Abstract
Sporadic colon cancer is caused predominantly by dietary factors. We have selected bile acids as a focus of this review since high levels of hydrophobic bile acids accompany a Western-style diet, and play a key role in colon carcinogenesis. We describe how bile acid-induced stresses cause cell death in susceptible cells, contribute to genomic instability in surviving cells, impose Darwinian selection on survivors and enhance initiation and progression to colon cancer. The most likely major mechanisms by which hydrophobic bile acids induce stresses on cells (DNA damage, endoplasmic reticulum stress, mitochondrial damage) are described. Persistent exposure of colon epithelial cells to hydrophobic bile acids can result in the activation of pro-survival stress-response pathways, and the modulation of numerous genes/proteins associated with chromosome maintenance and mitosis. The multiple mechanisms by which hydrophobic bile acids contribute to genomic instability are discussed, and include oxidative DNA damage, p53 and other mutations, micronuclei formation and aneuploidy. Since bile acids and oxidative stress decrease DNA repair proteins, an increase in DNA damage and increased genomic instability through this mechanism is also described. This review provides a mechanistic explanation for the important link between a Western-style diet and associated increased levels of colon cancer.
Introduction
Sporadic colon cancer is caused predominantly by dietary factors, most notably a high-fat diet accompanied by low intake of micronutrients (folate, niacin, zinc), antioxidants and other plant phytochemicals. A high intake of fat induces the release of bile acids from the gall bladder into the small intestine. Bacteria in the colonic lumen then convert conjugated and hydrophilic bile acids to unconjugated, hydrophobic bile acids such as deoxycholic acid and lithocholic acid (). These hydrophobic bile acids, deoxycholic acid in particular, have been reported to be multiple stress inducers and at high physiologic levels can create chaos within colon epithelial cells. These stresses include membrane perturbation, oxidative DNA damage, decrease in DNA repair proteins, mitotic stress, micronuclei formation, mitochondrial damage (metabolic stress) and endoplasmic reticulum (ER) stress ().
Figure 1 The primary bile acids, cholic acid and chenodeoxycholic acid, are formed in the liver from cholesterol and conjugated, usually with glycine or taurine. They are then secreted into the gall bladder. From the gall bladder, glyco-and tauro- conjugated primary bile acids are released into the duodenum to aid in the digestion of fats. In the terminal ileum and in the cecum, the primary bile acids are deconjugated. Further, in the cecum, they are dehydroxylated through the action of bacterial 7-alpha dehydroxylases. By this reaction, cholic acid and chenodeoxycholic acid are converted to the more genotoxic-hydrophobic bile acids deoxycholic acid and lithocholic acid, respectively.
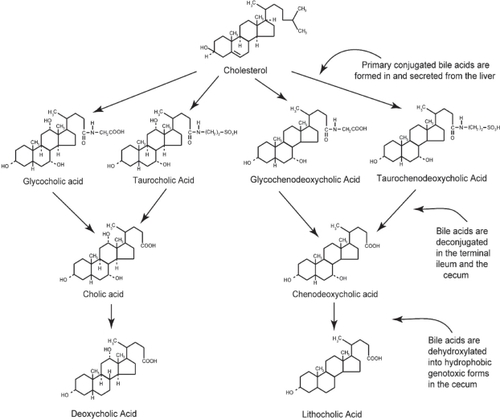
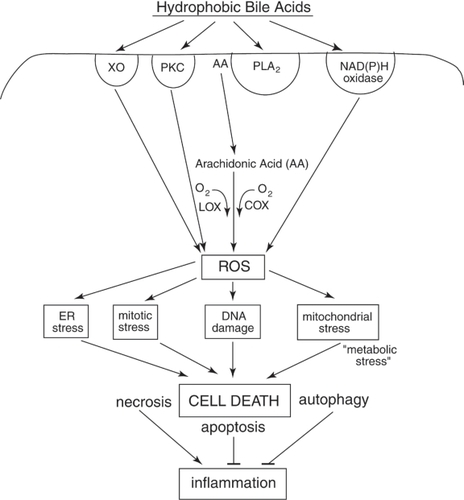
Figure 2 Schematic diagram indicating some of the hydrophobic bile acid (HBA)-induced signaling pathways that begin with the activation of surface enzymes, the subsequent generation of ROS, and the resultant stresses/damages that, if excessive, result in cell death. The most well documented deleterious effects of HBAs in colon cells are DNA damage, mitochondrial stress and ER stress. DNA damage results in the activation of mitotic checkpoint proteins leading to growth arrest. Mitochondrial damage results in the activation of caspases and the cleavage of multiple substrates in the cell. ER stress activates several pro-apoptotic molecules that result in cell death. Repair processes responsive to DNA damage, mitochondrial stress and ER stress can deplete the energy reserves of the cell, resulting in “metabolic stress”. Too much stress on the cell results in cell death through mechanisms that involve apoptosis, necrosis, and/or autophagy. HBA-induced mitotic stress can lead to abnormal cell division. Excessive mitotic stress may lead to mitotic catastrophe and cell death, although the details of this mode of cell death have not been well described. Necrotic cells induced by HBA may, especially, elicit an inflammatory response in vivo, and this could explain, in part, the induction of colitis by chronic feeding of HBA in mouse models. See text for details.
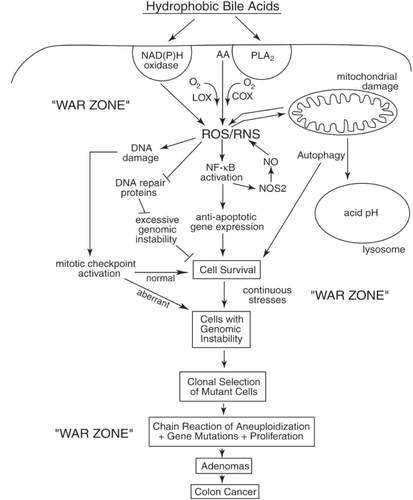
Although excessive stress will lead to apoptosis, this altruistic act of removing DNA-damaged cells for the survival of the organism becomes largely circumvented in cells with activated survival pathways. In this “war zone”,Citation1 stimulated by the presence of hydrophobic bile acids, mutated epithelial cells may act in a selfish manner,Citation2 like bacterial cells, and Darwinian selectionCitation3–Citation10 can come into play at the cellular level. The very stress-response pathways that result in cell survival (eg, activation of nuclear factor-kappa B [NF-κB], autophagy) () can become constitutively upregulated through clonal selection of mutated cells, and can serve to propagate cells even with unrepaired DNA damages and genomic instability. Although it has been known for decades (using animal models of carcinogenesis) that hydrophobic bile acids can promote colon cancer,Citation11–Citation14 the evidence is now convincing that bile acids are also carcinogens.Citation15–Citation17 Evidence that bile acids are carcinogens in human gastrointestinal (GI) cancers has been reviewed by Bernstein and colleagues.Citation15 Proof that hydrophobic bile acids cause mutation in vitro in epithelial cells of the GI tract was provided by the elegant studies of Jenkins and colleagues.Citation16,Citation17 Even though proliferation is essential to cancer development, it is not the high level of proliferation itself that is responsible for cancer. For example, adenocarcinomas are infrequent in the small intestine, although the proliferation rate of epithelial cells at this site is similar to that of the epithelial cells of the colon. The colon may be particularly susceptible to cancer because it contains tissues that are often exposed to unique stresses (eg, high levels of hydrophobic bile acids resulting from anaerobic bacterial metabolism). Since there may be insufficient time for DNA repair in this “war zone”,Citation18 the selection of mutated cells with a proliferative advantage may occur repeatedly. This proliferative advantage may take the form of apoptosis resistance and/or increased cell division.
Figure 3 Schematic diagram illustrating a probable sequence of events resulting from exposure to high levels of hydrophobic bile acids (HBAs) that accompany a high-fat diet. This exposure leads to HBA-induced generation of ROS/RNS, activation of survival pathways (eg, autophagy, NF-κB), the generation of cells with genomic instability (eg, mutations, aneuploidy) and clonal selection of mutant cells with survival and proliferative advantages. The end results are the production of adenomas that progress to colon cancer. The epithelial cells of the colon of a person on a Western-style (high-fat/low vegetable/low micronutrient) diet are probably in a persistant “war zone” (bombardment with HBA-induced ROS/RNS, presence of food carcinogens, toxins, etc.). Cells in different stages of progression to malignancy are thus persistently receiving damages to their genome, resulting in clones of cells that are selected for survival in the adverse environment of the colon. While cells in the previous population, if receiving excessive DNA damage, underwent cell death altruistically for the overall benefit of the organism, the new clones of cells may behave selfishly. The new clones may acquire resistance to apoptosis and undergo clonal selection on the basis of their survival advantage, even when their DNA is damaged and after their genomes become unstable. This allows progression to adenomas and colon cancer.
How, in particular, do bile acids contribute to colon carcinogenesis? Since somatic mutation rates are normally too low to account for the high incidence of colon cancers, it has been proposed that genomic instability (ie, a mutator phenotype) plays a major role in producing the plethora of random genomic changes needed to produce a malignancy.Citation19–Citation21 A mutator phenotype can explain the complex histopathology and behavior of tumors,Citation22–Citation24 including different tumor subsets,Citation25 divergence of clonal populations,Citation26 and different molecular pathwaysCitation27,Citation28 that may be selected during colon cancer progression and lead to different clinical outcomes.Citation29,Citation30 Genomic instability in cancer appears in three major forms: (i) aneuploidy, in which entire chromosomes are gained or lost, (ii) intrachromosomal instability, characterized by insertions, deletions, translocations, amplifications, and other forms all sharing the feature of utilizing DNA breakage as an early step, and (iii) point or oligobase mutations.Citation31 Genomic instability is a dynamic process in which the three major forms of genetic instability may co-exist at different stages of colon carcinogenesis, providing support for the chaotic roots of neoplastic development and continuous growth of aberrant clones.Citation32,Citation33
Mutations, aneuploidy or other chromosomal aberrations affecting genes that govern genetic stabilityCitation5,Citation34–Citation37 could be the cause of a mutator phenotype.Citation38,Citation39 The occurrence of a mutator phenotype would be made more likely by conditions that cause DNA damage, such as increased oxidative stress, due to persistent exposure to high levels of bile acids that accompanies a high-fat diet. Increased levels of oxidative stress caused by defective mitochondria have been proposed as a possible mechanism by which a “mutator phenotype” could arise.Citation40 As discussed below, bile acids cause oxidative DNA damage, double-strand DNA breaks (evidenced by activation of phosphorylated H2AXCitation41), aneuploidy,Citation16,Citation17 broken chromosomesCitation16,Citation17 and aberrant mitoses (unpublished data from our laboratory). In addition, high concentrations of bile acids in the lumen provide a persistent stressful environment that may select for mutated cells that can better survive in that environment (). Hydrophobic bile acids thus may serve to both initiate and propagate the mutator phenotype. Cycles of aneuploidizationCitation42 coupled with specific mutations (eg, APC, ras, p53) and large-scale structural alterations to chromosomes (eg, nonreciprocal translocations, inversions, deletions and insertions resulting in “genome scrambling”Citation43) may then give rise to a population of cells with multiple karyotypes and genotypes. The clonal selection of cells with particular karyotypes and/or mutations may then produce a “field defect” or “field cancerization” associated with gene expression changes. Frequently, one of the selected genotypes will be apoptosis resistance as found in the normal-appearing flat colonic mucosa of humansCitation44,Citation45 which is by definition a “survival genotype”. Within such a field defect, further mutations may then be selected for increased proliferation, altered cell-cell interactions and/or migratory behavior. On this basis a neoplastic clone can emerge (eg, as found in flat adenomas and polyps). These neoplastic survival karyotypes may undergo further mutation and selection to produce a cancer.
