to be responsible for mediating “Ca2+-sensitation”, a condition where smooth muscle constriction is enhanced in similar or lower levels of Ca2+ influx (CitationKimura et al 1996). Rho-kinase is of particular interest because it plays an important role in mediating vaso-constriction in the kidney (CitationRoos et al 2006). Additionally, RhoA/Rho-kinase regulates blood flow, GFR, and alters the function and structure of renal tubular epithelial cells and mesangial cells (CitationWakino et al 2005).
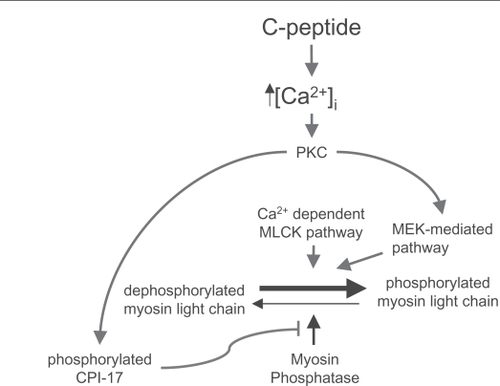
We have previously shown that the Rho-kinase inhibition with Y-27632 prevents the vasoconstrictive effects of C-peptide in isolated afferent arterioles, thus suggesting that C-peptide activates RhoA and consequently Rho-kinase in the renal vasculature (CitationNordquist et al 2008a). However, reduction of C-peptide-induced vasoconstriction by inhibition of Rho-kinase does not necessarily imply that C-peptide activates Rho-kinase (). C-peptide is known to increase PKC and MAPKs, both of which are capable of mediating contraction (via CPI-17 and caldesmon, respectively (CitationGerthoffer et al 1996; CitationWalsh et al 2007; CitationSakai et al 2007). Additionally, MAPK has been linked to phosphorylation of the regulatory subunit of the myosin light chain in the same location as myosin light chain kinase (CitationRoberts 2004). Moreover, Gαi-coupled receptors can activate integrin-linked kinase (ILK) through a PI3K-dependent pathway that results in phosphorylation of CPI-17 and the myosin light chain at the same site as myosin light chain kinase (CitationHuang et al 2006). Since inhibition of Rho-kinase allows for greater myosin phosphatase activity, which in turn will decrease the phosphorylation status of the regulatory subunit of the myosin light chain, it is possible that C-peptide induces phosphorylation of the myosin light chain through one of the aforementioned pathways, and that Y-27632 is only increasing the basal level of myosin phosphatase. Thus, more investigation is warranted into the link between C-peptide and RhoA/Rho-kinase activation in the renal vasculature.
Figure 2 Schematic illustrations of sequences of events leading to constriction. This schematic drawing illustrates possible downstream effects of C-peptide.
Concluding remarks
Thus far, the results from C-peptide studies display several unexplored research directions and opportunities for novel therapeutic interventions. It is clear that C-peptide possesses beneficial effects on diabetes-induced complications. This is supported by pancreas and islet of Langerhans transplantation in diabetic patients, restoring not only the patients’ insulin production, but also production of C-peptide. Thus, large scale clinical trials examining the benefits of C-peptide should be initiated in diabetic patients. In type 1 and stage 2 type 2 diabetes restoring circulating C-peptide is most likely going to be beneficial to the patient. However, the aforementioned bell-shaped concentration response curve may, paradoxically, help potentiate insulin resistance. In many cases mid to high nanomolar concentrations of C-peptide signalled less than low nanomolar concentrations, which could reduce the physiological effect of C-peptide and may explain why the increased levels of C-peptide in phase 1 type 2 diabetics is not beneficial. This apparent paradox must be more fully examined before C-peptide is administered to hyperinsulinemic type 2 diabetic patients. Additionally, if this paradox is physiologically relevant then the level of C-peptide administration would have to be carefully monitored, such as is done with insulin.
Finally, there are multiple basic and clinically relevant questions that remain unanswered. The identity of the receptor is clinically important, and the downstream signalling cascade, as currently known, does not fully explain amelioration of diabetes-induced complications. Further studies should be undertaken, investigating the intracellular effects of C-peptide. If the C-peptide receptor is a GPCR, then an agonist to the receptor can be generated. This is important since C-peptide may not be easily administered to patients, whereas a stable agonist may be able to be supplied as a once a day pill. Similarly, more fully understanding the cellular signaling of C-peptide may lead to novel interventions in diabetes associated diseases, such as nephropathy.