This review emphasizes the plethora of cellular stresses imposed by hydrophobic bile acids. Our review provides possible mechanisms by which a high-fat diet can lead to the development of sporadic colon cancers. The roles of mutations in critical genes (eg, APC, ras, p53) during colon carcinogenesis was previously addressed in the insightful discussions by the Vogelstein groupCitation46–Citation48 and will not be discussed in this review.
Hydrophobic bile acids generate cellular reactive oxygen species (ROS) and reactive nitrogen species (RNS) that damage DNA and proteins. These damages can lead to genomic instability (eg, point mutations and aneuploidy).Citation49 Point mutations, aneuploidy, and other gross structural alterations of chromosomes (eg, balanced or unbalanced translocations, dicentric chromosomes) appear to interact to produce the unstable and/or aberrant genomes associated with colon carcinogenesis. Aneuploidy is observed in most cancers, although it is not ordinarily found in colonic tumors that have a mismatch repair deficiency and display microsatellite instability.Citation50 A small subset of colorectal tumors display microsatellite instability, whereas the rest of colorectal tumors display numerical and/or structural chromosomal alterations as the most prominent outcome of genetic disruption.Citation51
Loss of heterozygosity (LOH) of a portion of a chromosome or a whole chromosome is already evident at the adenoma stage of colon carcinogenesisCitation52–Citation65 and is prominent at the colon cancer stage.Citation59,Citation60,Citation62,Citation63,Citation66–Citation68 A review of the literature indicates that at least 19 of the 22 human somatic chromosomes exhibit aneuploidy during the adenoma to colon cancer stage. The loss or gain of whole or portions of chromosomes involves thousands of genes.Citation69 This chromosomal imbalance could have dramatic effects on cellular homeostasis, including an increase in oxidative/redox state of the cell and the loss of key DNA repair and pro-apoptotic proteins. Approximately 11,000 genomic events were identified in sporadic colonic polyps using the technique of inter-(simple sequence repeat) polymerase chain reaction (PCR), indicating that genomic destabilization is an early event in sporadic tumor development.Citation31
Pre-neoplastic lesions smaller than adenomas are referred to as aberrant crypt foci (ACF).Citation70,Citation71 The ACF are not identified during routine colonoscopies since vital stains are often not used and biopsies are not usually taken of the flat colonic mucosa. However, human ACF have unequivocally been shown to exhibit genomic instability, including LOH.Citation72–Citation76 Since numerous genetic alterations and aberrant mitotic changes are also observed in the nonneoplastic colon of miceCitation77 and humansCitation78–Citation82 at high risk for colon cancer, biomarkers based on aberrant karyotypes have promise for identifying high-risk individuals. and are based on data from our laboratory and those of others and emphasize the role of hydrophobic bile acids in the induction of multiple stresses and genomic instability. We have selected bile acids as a focus of this review since bile acids play a key role in colon carcinogenesis. We describe how bile acid-induced stresses cause cell death in susceptible cells, contribute to genomic instability in surviving cells, impose Darwinian selection on survivors in a “war zone” and enhance initiation and progression to colon cancer. This review provides a mechanistic explanation for the important link between a Western-style diet and associated increased levels of colon cancer. However, other factors such as smoking, food-related carcinogens, long-chain fatty acids (eg, palmitic acid), excess iron, arsenic, viruses, bacteria, inflammatory cells, low dietary micronutrients, low omega-3 fatty acids, etc., may also contribute to genomic instability-based carcinogenesis.
Bile acids cause membrane perturbation
It has been known for over thirty years that bile salts perturb plasma membranes, and that deoxycholate is more membrane-damaging than cholate and its conjugates.Citation83 Binding of bile acids with rat colon and the resultant perturbation of membrane organization was later measured using 31P nuclear resonance spectroscopy.Citation84 Deoxycholate alters membrane composition, as evidenced by a significant upregulation in membrane cholesterol and phospholipids.Citation85 Bile acids also induce the modification and upregulation of caveolin-1 in a hydrophobicity-dependent manner, implying widespread receptor dysregulation.Citation85 These findings were mimicked by other hydrophobic molecules unrelated to bile acids, such as sodium lauryl sarcosine and cholesteryl hemisuccinate, strongly implicating hydrophobicity as an important determinant of bile acid effects on signaling.Citation85 Deoxycholate also perturbs membrane structure by alterating membrane microdomains.Citation86 Depletion of membrane cholesterol by treating cells with methyl-β-cyclodextrin suppressed deoxycholate-induced apoptosis, and staining for cholesterol with filipin showed that deoxycholate caused a marked rearrangement of this lipid in the membrane.Citation86 Additionally, fluorescence anisotropy studies indicated that deoxycholate caused a decrease in membrane fluidity consistent with the increase in membrane cholesterol content measured after deoxycholate treatment.Citation86
Bile acids activate surface enzymes, such as phospholipase A2 (PLA2),Citation87,Citation88 epidermal growth factor receptor (EGFR),Citation86,Citation89–Citation94 Fas receptor,Citation89 protein kinase C (PKC),Citation90 bile acid receptor M-BAR/TGR5 (G-protein-coupled receptor),Citation93 phospholipase C (PLC),Citation95 nicotinamide adenine dinucleotide phosphate (NAD(P)H) oxidaseCitation96,Citation97 and the Na+/K+ -ATPase.Citation97 We found that the surface enzymes, Na+/K+ -ATPase and NAD(P)H oxidase are involved in activating NF-κB,Citation97 a redox-sensitive transcription factor upregulated during the development of apoptosis resistance in colon cancer cells,Citation98 and associated with colon carcinogenesis. The activation of surface and other enzymes through membrane perturbation is, therefore, responsible for modulating signal-transduction pathways in colon cells.Citation85,Citation86,Citation89,Citation99
The fact that hydrophobic bile acids cause secretory diarrhea (diarrhea is the most common and debilitating symptom of patients with ulcerative colitis [a pre-neoplastic condition of the colon]), warrants a greater effort to understand how these bile acids induce changes in ion transport.Citation100 Taurodeoxycholic acid, for example, increases electrogenic Cl− secretion into the proximal colonic lumen.Citation101 This stimulated secretion is believed to occur via an increase in intracellular Ca++ concentration. Deoxycholic acid reduces transepithelial Na+ absorption by inhibiting amiloride-sensitive Na+ channels, and increasing secretion of Na+, K+, and Cl− ions in the distal colon.Citation102 Hydrophobic bile acids have been reported to act as calcium ionophores,Citation103,Citation104 most probably by partitioning bile acid/Ca++ complexes into the hydrophobic core of the membrane bilayerCitation104 and/or Ca++-transporting channels.Citation105 We have recently reported that high cytosolic Ca++ levels are a major factor in the activation of NF-κB in HCT-116 colon cells by bile acids.Citation97 Two agents (EGTA and Ruthenium Red (RuR)) used in that study prevented NF-κB activation by bile acids through at least two different mechanisms. EGTA does not cross cell membranes and chelates Ca++ ions in the extracellular milieu, thereby preventing Ca++ entry into the cell. RuR is an inorganic polycationic dye that inhibits Ca++ influx through voltage-sensitive calcium channels and blocks the release of Ca++ from the ER.
Bile acids cause oxidative and nitrosative stress in colon cells
Bile acids induce production of ROS and RNS in colon epithelial cells.Citation7,Citation106–Citation117 Bile acids generate ROS and RNS by several different pathways, as reviewed by Bernstein and colleaguesCitation15 and summarized here. Bile acids damage mitochondria and damaged mitochondria can “leak” electrons from the electron transport chain to form superoxide (O2−). ROS can also be generated by bile acid activation of PLA2, which releases arachidonic acid from the cell membrane. Arachidonic acid may then be acted on by cyclooxygenase and lipooxygenase to release ROS through partial reduction of O2, during the conversion of arachidonic acid to prostaglandins and leukotrienes.
Deoxycholate activates the redox-sensitive transcription factor NF-κB in colon epithelial cells.Citation97–Citation99,Citation110,Citation118–Citation124 When NF-κB is activated it can induce an increase in nitric oxide synthase 2 (NOS2), an enzyme that can generate micromolar quantities of NO. We have found that repeated exposure of HCT-116 colon epithelial cells to increasing concentrations of deoxycholate results in the consistent upregulation of S-nitrosylated proteins. Some of these post-translationally modified proteins were identified by mass fingerprinting analysis and found to include cytoskeletal proteins, metabolic enzymes, signaling proteins, chaperones, redox-related proteins and differentiation-related proteins.Citation114 We also found that feeding mice deoxycholate can induce inflammation in a mouse model,Citation115 and that the inflammation is mediated in large part by RNS, since colitis is markedly attenuated in NOS2 knockout mice.Citation116
DNA repair proteins can be modulated by oxidative stress and deoxycholate
The promoting and mutagenic effects of bile acids can be explained, in part, by a decrease in DNA repair proteins. Since oxidative stress is known to reduce expression of DNA repair proteins, bile acid-induced decreases in DNA repair proteins may also be through an oxidative mechanism. The cell types and experimental conditions in which oxidative stress decreases DNA repair proteins, and bile acids decrease proteins involved in 6 different DNA repair pathways are described below. Since DNA repair proteins often have multiple functions in the cell in addition to DNA repair, such as participation in apoptosisCitation125 and regulation of mitosisCitation126,Citation127 and checkpoint pathways,Citation128 a deleterious effect of bile acids on DNA repair protein expression can have important and varied implications for inducing genomic instability.
Oxidative stress causes a decrease in DNA repair proteins
Langie and colleagues,Citation129 working with human pulmonary epithelial cells A549, showed that oxidative stress caused a reduction in mRNA for the nucleotide excision repair (NER) enzyme ERCC1 as well as a reduction in NER capacity that correlated with level of ERCC1 mRNA. Feng and colleagues,Citation130 using the human colon epithelial cell line HCT-116 and the human hepatic fetal epithelial line CL-48, showed by in vitro DNA repair synthesis and host cell reactivation assays, that ROS also inhibit NER through production of malondialdehyde. This effect is likely mediated by malondialdehye-induced inactivation of DNA repair proteins. Furthermore, as shown in the human erythroleukemia HEL cell line, oxidative stress inactivates the mismatch DNA repair proteins MSH6 and Pms2 (but not MSH2 or MLH1).Citation131 In HeLa cells and the human Boleth lymphoblastoid cell line, the base excision repair enzyme OGG1 is degraded or reversibly inactivated following oxidative stress.Citation132,Citation133
In mitochondria, DNA polymerase γ is responsible for both mtDNA replication and repair. In an SV40-transformed human fibroblast cell line, the functional ability of the catalytic p140 subunit of DNA polymerase γ is reduced by oxidative stress.Citation134
Bile acids decrease DNA repair proteins
Short term exposure to deoxycholate decreases the expresson of the DNA repair proteins p53 and BRCA1
The enzyme p53 has multiple roles in the cell. In particular, it has roles in 6 different DNA repair pathways: (1) homologous recombinational repair, (2) nonhomologous end joining, (3) base excision repair, (4) nucleotide excision repair, (5) mismatch repair, and (6) transactivation of expression of O6-methyl-guanine-DNA-methyl-transferase for direct repair of alkyl adducts at the O6-methyl-guanine position.Citation125,Citation135 While treatment of colon HCT-116 cells with non-cytotoxic (200 μM) deoxycholate concentrations increases mRNA expression of the p53 gene, the level of p53 protein is decreased by 40% through proteasome-mediated degradation.Citation136 Although mutations in p53 are a late event in colon carcinogenesis, the persistent decrease in p53 function by deoxycholate may have similar consequences as that of a mutation. Work from our group has also shown that deoxycholate decreases BRCA1 at the protein and mRNA levels.Citation137 BRCA1 has a key role in recombinational repair of DNA damages, and also regulates the function of the centrosomes which carry out mitotic chromosome segregation through spindle organization.Citation127 Failure to adequately repair DNA damages can lead to replication errors and thus mutation; and failure of mitotic segregation can lead to aneuploidy. Since BRCA1 expression is also reduced in colon adenocarcinomas,Citation137 it is possible that the modulation of expression of DNA repair enzymes by dietary-related factors may, in part, be responsible.
Long term exposure of colon cells to deoxycholate selects for decreased protein or mRNA expression of DNA repair proteins
Three HCT-116 colon cell lines were developed in our laboratory for stable resistance to bile acids by repeated exposure to increasing concentrations of deoxycholate over a period of approximately 40 weeks.Citation98 These cell lines were found to have constitutively decreased protein expression of the DNA repair enzymes MSH3 (mismatch repair), Ku80 (non-homologous end joining), P36/MAT1 (nucleotide excision repair) and XPA (nucleotide excision repair).Citation138 These cell lines were also found to have decreased expression at the mRNA level for ATM (homologous recombinational repair), MSH3 (mismatch repair) and XRCC4 (non-homologous end joining).Citation98
Exposure of cholangiocytes to glycochenodeoxycholate (GCDA) decreases DNA repair enzymes
Komichi and colleaguesCitation139 exposed immortalized mouse cholangiocytes to 200 μM glycochenodeoxycholate (GCDC) for 4 weeks and then performed a microarray gene expression analysis. mRNA expression of OGG1 and MUTYH was downregulated 0.8-fold after exposure to CGDC compared to control cells.
Bile acids cause DNA damage in colon cells
Bile acids induce DNA damage in cells of the colon.Citation41,Citation90,Citation99,Citation108,Citation109,Citation115,Citation118,Citation140–Citation143 One type of induced damage in colon cells is oxidative DNA damage,Citation90,Citation108,Citation115,Citation143 suggesting that oxidative stress is a significant cause of the overall bile acid-induced DNA damage in the colon. Exposure of rat colon ex vivo with high physiologic concentrations of bile acids resulted in the activation of poly(ADP-ribose) polymerase (PARP).Citation99 PARP is activated by DNA strand breaks and attaches a polymer of ADP-ribose units to proteins which modulates their functional activity and assists in the opening up of the chromatin to allow for more efficient DNA repair.Citation144 It is probable that the increase in DNA strand breaks is mediated, in part, through the bile acid-induced increase in ROS and/or RNS. Treatment of human colonic biopsies ex vivo with deoxycholate also resulted in the activation of phosphorylated H2AX,Citation41 implying the generation of double-strand breaks in DNA.Citation145 Double-strand breaks can cause chromosome breakage with the generation of micronuclei upon exit from mitosis.Citation16,Citation17 Increased DNA damage may be caused by the bile acid-induced decrease in DNA repair enzymes (see Bile acids decrease DNA repair proteins, above).
Bile acids cause mutation
The repeated observation that bile acids cause DNA damage in colon cells suggests that bile acids also increase the incidence of mutation, since replication of a damaged DNA template strand frequently results in a replication mistake, and thus a mutation. Bile acids have been shown to induce mutation in esophageal cells in culture and in vivo in rat esophageal cells. When cultured esophageal cells were treated with deoxycholate, an increase in the frequency of GC to AT mutations in the p53 gene was observed.Citation16 In other experiments, Big Blue F1lacI transgenic rats were subjected to esophagoduodenostomy, a surgical procedure that increases duodenogastroesophageal reflux.Citation146 In these surgically altered rats, the frequency of mutant lacI cells of the esophageal mucosa was significantly greater than in the control rats that were not subjected to surgery. Thus, components of refluxate, such as bile acids, appear to cause mutation. Forty-six percent of the mutant esophageal cells were altered at CpG dinucleotide sites, and most of these mutations (61%) were C to T or G to A transitions. This pattern of mutation seen in the surgical model approximates that seen in human esophageal adenocarcinoma, suggesting that duodenogastroesophageal reflux is carcinogenic. In surgically treated Big Blue mice (rather than rats) that were altered to increase duodenogastroesophageal reflux, increased mutagenesis was also observed.Citation147 In another rat model of colon carcinogenesis based on exposure to azoxy-methane, deoxycholate increased both the incidence of colon tumors and the incidence of tumors with K-ras mutations.Citation148 This finding suggests that deoxycholate may induce K-ras mutations. In a sensitive bacterial flucturation test based on the Ames Salmonella test system, bile acids were also found to be mutagenic.Citation149
Hydrophobic bile acids induce aneuploidy and micronuclei formation
The induction of aneuploidy by deoxycholate was first reported 26 years ago by Assinder and UphallCitation150 in a heterozygous diploid strain of Aspergillus nidulans. The detection of aneuploidy was made possible by the unique orientation of mutations on the chromosomes. Each pair of homologues carried mutations, in repulsion, in at least 2 gene loci, those in linkage groups I, III, IV, and V being on opposite sides of the centromere. This orientation allowed a distinction between cross-over and nondisjunctional segregants (aneuploids). It was suggested that aneuploidy was a result of interference with the normal functioning of the mitotic apparatus through disruption of the nuclear membrane. Ferguson and ParryCitation151 confirmed that bile acids, including deoxycholate, were potent inducers of aneuploidy using the diploid yeast, Saccharomyces cerevisiae. The assay used to detect mitotic chromosome aneuploidy involved scoring of white cycloheximide resistant monosomic colonies. Albertini and colleaguesCitation152 determined that lithocholic acid induces mitotic chromosome loss in the diploid yeast strain, S. cerevisiae D61.M. The assay to detect mitotic chromosome loss involved scoring of segregants expressing three linked recessive markers, two of which were located close to the centromere on opposite arms of chromosome VII.
The evaluation of the induction of aneuploidy by bile salts in eukaryotic cells of higher organisms was neglected for almost 20 years, even though a role of bile acids in colon carcinogenesis was established in animal models in the 1970s. The first study of bile acid-induced aneuploidy in cells of multicellular organisms in vitro was reported by Jenkins and colleaguesCitation16 using human esophageal cell lines. In this elegant study, the frequency of deoxycholate-induced kinetocore-positive and -negative micronuclei was assessed as a measure of the level of aneuploidy, using the cytokinesis-block micro-nucleus assay.Citation153,Citation154 The rationale for this assay is based on data which indicate that aneuploid cells and cells with broken chromosomes will often produce micronuclei after exit from mitosis. The presence of kinetochore-negative micronuclei indicated that chromosome fragmentation occurred, probably as a result of deoxycholate-induced double strand breaks. The deoxycholate-induced increase in micronuclei was significantly reduced by the antioxidant, vitamin C, indicating the importance of diet in the possible prevention of genotoxicity. Unpublished data from our laboratory also indicate that deoxycholate can induce micronuclei in colon epithelial cells in vitro in addition to inducing aberrant mitoses (implying perturbation of the mitotic machinery). These unpublished results are consistent with published data from our laboratory that persistent exposure of cells to deoxycholate in vitroCitation98,Citation138 or in vivo in an animal model,Citation115 overall, results in the modulation of genes/proteins involved in chromosome maintenance and cell cycle progression (see Persistent exposure of colon cells to deoxycholate results in the modulation of expression of chromosomal maintenance/mitosis-related genes, below). These findings indicate that deoxycholate is a true carcinogen,Citation15 with important implications for dietary intervention strategies to prevent the initiation and/or progression of GI cancer.
A major stress induced by hydrophobic bile acids is the generation of ROS/RNS. Chronic exposure of cells to oxidative stress has been reported to result in increased genomic instability.Citation155,Citation156 An important source of ROS is damaged/mutated mitochondria.Citation40,Citation157 The mechanisms by which oxidative stress can lead to genomic instability are probably multifactorial, and include induction of double-strand breaks (resulting in broken chromosomes) and overriding the spindle checkpoint during cell cycle progression,Citation158 which can result in aneuploidy.