In conclusion, the field of C-peptide biology has accelerated greatly in the last 15 years. It is clear that C-peptide is biologically active, binds to cell surface receptors, and signals in a manner similar but unique from insulin. Physiologically, the effects are in some aspects similar to that of giving insulin to type 1 diabetic patients; however, C-peptide appears to restore insulin-treated type 1 diabetic patients to normal physiology. Given that C-peptide is a hormone co-secreted with insulin, and thus is absent in type 1 diabetes, the beneficial effects of C-peptide are not surprising.
Disclosure
The authors report no conflicts of interest.
References
- Al-RasheedNMWillarsGBBrunskillNJ2006C-peptide signals via Galpha i to protect against TNF-alpha-mediated apoptosis of opossum kidney proximal tubular cellsJ Am Soc Nephrol179869516510765
- BabIGavishHNamdar-AttarM1999Isolation of mitogenically active C-terminal truncated pentapeptide of osteogenic growth peptide from human plasma and culture medium of murine osteoblastic cellsJ Pept Res544081410563506
- BainJPlaterLElliottM2007The selectivity of protein kinase inhibitors: a further updateBiochem J40829731517850214
- BankNAynedjianHS1993Role of EDRF nitric oxide in diabetic renal hyperfiltrationKidney Int431306128315943
- BellTDDiBonaGFWangY2006Mechanisms for renal blood flow control early in diabetes as revealed by chronic flow measurement and transfer function analysisJ Am Soc Nephrol1721849216807404
- BlumerJBCismowskiMJSatoM2005AGS proteins: receptor-independent activators of G-protein signalingTrends Pharmacol Sci26470616084602
- BrownCDGhaliHSZhaoZ2005Association of reduced red blood cell deformability and diabetic nephropathyKidney Int6729530015610255
- BrownleeM2001Biochemistry and molecular cell biology of diabetic complicationsNature4148132011742414
- ChakrabartiSKhanZACukiernikM2004C-peptide and retinal microangiopathy in diabetesExp Diabesity Res591615198374
- CifarelliVLuppiPTseHM2008Human proinsulin C-peptide reduces high glucose-induced proliferation and NF-KB activation in vascular smooth muscle cellsAtherosclerosisonline first.
- CrespoPXuNSimondsWF1994Ras-dependent activation of MAP kinase pathway mediated by G-protein beta gamma subunitsNature369418208196770
- [DCCT] The Diabetes Control and Complications Trial Research Group1993The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research GroupN Engl J Med329977868366922
- De La TourDDRaccahDJannotMF1998Erythrocyte Na/K ATPase activity and diabetes: relationship with C-peptide levelDiabetologia41108049754827
- DitzelJStandlE1975The problem of tissue oxygenation in diabetes mellitusActa Med Scand Suppl5785968239528
- EscanoCSKeeverLBGutweilerAA2008Angiotensin II activates extracellular signal-regulated kinase independently of receptor tyrosine kinases in renal smooth muscle cells: implications for blood pressure regulationJ Pharmacol Exp Ther324344217911376
- FiorinaPFolliFZerbiniG2003aIslet transplantation is associated with improvement of renal function among uremic patients with type I diabetes mellitus and kidney transplantsJ Am Soc Nephrol142150812874470
- FiorinaPFolliFMaffiP2003bIslet transplantation improves vascular diabetic complications in patients with diabetes who underwent kidney transplantation: a comparison between kidney-pancreas and kidney-alone transplantation1Transplantation75129630112717219
- FiorinaPGremizziCMaffP2005aIslet transplantation is associated with an improvement of cardiovascular function in type 1 diabetic kidney transplant patientsDiabetes Care2813586515920052
- FiorinaPVenturiniMFolliF2005bNatural history of kidney graft survival, hypertrophy, and vascular function in end-stage renal disease type 1 diabetic kidney-transplanted patients: beneficial impact of pancreas and successful islet cotransplantationDiabetes Care2813031015920043