Persistent exposure of colon cells to deoxycholate results in the modulation of expression of chromosomal maintenance/mitosis-related genes
The induction of aneuploidy with the formation of micronuclei after cells exit mitosis may be caused by a number of defects during cell division. Defects such as insufficient or excessive sister chromatid cohesion,Citation159,Citation160 kinetochore-microtubule attachment defects,Citation161,Citation162 failure of DNA damage checkpoints,Citation163 failure of spindle checkpoints,Citation164–Citation166 telomere defectsCitation167,Citation168 and aberrant centrosomal organization and multiplicityCitation169,Citation170 may result in an incorrect number of chromosomes in daughter cells after mitosis. These defects coupled with chromosome breaks can produce an aberrant karyotype that may be carcinogenic. We, therefore, searched for the aberrant expression of mitosis-related genes in colon epithelial cells and colonic tissues that were modulated by deoxycholate. cDNA microarray () and proteomic analyses () of HCT-116 cell lines persistently exposed to increasing concentrations of deoxycholate in vitroCitation98,Citation138 and cDNA microarray analysis of colonic tissue of wild-type mice receiving dietary supplementation with deoxycholate in vivoCitation115 () indicate that deoxycholate modulates the expression of numerous genes associated with mitosis and chromosome maintenance. The list includes 71 genes/proteins associated with cohesion and segregation of chromosomes, telomeres, kinetochore structure, spindle assemby and function, centrosome regulation, various aspects of the cell cycle (metaphase, anaphase), cell cycle progression (kinases, phosphatases, ubiquitin-conjugating enzymes), cell cycle checkpoints and cytokinesis (–).
Table 1 Fold reduction/induction of mRNA levels of chromosomal maintenance/mitosis related genes in HCT-116 colon cancer cells persistently exposed to deoxycholate
Table 2 Statistically significant increases/decreases in protein levels of chromosomal maintenance/mitosis related genes in HCT-116R colon cancer cells persistently exposed to deoxycholate
Table 3 Fold reduction/induction of mRNA Levels of mitosis-related genes in colon of mice fed a diet supplemented with deoxycholate
Since the microarray study was published six years ago,Citation98 recently identified gene sequences related to mitosis and chromosome maintenance have been added to to bring the 2002 study up to date.
In summary, persistent upregulation and/or downregulation of key proteins of the division process by deoxycholate, coupled with decreased DNA repair and increased DNA damage, may lead to genomic instability, including mutator phenotypes. Recent studies have indicated that 1) the spindle proteins, Aurora A and BUB1B, are aberrantly expressed in dysplastic mucosa of patients with longstanding ulcerative colitis,Citation171 a pre-neoplastic condition), 2) both Aurora A and BUB1B undergo a shift in subcellular localization during malignant transformation,Citation172 3) a reduced level of the spindle checkpoint protein, BUB1B, is associated with aneuploidy in colorectal cancers,Citation173 and 4) chromatid cohesion defects may underlie chromosome instability in human colorectal cancers.Citation174 It is an intriguing possibility that these defects occur very early during colon carcinogenesis, and may be caused, in part, by dietary-related factors, such as high levels of endogenous bile acids, coupled with polymorphisms in mitotic checkpoint genes.Citation175
Oxidative stress as an important cause of genomic instability
The induction of kinetochore-positive and -negative micronuclei by bile acids may result from aneuploidy (involving the loss and gain of whole chromosomes) or portions of chromosomes, resulting from unrepaired double-strand breaks. This bile acid-induced induction of genomic instability is most probably caused by oxidative stress directed at parts of the mitotic machinery, checkpoint proteins and/or direct oxidative damage to DNA that results in double-strand breaks. The only studies that addressed this issue in the gastrointestinal tract are those of Jenkins and colleagues,Citation16,Citation17 who evaluated several antioxidants (eg, vitamin C, resveratrol, EGCG) to determine their effectiveness at reducing deoxycholate-induced micronuclei formation in esophageal cells. Of the antioxidants tested, only vitamin C had a significant effect on reducing micronuclei formation. Since bile acids damage mitochondria, resulting in an increase in mitochondrial ROS, and ROS derived from damaged mitochondria are known to induce genomic instability,Citation157 various antioxidants specifically directed at mitochondria should be tested for their effectiveness at reducing bile acid-induced genomic instability.
Since this area of bile acid research is just beginning to be explored, we have reviewed the literature involving other cell types (beyond those of the GI tract) and found that oxidative stress plays a definite role in the induction of genomic instability.Citation40,Citation156,Citation157,Citation176–Citation178 Aneuploidy may often reflect defects in mitotic segregation in cancer cells.Citation50 The faithful cohesion and separation of chromosomes are controlled by a large number of proteins, some of which have a checkpoint function.Citation180–Citation185 Increased production of ROS has been reported to induce aneuploidy by impairing the spindle check-point function.Citation158 The spindle checkpoint is the safeguard mechanism that halts anaphase onset until the mitotic spindle has been assembled and metaphase chromosomes properly attached. At the metaphase-anaphase transition, the APC (anaphase-promoting complex) initiates the separation of sister chromatids by catalyzing the ubiquitination of securin, a protein that prevents the proteolysis of the cohesin complex and the subsequent segregation of the chromosomes, in the securin-separase complex.Citation186 The APC11 subunit functions as the catalytic core of the APC complex and contains a RING-H2-finger domain, which includes one histidine and seven cysteines residues that coordinate two Zn2+ ions. The RING-H2-finger domain is a target of hydrogen peroxide, which induces the release of of bound zinc as a result of the oxidation of cysteine residues.Citation187 The oxidation of APC11 reduces the normal functions of the unoxidized APC11 protein, resulting in the inhibition of ubiquitination and degradation of cyclin B1, and the degradation of securin. Other chromosomal aberrations, such as translocations and dicentric chromosomes, may be caused by accumulation of double-strand breaks induced by oxidative damage, as in Werner syndrome cells.Citation188 Based on the data from our laboratory that shows the modulation of 71 genes involved in chromosome maintenance, chromatid cohesion, kinetochore-microtubule attachments, chromatid separation, cytokinesis, etc. by deoxycholate (see –), some of these proteins may be oxidatively damaged and/or targeted for destruction by deoxycholate. However, no studies have yet addressed this important mechanism of bile acid-induced damage to dividing cells. Thus, in colon carcinogenesis, the persistent increase in oxidative stress in epithelial cells caused by excessive exposure to bile acids may induce mitotic defects very early in the sequence of events that lead to colon cancer. Hydrophobic bile acids damage mitochondria, and increased levels of oxidative stress caused by defective mitochondria have been proposed as a possible mechanism by which a “mutator phenotype” could arise.Citation40
Bile acids induce ER stress
The induction of ER stress by hydrophobic bile acids is evidenced by an increase in Gadd153/CHOP,Citation142,Citation189,Citation190 a pro-apoptotic transcription factor,Citation191–Citation197 and an increase in GRP78,Citation98,Citation190 a molecular chaperone that protects against ER stress.Citation197,Citation198 Furthermore, hydrophobic bile acids dilate the cisternae of the endoplasmic reticulum (identified using transmission electron microscopyCitation199). The mechanisms by which bile acids cause ER stress probably include the generation of ROS that may damage the ER membrane and/or cause protein unfolding, and the release of Ca++ from the ER through the activation of inositol triphosphate and ryanodine receptors.Citation200 Excessive ER stress may lead to cell death in the form of apoptosis and/or necrosis. Since deoxycholate induces the formation of nitrotyrosine residues in proteins of colon cells,Citation112 ER stress may also be mediated through the generation of RNS.Citation195,Citation201 Persistent ER stress may then select for cells that exhibit activated ER stress-responsive survival pathways important in colon carcinogenesis. We have shown that ER stress-related survival proteins are consitutively upregulated in colon cancer cells that are resistant to deoxycholate-induced apoptosis, and include Grp78,Citation98 S-nitrosylated Grp78Citation114 and S-nitrosylated ORP150,Citation114 another inducible ER stress-related chaperone protein.Citation202 The observed constitutive increase in autophagic proteinsCitation98 observed in these resistant cells may also serve to protect against ER stress, although this area of research has not been fully explored.
Consequences of bile acid-induced mitochondrial damage: metabolic stress, autophagy, apoptosis, and necrosis
Bile acid-induced DNA damage, ER stress, mitotic stress, and mitochondrial damage may all lead to cell death through various processes including apoptosis, necrosis, autophagic cell death and/or mitotic catastrophe, depending on the level of combined stresses. Of all organelles, mitochondria are most central to the three main cell death pathways, apoptosis, necrosis, and autophagic cell death.Citation203 Apoptosis as a mechanism of mitochondrially-mediated bile acid-induced cell death has been studied the most in cells of the GI tract, as discussed below.
Mitochondria generate most of the ATP used for energy-consuming processes within the cell. Moreover, mitochondria play an important role in the regulation of other cellular processes such as autophagy and apoptosis. Autophagy is a catabolic process involving the degradation of a cell’s own components through the activation of lysosomes. In response to stresses induced by hydrophobic bile acids, autophagy is initiated (unpublished data from our laboratory) as the first mechanism to cope with damage when mitochondrial injury is limited. The damaged mitochondria are sequestered within lysosomes, thereby preventing the release of apoptosis-inducing factors, such as cytochrome c, apoptosis-inducing factor (AIF), and Smac/DIABLO. As more mitochondria are damaged and undergo the mitochondrial membrane permeability transition in response to bile acids, the pro-apoptotic proteins are released from the mitochondria.Citation204 These releases result in the formation of the apoptosome, caspase activation, degradation of vital proteins in the cell and, finally, apoptosis. Apoptosis can be triggered by two distinct signaling cascades, the extrinsic and intrinsic pathways. The extrinsic pathway involves the activation of specific cell surface receptors such as Fas/CD95 and the tumor necrosis factor receptor (TNFR), while the intrinsic pathway is primarily regulated by mitochondria. Importantly, in contrast to hepatocytes and hepatic cancer cell lines, in which hydrophobic bile acids induce apoptosis by an extrinsic pathway, deoxycholate induces apoptosis in colonic epithelial cells in a CD95-independent pathway.Citation205 Bile acid-induced mitochondrial damage may initially increase autophagy to rid cells of damaged organelles. As stress continues, apoptosis or autophagic cell death may follow. If, however, the stresses on mitochondria are initially severe, oxidative phosphorylation fails, NAD+ levels drop, ATP levels are rapidly depleted, resulting in loss of ionic control,Citation206 followed by cell swelling and lysis, characteristic features of necrosis.Citation207–Citation211 Apoptotic cell death induced by deoxycholate has been frequently reported in colon epithelial cells.Citation41,Citation44,Citation45,Citation98,Citation99,Citation110,Citation112,Citation118,Citation199,Citation205,Citation212–Citation224 Therefore, an understanding of bile acid-induced alterations in mitochondrial function are of great interest. At least two major alterations occur during bile acid-induced mitochondrial dysfunction. These are (1) disruption of electron transport leading to generation of ROS, loss of mitochondrial membrane potential (MMP), decrease in oxidative phosphorylation, and decrease in ATP production, and (2) release of mitochondrial proteins that trigger activation of caspases. Bile acids were shown to be involved in all three processes.
The exact mechanism by which bile acids disrupt electron transport and decrease oxidative phosphorylation has yet to be defined.Citation225–Citation227 Experiments performed with mitochondria isolated from hepatocytes clearly demonstrate that hydrophobic bile acids induced perturbations in mitochondrial bioenergetics.Citation225,Citation226 Specifically, complex I and complex III of the mitochondrial electron transport chain were inhibited by treatment with10 μM bile acid. Complex IV was inhibited at higher concentrations of bile acids.Citation225 The authors speculated that the inhibition of the electron transport complexes may be due to bile acid incorporation into the mitochondrial membranes resulting either in direct toxicity or alteration in lipid composition. When they tested this hypothesis, they found that, indeed, bile acids were incorporated into the mitochondrial membranes. Furthermore, they found that the hydrophobic bile acid, chenodeoxycholic acid, induces alteration in the lipid composition of the mitochondrial membrane, while no alterations were found after treatment with lithocholic acid.Citation225 However, it is possible that the changes in lipid composition induced by lithocholic acid were below the limit of detection used in the study.Citation225 Membrane structural changes observed using spin-labelling techniques and electron paramagnetic resonance (EPR) spectroscopy analysis also support the involvement of mitochondria in bile acid-induced apoptosis.Citation228 These mitochondrial structural changes include modified lipid polarity and fluidity, altered protein order and increased oxidative injury.Citation228
Hydrophobic bile acids cause mitochondrial oxidative stressCitation97,Citation110,Citation117,Citation228,Citation229 leading to mitochondrial swellingCitation199 and formation of megamitochondria.Citation110 The excessive generation of ROS that overwhelms the cells’ antioxidant defenses is most probably responsible for the death of colonic epithelial cells,Citation230 and is consistent with the role of oxidative stress in mediating apoptosis in many other types of cells.Citation231 Importantly, bile acid-induced apoptosis in HT-29 colon cells is dramatically reduced if mitochondria are protected against ROS by inhibiting complexes I and II of the electron transport chain.Citation110 Later studies indicated, moreover, that perturbation of the mitochondrial electron transport chain, in general, and uncoupling of oxidative phosphorylation, attenuates deoxycholate-induced apoptosis.Citation199 The mechanism by which this perturbation of mitochondrial function results in apoptotis resistance is not clearly understood. Another effect of hydrophobic bile acids on mitochondria is rapid loss of mitochondrial membrane potential (MMP).Citation110,Citation199,Citation205 For example, treatment of SW480 cancer cell line with 0.5 mM deoxycholic acid induced loss of MMP in 5 minutes.Citation205 This loss of MMP correlates with the induction of ROS.
The second major effect of hydrophobic bile acids is to induce rapid release of pro-apoptotic proteins in colon cancer cells, such as cytochrome c, which leads to the activation of caspases.Citation218 Incubation of HT-29 and HCT-116 colon cells with deoxycholate resulted in the cleavage of procaspases and the activation of effector caspases 2, 3, 7 and 8,Citation110,Citation222 while caspase-1 proinflammatory caspase was not cleaved.Citation222 Caspases 9 and 3 are rapidly activated by deoxycholate, while caspase 8 activation is a later event.Citation110,Citation218,Citation222 Caspase-6 is of particular importance to deoxycholate-induced apoptosis of colon epithelial cells, since the antiapoptotic nitric oxide-targeted enzyme, guanylate cyclase-α1, is specifically cleaved by caspase-6, resulting in apoptosis.Citation224 Apoptosis in HCT116 cells is not induced simply by a detergent effect, since sodium dodecyl sulfate (SDS), a detergent that is structurally distinct from bile acids, does not induce apoptosis.Citation218 Although protein synthesis inhibitors have been shown to protect against programmed cell death, de novo synthesis of proteins does not appear to be necessary for bile acid-induced apoptosis.Citation218 Altogether, these data suggest that in contrast to the extrinsic induction of apoptosis, as seen in hepatocytes, hydrophobic bile acids primarily activate the intrinsic mitochondrial pathway of apoptosis in colon cancer cell lines.
How exactly do bile acids interact with mitochondria to perturb their function? Hydrophobic bile acids may directly affect mitochondria, leading to disruption of the electron transport chain and increased production of mitochondrial ROS. Alternatively, mitochondria can be damaged indirectly by extramitochondrial ROS produced by plasma membrane bound enzymes targeted by bile acids, such as NAD(P)H oxidaseCitation96 (). The importance of NAD(P)H to deoxycholate-induced apoptosis was ascertained by the use of an inhibitor of NAD(P)H oxidase, diphenyleneiodonium (DPI).Citation232 DPI attenuated overall ROS production and prevented the occurrence of deoxycholate-induced apoptosis, assessed using translocated membrane phospholipid and nuclei containing condensed chromatin.Citation232 Interestingly, NADPH oxidase levels are elevated in colon cancer cell lines as well as in the tissues from patients with colon cancer.Citation233–Citation236 Another extramitochondrial source of ROS occurs when molecular oxygen is partially reduced during the metabolism of arachidonic acid by the cyclooxygenase (COX) and lipoxy-genase (LOX) pathways (). Another indirect effect of bile acids on mitochondria is the release of arachidonic acid from phospholipids following activation of PLA2 by bile acids.Citation87 Arachidonic acid induces inhibition of mitochondrial complex I and III and causes an increase in mitochondrial ROS production.Citation237 Hydrophobic bile acids may also trigger the formation of ceramide,Citation238 which can damage mitochondria and induce apoptosis. The ceramide-induced disruption of mitochondrial function and induction of apoptosis is caused, in part, by modulation of gene expression of members of the Bcl-2 family of apoptosis-related proteins.Citation239
Persistent exposure to deoxycholate selects for apoptosis resistance
Repeated long-term exposure of colon epithelial cells to high physiologic concentrations of bile acids may allow preferential survival of cells that are resistant to induction of apoptosis by bile acids. Such apoptosis resistant cells could arise and proliferate by the processes of mutation and Darwinian selection (). Apoptosis-resistance in the colon appears to arise in settings where the colonic mucosa is chronically exposed to high physiologic levels of bile acids. Magnuson and colleaguesCitation240 reported that chronic feeding of 0.2% cholic acid (added to the AIN-76 diet) to azoxymethane (AOM)-treated Sprague-Dawley rats for 18 weeks resulted in a statistically significant reduction in the number of apoptotic bodies in ACF after an acute low dose of AOM, compared with normal-appearing crypts. Both normal and ACF crypts had fewer apoptotic bodies per 100 cells than crypts from rats fed the control diet.
Work from our laboratory indicated that in vitro exposure of apoptosis-sensitive colon epithelial cells (HCT-116) for approximately 40 weeks resulted in the generation of stable apoptosis-resistant cell populations.Citation98 We also evaluated the nonneoplastic flat colonic mucosa of patients in different colon cancer risk groups for apoptosis competence using an ex vivo live cell bioassay.Citation44,Citation45,Citation212,Citation213,Citation241 The apoptosis-inducing agent used in this bioassay was deoxycholate. The nonneo-plastic colonic mucosa of patients with colon cancer had a significantly reduced apoptotic index compared to that of low-risk and normal patients. The exact etiologic agents that were responsible for the development of apoptosis resistance are not known, but hydrophobic bile acids, other agents that generate ROS/RNS, and actual components of a high-fat diet may have been key players.
Bile acids, inflammation and colon cancer
Numerous epidemiological studies have shown that fecal bile acid concentrations are increased in populations with a high incidence of colon cancer, suggesting an etiological linkage.Citation242–Citation251 We have shown that supplemental feeding of deoxycholate in a mouse model can cause colitis in association with markers of oxidative stress.Citation115 An important link between high bile acid concentrations and colon cancer may, therefore, be the induction of inflammation, a condition that is associated with many cancers of the GI tract. Bile acids may induce inflammation through a disruption of tight junctions between colonic epithelial cellsCitation115 and increase in transepithelial permeability,Citation88 providing a means for entry of pro-inflammatory bacterial species.Citation252 In addition, bile acids may cause dysregulation of cytokine expression through the constitutive activation of NF-κB,Citation98 a redox-associated transcription factor, that contributes to apoptosis resistance and a persistent increase in pro-inflammatory proteins.
Field defects in the colon
A “field defect” or “field of cancerization” is a region of tissue that precedes and predisposes to the development of cancer.Citation253 Field defects are of interest because they provide insight into the early events of progression to cancer and may lead to clinically useful biomarkers of cancer risk. It is generally considered that a solid tumor is initiated by a series of somatically inherited changes (ie, aneuploidy, mutations or transmissible epigenetic events (epimutations), such as methylation of CpG islandsCitation254). Aneuploidy, mutations and epimutations are considered to contribute to early progression, usually by causing a somatically inherited proliferative advantage relative to surrounding cells. Furthermore, a mutator phenotype or genetic instability may accelerate the process of mutation followed by Darwinian selection.Citation19–Citation21 In a normal population of dividing cells within a tissue, a cell may acquire a proliferative advantage through aneuploidy, mutation or epimutation. Such a cell will tend to undergo clonal expansion and replace neighboring cells, thus giving rise to a patch of abnormal cells. Within this patch, a cell might acquire a second aneuploidy, mutation or epimutation that provides a proliferative advantage compared to other cells within the established patch. This cell may then expand clonally forming a secondary patch within the first patch. Within this new patch, the process of mutation and Darwinian selection may be repeated several more times over a long period, perhaps decades, until a malignant cell arises that clonally expands into a cancer. If solid tumors generally arise by such a process, then tumors should ordinarily be accompanied by a field defect in associated tissue that may appear histologically normal.
The most common type of colorectal cancer, sporadic adenocarcinoma, develops by a pathway that appears to proceed through the following stages: normal flat mucosa, development of fields of defective flat mucosa, aberrant crypt foci, microadenomatous fields, adenoma with low grade dysplasia, adenoma with high grade dysplasia, and adenocarcinoma. However, currently, it is unclear whether all sporadic adenocarcinomas progress through each of these stages.