- FlattPRSwanston-FlattSKHamptonSM1986Specific binding of the C-peptide of proinsulin to cultured B-cells from a transplantable rat islet cell tumorBiosci Rep619393013335
- ForstTKuntTPohlmannT1998Biological activity of C-peptide on the skin microcirculation in patients with insulin-dependent diabetes mellitusJ Clin Invest1012036419593759
- GerthofferWTYambolievIAShearerM1996Activation of MAP kinases and phosphorylation of caldesmon in canine colonic smooth muscleJ Physiol495Pt 35976098887769
- GrunbergerGQiangXLiZ2001Molecular basis for the insulino-mimetic effects of C-peptideDiabetologia4412475711692173
- HansenAJohanssonBLWahrenJ2002C-peptide exerts beneficial effects on myocardial blood flow and function in patients with type 1 diabetesDiabetes5130778212351450
- HaugeCFrodinM2006RSK and MSK in MAP kinase signallingJ Cell Sci119Pt 153021316868029
- HawesBELuttrellLMvan BiesenT1996Phosphatidylinositol 3-kinase is an early intermediate in the G beta gamma-mediated mitogen-activated protein kinase signaling pathwayJ Biol Chem2711213368647803
- HeHShehanBPBarnhamKJ2004Biological activity and ferric ion binding of fragments of glycine-extended gastrinBiochemistry43118536115362871
- HedinKEBellMPHuntoonCJ1999Gi proteins use a novel beta gamma- and Ras-independent pathway to activate extracellular signal-regulated kinase and mobilize AP-1 transcription factors in Jurkat T lymphocytesJ Biol Chem274199922000110391949
- HenrikssonMJohanssonJMoedeT2006Proinsulin C-peptide and insulin: Limited pattern similarities of interest in inter-peptide interactions but no C-peptide effect on insulin and IGF-1 receptor signalingCell Mol Life Sci6330556017115117
- HuangDYRichterKBreidenbachA2002Human C-peptide acutely lowers glomerular hyperfiltration and proteinuria in diabetic rats: a dose-response studyNaunyn Schmiedebergs Arch Pharmacol365677311862335
- HuangJMahavadiSSriwaiW2006Gi-coupled receptors mediate phosphorylation of CPI-17 and MLC20 via preferential activation of the PI3K/ILK pathwayBiochem J39619320016472257
- IdoYVindigniAChangK1997Prevention of vascular and neural dysfunction in diabetic rats by C-peptideScience27756369228006
- IkenagaHBastJPFalletRW2000Exaggerated impact of ATP-sensitive K+ channels on afferent arteriolar diameter in diabetes mellitusJ Am Soc Nephrol11119920710864575
- JensenTBjerre-KnudsenJFeldt-RasmussenB1989Features of endothelial dysfunction in early diabetic nephropathyLancet1863646132563840
- JohanssonBLLindeBWahrenJ1992aEffects of C-peptide on blood flow, capillary diffusion capacity and glucose utilization in the exercising forearm of type 1 insulin-dependent diabetic patientsDiabetologia351211518
- JohanssonBLSjobergSWahrenJ1992bThe influence of human C-peptide on renal function and glucose utilization in type 1 insulin-dependent diabetic patientsDiabetologia3521218
- JohanssonBLBorgKFernqvist-ForbesE2000Beneficial effects of C-peptide on incipient nephropathy and neuropathy in patients with Type 1 diabetes mellitusDiabet Med173181910784221
- JohanssonBLWahrenJPernowJ2003C-peptide increases forearm blood flow in patients with type 1 diabetes via a nitric oxide-dependent mechanismAm J Physiol Endocrinol Metab2854E8647012799312
- JuncosLAItoS1993Disparate effects of insulin on isolated rabbit afferent and efferent arteriolesJ Clin Invest924198158408651
- KalantariniaKAwadASSiragyHM2003Urinary and renal interstitial concentrations of TNF-alpha increase prior to the rise in albuminuria in diabetic ratsKidney Int64412081312969138
- KamikawaAIshiiTShimadaK2008Proinsulin C-peptide abrogates type-1 diabetes-induced increase of renal endothelial nitric oxide synthase in ratsDiabetes Metab Res Rev24331818088079