The morphologically normal appearing colonic mucosa of patients with colorectal neoplasia has been found by proteomic analysis to have numerous changes in protein expression.Citation255 Alterations of gene expression that occur in the normal appearing colonic mucosa of human colon cancer patients correspond to alterations observed in the normal appearing colonic mucosa of cancer prone APCmin mice.Citation256 The promoter of the DNA repair gene O6-methylguanine-DNA methyltransferase (MGMT) is methylated in 46% of colonic tumors. Within this group of tumors with MGMT methylation, 94% of tissue samples from apparently normal mucosa associated with these tumors also had MGMT promoter methylation.Citation257 Often MGMT methylation was detected as far away as 10 cm from the tumor, indicating a field defect. Also patients with resected adenomas of the colon or rectum had altered expression of O-acetylated sialic acids in their “uninvolved” colon and rectal mucosa compared to sialic acids of colon or rectal mucosa of patients without colon tumors.Citation258 These findings indicate a field defect of sialic acid expression in the patients with colorectal cancer. Since a decrease in the level of O-acetylated sialic acids on the surface of colon epithelial cells is found to be associated with the early stages of colorectal cancer,Citation259 it is considered a risk factor. This decrease in the level of O-acetylated sialic acids may lead to an increase in the expression of sialyl LewisX, a tumor-associated antigen related to the progression of colorectal cancer cells to metastasis.Citation260
An imbalance of proliferation and apoptosis in the left colon and sigmoid/rectum was found in patients who had a resected large adenoma (≥1.5 cm) compared to patients who never had a tumor.Citation261 Several lines of evidence indicate that colorectal cancers often arise in a field of apoptosis-resistant cells. The Bcl-xL protein inhibits apoptosis, in part, by inhibiting release of cytochrome c from the mitochondria. The expression of Bcl-xL was found to be increased at 1 cm and 10 cm away from colorectal adenocarcinomas in the non-neoplastic colorectal mucosa, suggesting the presence of an apoptosis-resistant field.Citation262 The prototypic anti-apoptotic protein, Bcl-2, also behaves similarly.Citation263 Expression of the antiapoptotic survivin mRNA in colon tumors predicts poor patient survival due to subsequently recurrent colorectal tumors.Citation264 The normal appearing mucosa of about half of patients with survivin-positive tumors also express this mRNA. This finding suggests that survivin- positive tumors often develop in a normal appearing, but survivin- positive, apoptosis-resistant field. When rats are fed a diet containing 0.2% cholic acid, their colon crypt cells develop increased resistance to apoptosis.Citation240 Apoptosis-resistant crypts may then spread through the flat colonic mucosa by crypt fission.Citation265 These various observations suggest that repeated exposure of the colon to high levels of bile acids as a result of a Western style diet cause Darwinian selection for cells resistant to induction of apoptosis, and give rise to fields of apoptosis-resistant epithelium that may further evolve to malignancy.
Work from our laboratory on biomarkers associated with colon cancer risk indicate that, in addition to loss of apoptosis competence, lectin staining using DBA (Dolichos biflorus agglutinin) (a measure of epithelial cell differentiation), cytochrome c oxidase subunit I (CcOI) (a mitochondrial DNA-encoded subunit of CcO) and Pms2 (a DNA repair protein) were all significantly decreased in the normal-appearing flat mucosa of patients with colon cancer compared to that of control subjects.Citation213,Citation241,Citation266
Scalmati and Lipkin Citation267 reviewed numerous studies on cell proliferation pattern in patients with colon adenomas or cancer. Expansion of the proliferative compartment (a lumenward displacement of the zone of active cell proliferation in the crypts) occurred along the entire colon of individuals, indicating that there was increased risk throughout the colon, no matter where the actual lesion was located. We also determined that apoptosis resistance in the nonneoplastic flat mucosa of patients with colon cancer was present in several regions of the colon far removed from the tumor site,Citation45 indicating a generalized field defect of apoptosis resistance, albeit “patchy” in nature,Citation213 throughout the colon.
A recent study indicated that the consumption of a Western style diet plays a crucial role in the progression of a field defect to colon cancer.Citation268 Among 1009 patients with resected stage III colon cancer, colon cancer recurred in 324 patients in a median of 5.3 years. This recurrence suggests the frequent presence of a field defect. The patients had all undergone fluorouracil-based adjuvant chemotherapy after surgery, and were evaluated for diet during and 6 months after adjuvant chemotherapy. The rate of cancer recurrence was almost threefold greater among patients in the highest quintile of Western style diet consumption compared to those in the lowest quintile of Western style diet consumption.
Prevention of bile acid-induced stresses and genomic instability: importance of butyrate
We have reviewed evidence that deoxycholate induces cellular stresses in epithelial cells of the GI tract and that bile acids play a role in colon carcinogenesis. Although hydrophobic bile acids were once believed to be mainly tumor promoters, the bile acid induction of DNA damage and aneuploidy indicates that they are also genotoxins relevant to the initiation and progression of the neoplastic process. It is well documented that phytosterols and other types of antioxidants, micronutrients and dietary fiber have antineoplastic effects. An overall discussion of dietary-related colon cancer chemopreventive agents is beyond the scope of this review. However, we will focus on the short-chain fatty acid, butyrate, since previous studies have shown that butyrate interferes with many deleterious effects of hydrophobic bile acids such as increased proliferation,Citation269–Citation272 and has been shown to have anti-genotoxic effects on deoxycholateCitation143 and H2O2-inducedCitation273,Citation274 genotoxicity in colon epithelial cells. Furthermore, butyrate affects many signal-transduction pathways and histological parameters associated with colon carcinogenesis.Citation272,Citation275–Citation286
Butyrate is a 4-carbon fatty acid that is an end product of bacterial fermentation of dietary fiber and occurs at concentrations of 2–10 mM in the colon.Citation287 Certain intestinal bacterial species (eg, Eubacterium rectale, Faecalibacterium prausnitzii, Roseburia, Coprococcus) are butyrate producersCitation288–Citation290 and beneficial to colon cancer prevention, whereas others, such as Enterococcus faecalis (extracellular superoxide and H2O2-producing bacterial speciesCitation291,Citation292) and Desulfovibrio (sulfate-reducing bacteria which produces hydrogen sulfide) are deleterious in that they cause DNA damage and chromosomal instability.Citation291–Citation295 In a recent study that quantified specific bacterial species using real-time PCR in the feces of healthy control volunteers and patients with colon cancer, it was found that the butyrate-producing bacteria species were significantly decreased and the superoxide-producing bacterial species were significantly increased in the feces of colon cancer patients compared to control subjects.Citation290 It is significant that the feces of patients with ulcerative colitis, a pre-neoplastic inflammatory condition of the colon, uniformly contain sulfate-reducing bacteria. Sulfide substantially increases the proliferative index of the crypts in mucosal biopsies, and this increase is prevented by the presence of butyrate.Citation296 A general consequence of the fermentation of fiber is to reduce the pH of the gut lumen, thereby affecting bacterial metabolism and competitionCitation297 and reducing the bacteria-mediated formation of hydrophobic bile acids.Citation298 These shifts in bacterial species may be important determinants of the risk of colon cancer.
It is significant that butyrate can protect against oxidative DNA damage induced by deoxycholate ex vivo in colono-cytes derived from human biopsies.Citation143 The processes in the colon that are affected by butyrate include a suppression of proliferation,Citation271,Citation277,Citation299 induction of apoptosis,Citation214,Citation215 induction of differentiation,Citation275,Citation276,Citation278 a reduction in inflammation,Citation280,Citation286 suppression of NF-κB activation,Citation300 increase in xenobiotic metabolizing enzymesCitation301,Citation302 and a decrease in DNA damage and genomic instability.Citation143,Citation273,Citation274 All of these processes affect colon carcinogenesis in a profound way.Citation280,Citation303
In our studies of field defects in the human colon, we observed that in the nonneoplastic flat mucosa of patients with colon cancer there is an increase in apoptosis resistance,Citation44,Citation45,Citation212,Citation213 loss of differentiation,Citation213 decrease in mitochondrial gene expression (eg, CcOI)Citation266 and a decrease in PMS2, a mismatch repair protein.Citation241 It is significant that butyrate has opposite effects. It can increase differentiation, induce apoptosis, increase CcOI levels,Citation276 and has more potent antineoplastic effects on colon cancer cells defective in mismatch repair.Citation304
Future directions
We have just begun to understand the role of hydrophobic bile acids in inducing DNA damage and genomic instability in colon epithelial cells, and potential strategies to reduce these deleterious effects. Some key areas that require further experimentation include: 1) the role of oxidative stress as mediators of the effect of bile acids on the reduction of DNA repair proteins (eg, Ku80, MSH3), tumor suppressors (eg, p53) and spindle checkpoint proteins (eg, Mad2); 2) the role of oxidative stress in the deoxycholate-induced induction of micronuclei and aberrant mitoses; 3) the ability of deoxycholate to induce aberrant mitoses and aneuploidy in vivo using mouse models; 4) the possible synergistic effect of bile acids and environmental agents (eg, nicotine from tobacco smoking, arsenic, food carcinogens) in inducing DNA damage and genomic instability; and 5) the effectiveness of dietary-related nutrients and co-factors (eg, zinc, niacin, selenium compounds, plant polyphenols, butyrate) in preventing the genotoxic effects of hydrophobic bile acids both in vitro using cell culture models and in vivo using mouse models. The results from these types of experiments will have relevance to other cancers of the GI tract (esophagus, stomach, liver, pancreas) in which hydrophobic bile acids have a potential etiologic role.
Summary
In this review, we have described the likely major mechanisms by which hydrophobic bile acids can induce stresses on cells, which can then result in colon carcinogenesis. Persistent exposure of colon epithelial cells to hydrophobic bile acids can result in the development of apoptosis resistance and the modulation of numerous genes/proteins associated with chromosome maintenance and mitosis. Hydrophobic bile acids also damage the genome through multiple mechanisms, including oxidative DNA damage, p53 and other mutations, micronuclei formation and aneuploidy, all major components of genomic instability. These findings link dietary-related factors to the development of genomic instability, extend our understanding of the underlying causes of colon cancer, and may open up new avenues for hypothesis-driven biomarker development to assess colon cancer risk.
Acknowledgements
This work was supported in part by NIH 5 R01 CA119087, Arizona Biomedical Research Commission Grant #0803, VA Merit Review Grant 0142 of the Southern Arizona Veterans Affairs Health Care System and Biomedical Diagnostics and Research, Inc., Tucson, Arizona, USA.
References
- BreivikJDon’t stop for repairs in a war zone: Darwinian evolution unites genes and environment in cancer developmentProc Natl Acad Sci U S A2001985379538111344279
- IsraelLTumor progression: Random mutations or an integrated survival response to cellular stress conserved from unicellular organisms?J Theoret Biol19961783753808733476
- BreivikJGaudernackGCarcinogenesis and natural selection: a new perspective to the genetics and epigenetics of colorectal cancerAdv Cancer Res19997618721210218102
- BreivikJGaudernackGGenomic instability, DNA methylation, and natural selection in colorectal carcinogenesisSemin Cancer Biol1999924525410448112
- CahillDPKinzlerKWVogelsteinBLengauerCGenetic instability and Darwinian selection in tumoursTrends Cell Biol19999M57M6010611684
- BreivikJThe evolutionary origin of genetic instability in cancer developmentSemin Cancer Biol200515516015613288
- GorringeKLChinS-FPharaoahPEvidence that both genetic instability and selection contribute to the accumulation of chromosome alterations in cancerCarcinogenesis20052692393015677628
- TsafrirDBacolodMSelvanayagamZRelationship of gene expression and chromosomal abnormalities in colorectal cancerCancer Res2006662129213716489013
- BreivikJCancer–evolution withinInt J Epidemiol2006351161116216990284
- VineisPBerwickMThe population dynamics of cancer: a Darwinian perspectiveInt J Epidemiol2006351151115916987840
- NarisawaTMagadiaNEWeisburgerJHWynderELPromoting effect of bile acids on colon carcinogenesis after intrarectal instillation of N-methyl-N’-nitro-N-nitrosoguanidine in ratsJ Natl Cancer Inst197453109310974427390
- ReddyBSNarasawaTWeisburgerJHWynderELPromoting effect of sodium deoxycholate on colon adenocarcinomas in germ-free ratsJ Natl Cancer Inst1976564414421255778
- ReddyBSWatanabeKWeisburgerJHWynderELPromoting effect of bile acids in colon carcinogenesis in germ-free and conventional F344 ratsCancer Res19773732383242884672
- McSherryCKCohenBIBokkenheuserVDMosbachEHWinterJEffects of calcium and bile acid feeding on colon tumors in the ratCancer Res198949603960432790818
- BernsteinHBernsteinCPayneCMDvorakovaKGarewalHBile acids as carcinogens in human gastrointestinal cancersMutat Res2005589476515652226
- JenkinsGJD’SouzaFRSuzenSHDeoxycholic acid at neutral and acid pH, is genotoxic to oesophageal cells through the induction of ROS: The potential role of anti-oxidants in Barrett’s oesophagusCarcinogenesis20072813614216905748
- JenkinsGJCroninJAlhamdaniAThe bile acid deoxycholic acid has a non-linear dose response for DNA damage and possibly NF-{kappa}B activation in oesophageal cells, with a mechanism of action involving ROSMutagenesis20082339940518515815
- BreivikJGaudernackGResolving the evolutionary paradox of genetic instability: a cost-benefit analysis of DNA repair in changing environmentsFEBS Lett200456371215063714
- LoebKRLoebLAGenetic instability and the mutator phenotype. Studies in ulcerative colitisAm J Pathol19991541621162610362784
- BielasJHLoebKRRubinBPTrueLDLoebLAHuman cancers express a mutator phenotypeProc Natl Acad Sci U S A2006103182381824217108085
- LoebLABielasJHBeckmanRACancers exhibit a mutator phenotype: clinical implicationsCancer Res20086835313557
- BignoldLPThe mutator phenotype theory can explain the complex morphology and behavior of cancersCell Mol Life Sci20025995095812169025
- BignoldLPThe mutator phenotype theory of carcinogenesis and the complex histopathology of tumours: support for the theory from the independent occurrence of nuclear abnormality, loss of specialisation and invasiveness among occasional neoplastic lesionsCell Mol Life Sci20036088389112827278
- BignoldLPVariation, “evolution”, immortality and genetic instabilities in tumour cellsCancer Lett200725315516917250959
- Abdel-RahmanWMKatsuraKRensWSpectral karyotyping suggests additional subsets of colorectal cancers by pattern of chromosome rearrangementProc Natl Acad Sci U S A2001982538254311226274
- MasramonLVendrellTrafaGGenetic instability and divergence of clonal populations in colon cancer cells in vitroJ Cell Sci20061191477148216551697
- HermsenMPostmaCBaakJColorectal adenoma to carcinoma progression follows multiple pathways of chromosomal instabilityGastroenterology20021231109111912360473
- RowanAHalfordSGaasenbeekMRefining molecular analysis in the pathways of colorectal carcinogenesisClin Gastroenterol Hepatol200531115112316271343
- RisquesR-AMorenoVMarcuelloERedefining the significance of aneuploidy in the prognostic assessment of colorectal cancerLab Invest20018130731511310824
- RisquesR-AMorenoVRibasMGenetic pathways and genome-wide determinants of clinical outcome in colorectal cancerCancer Res2003637206721414612515
- StolerDLChenNBasikMThe onset and extent of genomic instability in sporadic colorectal tumor progressionProc Natl Acad Sci U S A199996151211512610611348
- SchneiderBLKulesz-MartinMDestructive cycles: the role of genomic instability and adaptation in carcinogenesisCarcinogenesis2004252033204415180945
- GagosSIrminger-FingerIChromosome instability in neoplasia: chaotic roots to continuous growthInt J Biochem Cell Biol2005371014103315743675
- BignoldLPInitiation of genetic instability and tumour formation: a review and hypothesis of a nongenotoxic mechanismCell Mol Life Sci2003601107111712861379
- SieberOMHeinimannKTomlinsonIPMGenomic instability – the engine of tumorigenesis?Nature Rev20033701708
- HanksSColemanKReidSConstitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1BNat Genet2004361159116115475955
- WooRAPoonRYCGene mutations and aneuploidy. The instability that causes cancerCell Cycle200431101110315326384
- LiRSonikAStindlRRasnickDDuesbergPAneuploidy vs gene mutation hypothesis of cancer: Recent study claims mutation but is found to support aneuploidyProc Natl Acad Sci U S A2000973236324110725343
- RajagopalanHNowakMAVogelsteinBLengauerCThe significance of unstable chromosomes in colorectal cancerNature Rev20033695701
- SlaneBGAykin-BurnsNSmithBJMutation of succinate dehydrogenase subunit C results in increased O2.−, oxidative stress and genomic instabilityCancer Res2006667615762016885361
- HolubecHPayneCMBernsteinHAssessment of apoptosis by immunohistochemical markers compared to cellular morphology in ex vivo-stressed colonic mucosaJ Histochem Cytochem20055322923515684335
- DuesbergPLiRMultistep carcinogenesis. A chain reaction of aneuploidizationsCell Cycle2003220221012734426
- PihanGDoxseySMutations and aneuploidy: Co-conspirators in cancer?Cancer Cell20034899412957283
- PayneCMBernsteinHBernsteinCGarewalHRole of apoptosis in biology and pathology: resistance to apoptosis in colon carcinogenesisUltrastruct Pathol1995192212487571081
- BernsteinCBernsteinHGarewalHA bile acid-induced apoptosis assay for colon cancer risk and associated quality control studiesCancer Res19995923535710344743
- FearonERVogelsteinBA genetic model for colorectal tumorigenesisCell1990617597672188735
- KinzlerKWVogelsteinBLessons from hereditary colorectal cancerCell1996871591708861899
- LengauerCKinzlerKWVogelsteinBGenetic instabilities in human cancersNature19983966436499872311
- GeiglJBObenaufACSchwarzbraunTDefining ‘chromosomal instability.’Trends Genet200824646918192061
- RajagopalanHLengauerCAneuploidy and cancerNature20044323384115549096
- RibasMMasramonLAizaGCapellaGMiroRPeinadoMAThe structural nature of chromosomal instability in colon cancer cellsFASEB J20031728929112475895
- ParaskevaCCorfieldAPHarperSHagueAAudcentKWilliamsACColorectal carcinogenesis: sequential steps in the in vitro immortalization and transformation of human colonic epithelial cells (review)Anticancer Res199010118912002241098
- GriffioenGCornelisseCJVerspagetHWAssociation of aneuploidy in index adenomas with metachronous colorectal adenoma development and a comparisonCancer199270203520431394033
- BommeLBardiGPandisNFengerCKronbergOHeimSCytogenetic analysis of colorectal adenomas: Karyotypic comparisons of synchronous tumorsCancer Genet Cytogenet199810666719772912
- BommeLLotheRABardiGFengerCKronbergOHeimSAssessments of clonal composition of colorectal adenomas by FISH analysis of chromosomes 1, 7, 13, 20Int J Cancer20019281682311351301
- ShihIMZhouWGoodmanSNLengauerCKinzlerKWVogelsteinBEvidence that genetic instability occurs at an early stage of colorectal tumorigenesisCancer Res2001618181211221861
- NowakMAKomarovaNLSenguptaAThe role of chromosomal instability in tumor initiationProc Natl Acad Sci U S A200299162261623112446840
- HeQJZengWFShamJSRecurrent genetic alterations in 26 colorectal carcinomas and 21 adenomas from Chinese patientsCancer Genet Cytogenet200314411211812850373
- RichterHSlezakPWalchADistinct chromosomal imbalances in nonpolypoid and polypoid colorectal adenomas indicate different genetic pathways in the development of colorectal neoplasmsAmer J Pathol200316328729412819033
- SugaiTTakahashiHHabanWAnalysis of genetic alterations, classified according to their DNA ploidy pattern, in the progression of colorectal adenomas and early colorectal carcinomasJ Pathol200320016817612754737
- GiarettiWMolinuSCeccarelliJPrevostoCChromosomal instability, aneuploidy, and gene mutations in human sporadic colorectal adenomasCell Oncol20042630130515623940
- PostmaCHermsenMAJACoffaJChromosomal instability in flat adenomas and carcinomas of the colonJ Pathol200520551452115685687
- GradeMBeckerHLierschTRiedTGhadimiBMMolecular cytogenetics: Genomic imbalances in colorectal cancer and their clinical impactCellular Oncol2006287184
- IshiguroKYoshidaTYagishitaHNumataYOkayasuTEpithelial and stromal genetic instability contributes to genesis of colorectal adenomasGut20065569570216354798
- MartinESTononGSinhaRCommon and distinct genomic events in sporadic colorectal cancer and diverse cancer typesCancer Res200767107361074318006816
- OlivaMRRipoliFMunizPGenetic alterations and oxidative metabolism in sporadic colorectal tumors from a Spanish communityMol Carcinogen199718232243
- BlakerHGrafMRiekerRJOttoHFComparison of losses of heterozygosity and replication errors in primary colorectal carcinomas and corresponding liver metastasesJ Pathol199918825826210419592
- ThiagalingamSKakenSWillsonJKVMechanisms underlying losses of heterozygosity in human colorectal cancersProc Natl Acad Sci U S A2001982698270211226302
- RasnickDDuesbergPHHow aneuploidy affects metabolic control and causes cancerBiochem J199934062163010359645
- BirdRPObservation and quantification of aberrant crypts in the murine colon treated with a colon carcinogen: preliminary findingsCancer Lett1987371471513677050