- KatadaTMUi1982ADP ribosylation of the specific membrane protein of C6 cells by islet-activating protein associated with modification of adenylate cyclase activityJ Biol Chem257127210167200979
- KatzAWuDSimonMI1992Subunits beta gamma of heterotrimeric G protein activate beta 2 isoform of phospholipase CNature360640568691465134
- KimuraKMItoMAmano1996Regulation of myosin phosphatase by Rho and Rho-associated kinase Rho-kinaseScience273527224588662509
- KitamuraTKimuraKJungBD2001Proinsulin C-peptide rapidly stimulates mitogen-activated protein kinases in Swiss 3T3 fibroblasts: requirement of protein kinase C, phosphoinositide 3-kinase and pertussis toxin-sensitive G-proteinBiochem J355Pt 1123911256956
- KitamuraTKimuraKJungBD2002Proinsulin C-peptide activates cAMP response element-binding proteins through the p38 mitogen-activated protein kinase pathway in mouse lung capillary endothelial cellsBiochem J366Pt 37374412059784
- KitamuraTKimuraKMakondoK2003Proinsulin C-peptide increases nitric oxide production by enhancing mitogen-activated protein-kinase-dependent transcription of endothelial nitric oxide synthase in aortic endothelial cells of Wistar ratsDiabetologia4612169870514586499
- KitazawaMShibataYHashimotoS2006Proinsulin C-peptide stimulates a PKC/IkappaB/NF-kappaB signaling pathway to activate COX-2 gene transcription in Swiss 3T3 fibroblastsJ Biochem13961083816788059
- KleeCBCrouchTHKrinksMH1979Calcineurin: a calcium- and calmodulin-binding protein of the nervous systemProc Natl Acad Sci USA761262703293720
- KobayashiYNaruseKHamadaY2005Human proinsulin C-peptide prevents proliferation of rat aortic smooth muscle cells cultured in high-glucose conditionsDiabetologia4811239640116195866
- KochWJHawesBEAllenLF1994Direct evidence that Gi-coupled receptor stimulation of mitogen-activated protein kinase is mediated by G beta gamma activation of p21rasProc Natl Acad Sci USA912612706107809106
- KomersRAllenTJCooperME1994Role of endothelium-derived nitric oxide in the pathogenesis of the renal hemodynamic changes of experimental diabetesDiabetes4310119077926287
- KuntTSchneiderSPfutznerA1999The effect of human proinsulin C-peptide on erythrocyte deformability in patients with Type I diabetes mellitusDiabetologia4244657110230651
- L’AllemainGPouyssegurJWeberMJ1991p42/mitogen-activated protein kinase as a converging target for different growth factor signaling pathways: use of pertussis toxin as a discrimination factorCell Regul2867584
- LeaJPSandsJMMcMahonSJ1994Evidence that the inhibition of Na+/K+-ATPase activity by FK506 involves calcineurinKidney Int463647527527873
- LeeJCDowningSE1979Coronary dynamics and myocardial metabolism in the diabetic newborn lambAm J Physiol2372H11824464102
- LeeTCBarshesNRAgeeEE2006The effect of whole organ pancreas transplantation and PIT on diabetic complicationsCurr Diab Rep64323716879786
- LiHXuLDunbarJC2004Effects of C-peptide on expression of eNOS and iNOS in human cavernosal smooth muscle cellsUrology643622715351621
- LiZGZhangLSimaAA2003C-peptide enhances insulin-mediated cell growth and protection against high glucose-induced apoptosis in SH-SY5Y cellsDiabetes Metab Res Rev1953758512951645
- LindahlENymanUMellesE2007Cellular internalization of proinsulin C-peptideCell Mol Life Sci6444798617279313
- LindstromKJohanssonCJohnssonE1996Acute effects of C-peptide on the microvasculature of isolated perfused skeletal muscles and kidneys in ratActa Physiol Scand156119258866882
- Lopez-IlasacaMCrespoPPelliciPG1997Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI 3-kinase gammaScience275529839478994038
- LuttrellLMHawesBEvan BiesenBE1996Role of c-Src tyrosine kinase in G protein-coupled receptor- and Gbetagamma subunit-mediated activation of mitogen-activated protein kinasesJ Biol Chem2713219443508702633
- MaezawaYYokoteKSonezakiK2006Influence of C-peptide on early glomerular changes in diabetic miceDiabetes Metab Res Rev2243132216389646
- MashimoHGoyalRK1999Lessons from genetically engineered animal models. IV. Nitric oxide synthase gene knockout miceAm J Physiol2774Pt 1G7455010516139
- MeyerJAFroelichJMReidGE2008Metal-activated C-peptide facilitates glucose clearance and the release of a nitric oxide stimulus via the GLUT1 transporterDiabetologia511758217965850
- MoriwakiYInokuchiTYamamotoA2007Effect of TNF-alpha inhibition on urinary albumin excretion in experimental diabetic ratsActa Diabetol4442151817767370
- NordquistLMoeESjoquistM2007The C-peptide fragment EVARQ reduces glomerular hyperfiltration in streptozotocin-induced diabetic ratsDiabetes Metab Res Rev2340040517103462
- NordquistLLaiEYSjoquistM2008aProinsulin C-peptide constricts glomerular afferent arterioles in diabetic mice. A potential renoprotective mechanismAm J Physiol Regul Integr Comp Physiol294R8364118077505
- NordquistLLaiEYSjoquistM2008bC-peptide constricts pancreatic islet arterioles in diabetic, but not normoglycaemic miceDiabetes Metab Res Rev24165818157793
- OhtomoYAperiaASahlgrenB1996C-peptide stimulates rat renal tubular Na+, K+-ATPase activity in synergism with neuropeptide YDiabetologia3921992058635672
- OhtomoYBergmanTJohanssonBL1998Differential effects of proinsulin C-peptide fragments on Na+, K+-ATPase activity of renal tubule segmentsDiabetologia413287919541168
- PortelliMRenziG1973Pentapeptide derivatives of the C-terminal tetrapeptide of gastrinFarmaco Sci284316224698887
- RaccahDFabreguettsCAzulayJP1996Erythrocyte Na+-K+-ATPase activity, metabolic control, and neuropathy in IDDM patientsDiabetes Care19656488725852
- RamasharmaKYamashiroDLiCH1988Human follicular gonadotropin releasing peptide analogs. Evaluation of biological in vitro and immunological activityInt J Pept Protein Res326419243149951
- RebsomenLTsimaratosM2005Association of reduced red blood cell deformability and diabetic nephropathyKidney Int6752066author reply 2066–715840062
- RiglerRPramanikAJonassonP1999Specific binding of proinsulin C-peptide to human cell membranesProc Natl Acad Sci USA9623133182310557318
- RobertsRE2004The role of Rho kinase and extracellular regulated kinase-mitogen-activated protein kinase in alpha2-adrenoceptor-mediated vasoconstriction in the porcine palmar lateral veinJ Pharmacol Exp Ther311742715231868
- RoosMHvan RodijnenWFvan LambalgenAA2006Renal micro-vascular constriction to membrane depolarization and other stimuli: pivotal role for rho-kinasePflugers Arch452471716523358
- SakaiHChibaYMisawaM2007Role of Rho kinase in endothelin-1-induced phosphorylation of CPI-17 in rat bronchial smooth musclePulm Pharmacol Ther20734917071121
- SallstromJCarlssonPOFredholmBB2007Diabetes-induced hyperfiltration in adenosine A1-receptor deficient mice lacking the tubu-loglomerular feedback mechanismActa Physiol Oxf190253917581137
- SamnegardBBrundinT2001Renal extraction of insulin and C-peptide in man before and after a glucose mealClin Physiol211647111318824
- SamnegardBJacobsonSHJaremkoG2001Effects of C-peptide on glomerular and renal size and renal function in diabetic ratsKidney Int6012586511576340
- SamnegardBJacobsonSHJohanssonBL2004C-peptide and captopril are equally effective in lowering glomerular hyperfiltration in diabetic ratsNephrol Dial Transplant1913859115004258
- SamnegardBJacobsonSHJaremkoG2005C-peptide prevents glomerular hypertrophy and mesangial matrix expansion in diabetic ratsNephrol Dial Transplant20532815665028
- SandowJKonigW1979Studies with fragments of a highly active analogue of luteinizing hormone releasing hormoneJ Endocrinol8117582379261
- SariRBalciMK2005Relationship between C peptide and chronic complications in type-2 diabetes mellitusJ Natl Med Assoc9711131816173326
- SchmidtCHocherlKSchwedaF2007Regulation of renal glucose transporters during severe inflammationAm J Physiol Renal Physiol292F8041117032938
- SchonwasserDCMaraisRMMarshallCJ1998Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypesMol Cell Biol1879089447975
- SimaAALiZG2005The effect of C-peptide on cognitive dysfunction and hippocampal apoptosis in type 1 diabetic ratsDiabetes54149750515855338
- SpragueRSEllsworthMLStephensonLH1996ATP: the red blood cell link to NO and local control of the pulmonary circulationAm J Physiol2716Pt 2H2717228997335
- SpragueRSOlearczykJJSpenceDM2003Extracellular ATP signaling in the rabbit lung: erythrocytes as determinants of vascular resistanceAm J Physiol Heart Circ Physiol285H69370012689860
- StoyanovBVoliniaSHanchT1995Cloning and characterization of a G protein-activated human phosphoinositide-3 kinaseScience26969037624799
- SutherlandCLeightonIACohenP1993Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signallingBiochem J296Pt 115198250835
- ThomsonSCDengABaoD2001Ornithine decarboxylase, kidney size, and the tubular hypothesis of glomerular hyperfiltration in experimental diabetesJ Clin Invest1072172411160138
- TsimaratosMRogerFChabardesD2003C-peptide stimulates Na+,K+-ATPase activity via PKC alpha in rat medullary thick ascending limbDiabetologia461243112637991
- WakinoSKandaTHayashiK2005Rho/Rho kinase as a potential target for the treatment of renal diseaseDrug News Perspect186394316491166
- WalcherDBabiakCPoletekP2006C-Peptide induces vascular smooth muscle cell proliferation: involvement of SRC-kinase, phos-phatidylinositol 3-kinase, and extracellular signal-regulated kinase 1/2Circ Res991181717068290
- WallerathTKuntTForstT2003Stimulation of endothelial nitric oxide synthase by proinsulin C-peptideNitric Oxide99510214623175
- VallonVBlantzRCThomsonS1995Homeostatic efficiency of tubulo-glomerular feedback is reduced in established diabetes mellitus in ratsAm J Physiol2696Pt 2F876838594883
- WalshMPSusnjarMDengJ2007Phosphorylation of the protein phosphatase type 1 inhibitor protein CPI-17 by protein kinase CMethods Mol Biol3652092317200564
- WangYXBrooksDPEdwardsRM1993Attenuated glomerular cGMP production and renal vasodilation in streptozotocin-induced diabetic ratsAm J Physiol2645Pt 2R95268388664
- VenturiniMFiorinaPMaffiP2006Early increase of retinal arterial and venous blood flow velocities at color doppler imaging in brittle type 1 diabetes after islet transplant aloneTransplantation811274716699454
- WestREJrMossJVaughanM1985Pertussis toxin-catalyzed ADP-ribosylation of transducin. Cysteine 347 is the ADP-ribose acceptor siteJ Biol Chem26014428303863818
- ViardPExnerTMaierU1999Gbetagamma dimers stimulate vascular L-type Ca2+ channels via phosphoinositide 3-kinaseFaseb J136859410094929
- WuDKatzASimonMI1993Activation of phospholipase C beta 2 by the alpha and beta gamma subunits of trimeric GTP-binding proteinProc Natl Acad Sci U S A9052973018389480
- YajimaHKaiYOgawaH1977Structure-activity relationships of gastrointestinal hormones: motilin, GIP, and [27-TYR]CCK-PZGastroenterology724Pt 27936190081
- YoungLHIkedaYScaliaR2000C-peptide exerts cardioprotective effects in myocardial ischemia-reperfusionAm J Physiol Heart Circ Physiol279H1453911009429
- ZhongZKotovaODavidescuA2004C-peptide stimulates Na+, K+-ATPase via activation of ERK1/2 MAP kinases in human renal tubular cellsCell Mol Life Sci6127829015549182
- ZhongZDavidescuAEhrenI2005C-peptide stimulates ERK1/2 and JNK MAP kinases via activation of protein kinase C in human renal tubular cellsDiabetologia481879715624099