- PretlowTPBarrowBJAshtonWSAberrant crypts: putative preneoplastic foci in human colonic mucosaCancer Res199151156415671997197
- AugenlichtLHRichardsCCornerGPretlowTPEvidence for genomic instability in human colonic aberrant crypt fociOncogene199612176717728622897
- SiuIMRobinsonDRSchwartzSThe identification of monoclonality in human aberrant crypt fociCancer Res5963669892186
- LuoLLiBPretlowTPDNA alterations in human aberrant crypt foci and colon cancers by random primed polymerase chain reactionCancer Res2003636166616914559798
- PretlowTPPretlowTGMutant KRAS in aberrant crypt foci (ACF): Initiation of colorectal cancer?Biochim Biophys Acta20051756839616219426
- LuoLShenGQStifflerKAWangQKPretlowTGPretlowTPLoss of heterozygosity in human aberrant crypt foci (ACF), a putative precursor of colon cancerCarcinogenesis2006271153115916474178
- NambiarPRNakanishiMGuptaRGenetic signatures of high- and low-risk aberrant crypt foci in a mouse model of sporadic colon cancerCancer Res2004646394640115374946
- SteinbeckRG 1997Imbalance of morphologically addressed telophases reflects interphase DNA aneuploidy in tumorigenesisEur J Histochem1997412432549491310
- SteinbeckRGThe DNA content of chromosome division figures and interphase nuclei classifies ulcerative colitisEur J Cancer1998341751819624254
- RabinovitchPSDziadonSBrentnallTAPancolonic chromosomal instability precedes dysplasia and cancer in ulcerative colitisCancer Res1999595148515310537290
- WillenbucherRFAustDEChangCGGenomic instability is an early event during the progression pathway of ulcerative-colitis-related neoplasiaAmer J Pathol19991541825183010362807
- TakayamaTMiyanishiKHayashiTAberrant crypt foci: detection, gene abnormalities, and clinical usefulnessClin Gastroenterol Hepatol200537 Suppl 1S42S4516012995
- VyvodaOSColemanRHoldsworthGEffects of different bile salts upon the composition and morphology of a liver plasma membrane preparation. Deoxycholate is more membrane damaging than cholate and its conjugatesBiochim Biophys Acta19774656876836833
- SugimotoYSaitoHTabetaRBinding of bile acids with rat colon and resultant perturbation of membrane organization as studied by uptake measurement and 31P nuclear magnetic resonance spectroscopyGann1984757988086500237
- AkareSMartinezJDBile acid induces hydrophobicity-dependent membrane alterationsBiochim Biophys Acta20051735596715951237
- Jean-LouisSAkareSAliMAMashEAJrMeuilletEMartinezJDDeoxycholic acid induces intracellular signaling through membrane perturbationsJ Biol Chem2006281149481496016547009
- DeRubertisFRCravenPASaitoRBile salt stimulation of colonic epithelial proliferation. Evidence for involvement of lipoxygenase productsJ Clin Invest198474161416246438153
- ArakiYKatohTOgawaABile acid modulates transepithelial permeability via the generation of reactive oxygen species in the Caco-2 cell lineFree Radic Biol Med20053976978016109307
- QiaoLStuderELeachKDeoxycholic acid (DCA) causes ligand-independent activation of epidermal growth factor receptor (EGFR) and Fas receptor in primary hepatocytes: inhibition of EGFR/mitogen-activated protein kinase-signaling module enhances DCA-induced apoptosisMol Biol Cell2001122629264511553704
- BoothLABiltonRFGenotoxic potential of the secondary bile acids: a role for reactive oxygen species, In: Arouma OI, Halliwell B (editors)DNA and Free Radicals: Techniques, Mechanisms and ApplicationsLondonOICA International1998161177
- WerneburgNWYoonJHHiguchiHGoresGJBile acids activate EGF receptor via a TGF-alpha-dependent mechanism in human cholangiocyte cell linesAmer J Physiol Gastrointest Liver Physiol2003285G31G3612606307
- MerchantNBRogersCMTrivediBMorrowJCoffeyRJLigand-dependent activation of the epidermal growth factor receptor by secondary bile acids in polarizing colon cancer cellsSurgery200513841542116213893
- YasudaHHirataSInoueKMashimaHOhnishiHYoshibaMInvolvement of membrane-type bile acid receptor M-BAR/TGR5 in bile acid-induced activation of epidermal growth factor receptor and mitogen-activated protein kinases in gastric carcinoma cellsBiochem Biophys Res Comm200735415415917214962
- RaimondiFSantoroPBaroneMVBile acids modulate tight junction structure and barrier function of Caco-2 monolayers via EGFR activationAmer J Physiol Gastrointest Liver Physiol2008294G906G91318239063
- NakanishiHTakeyamaYOhyanagiHSaitohYTakaiYMode of stimulatory action of deoxycholate in signal transduction system of isolated rat pancreatic aciniBiochem Biophys Res Comm19901701111181695504
- ReinehrRBeckerSKeitelVEberleAGrether-BeckSHaussingerDBile salt-induced apoptosis involves NADPH oxidase isoform activationGastroenterol2005129200931
- PayneCMWeberCCrowley-SkillicornCDeoxycholate induces mitochondrial oxidative stress and activates NF-κB through multiple mechanisms in HCT-116 colon epithelial cellsCarcinogenesis20072821522216887864
- Crowley-WeberCLPayneCMGleason-GuzmanMDevelopment and molecular characterization of HCT-116 cell lines resistant to the tumor promoter and multiple stress-inducer, deoxycholateCarcinogenesis2002232063208012507930
- PayneCMCrowleyCWasho-StultzDThe stress-response proteins poly(ADP-ribose) polymerase and NF-κB protect against bile salt-induced apoptosisCell Death Differ199856233610200517
- PayneCMFassRBernsteinHPathogenesis of diarrhea in the adult: diagnostic challenges and life-threatening conditionsEur J Gastroenterol Hepatol2006181047105116957509
- MoschettaAPortincasaPDebellisLBasolateral Ca2+-dependent K+-channels play a key role in Cl− secretion induced by taurodeoxy-cholate from colon mucosaBiol Cell20039511512212799067
- MauricioACSlawikMHeitzmannDDeoxycholic acid (DOC) affects the transport properties of distal colonPflugers Arch200043953254010764211
- OelbergDGWangLBSackmanJWAdcockEWLesterRDubinskyWPBile salt-induced calcium fluxes in artificial phospholipid vesiclesBiochim Biophys Acta19889372892992827771
- ZimniakPLittleJMRadominskaAOelbergDGAnwerMSLesterRTaurine-conjugated bile acids act as Ca2+ ionophoresBiochemistry199130859886041832296
- TapiaRVelascoIRuthenium red as a tool to study calcium channels, neuronal death and the function of neural pathwaysNeurochem Int1997301371479017661
- CravenPAPfanstielJDeRubertisFRRole of reactive oxygen in bile salt stimulation of colonic epithelial proliferationJ Clin Invest1986778508593005368
- CasellasFMourelleMPapoMBile acid induced colonic irritation stimulates intracolonic nitric oxide release in humansGut1996387197238707118
- VenturiMHamblyRJGlinghammerBRafterJJRowlandIRGenotoxic activity in human faecal water and the role of bile acids: a study using the alkaline comet assayCarcinogenesis199718235323599450481
- BoothLAGilmoreITBiltonRFSecondary bile acid induced DNA damage in HT29 cells: are free radicals involved?Free Rad Res199726135144
- Washo-StultzDCrowley-WeberCLDvorakovaKRole of mitochondrial complexes I and II, reactive oxygen species and arachidonic acid metabolism in deoxycholate-induced apoptosisCancer Lett200217712914411825660
- LechnerSMuller-LadnerUSchlottmannKBile acids mimic oxidative stress induced upregulation ot thioredoxin reductase in colon cancer cell linesCarcinogenesis2002231281128812151345
- Washo-StultzDHoglenNBernsteinHBernsteinCPayneCMRole of nitric oxide and peroxynitrite in bile salt-induced apoptosis: Relevance to colon carcinogenesisNutr Cancer19993318018810693173
- Crowley-WeberCLDvorakovaKCrowleyCNicotine increases oxidative stress, activates NF-κB and GRP78, induces apoptosis and sensitizes cells to genotoxic/xenobiotic stresses by a multiple stress inducer, deoxycholate: relevance to colon carcinogenesisChemico-Biological Interact20031455366
- Dall’AgnolMBernsteinCBernsteinHGarewalHPayneCMIdentification of S-nitrosylated proteins after chronic exposure of colonic epithelial cells to deoxycholateProteomics200661654166216404723
- BernsteinHHolubecHBernsteinCUnique dietary-related mouse model of colitisInflamm Bowel Dis2006122789316633050
- BernsteinHHolubecHBernsteinCDeoxycholate-induced colitis is markedly attenuated in Nos2 knock-out mice in association with modulation of gene expression profilesDig Dis Sci20075262864217253130
- FangYHanSIMitchellCBile acids induce mitochondrial ROS, which promote activation of receptor tyrosine kinases and signaling pathways in rat hepatocytesHepatology20044096197115382121
- GlinghammarBInoueHRafterJJDeoxycholic acid causes DNA damage in colonic cells with subsequent induction of caspases, COX-2 promoter activity and transcription factors NF-κB and AP-1Carcinogenesis20022383984512016158
- MuhlbauerMAllardBBosserhoffAKDifferential effects of deoxycholic acid and taurodeoxycholic acid on NF-κB signal transduction and IL-8 gene expression in colonic epithelial cellsAm J Physiol Gastrointest Liver Physiol2004286G1000G100814726307
- LeeDKParkSYBaikSKDeoxycholic acid-induced signal transduction in HT-29 cells: role of NF-kappa B and interleukin 8Korean J Gastroenterol20044317618515034288
- ShahSAVolkovYArfinQAbdel-LatifMMKelleherDUrso-deoxycholic acid inhibits interleukin beta 1 and deoxycholic acid-induced activation of NF-kappaB and AP-1 in human colon cancer cellsInt J Cancer200611853253916106402
- ShahSAMahmudNMftahMRocheHMKelleherDChronic but not acute linoleic acid treatment inhibits deoxycholic acid-induced protein kinase C and nuclear factor-kappaB activation in human colorectal cancer cellsEur J Cancer Prev20061512513316523009
- FangCDeanJSmithJWA novel variant of ileal bile acid binding protein is up-regulated through nuclear factor-kappaB activation in colorectal adenocarcinomaCancer Res2007679039904617909007
- RaufmanJPShantJGuoCYRoySChengKDeoxycholyltaurine rescues human colon cancer cells from apoptosis by activating EGFR-dependent PI3K/Akt signalingJ Cell Physiol200821553854918064605
- BernsteinCBernsteinHPayneCMGarewalHDNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesisMutat Res200251114517812052432
- ZhangSHemmerichPGrosseFCentrosomal localization of DNA damage checkpoint proteinsJ Cell Biochem200710145146517171639
- SankaranSCroneDEPalazzoREParvinJDAurora-A kinase regulates breast cancer – associated gene 1 inhibition of centrosome-dependent microtubule nucleationCancer Res200767111861119418056443
- BarzilaiAYamamotoK-IDNA damage responses to oxidative stressDNA Repair200431109111515279799
- LangieSAKnaapenAMHoubenJMThe role of glutathione in the regulation of nucleotide excision repair during oxidative stressToxicol Lett200716830230917207589
- FengZHuWMarnettLJTangMSMalondialdehyde, a major endogenous lipid peroxidation product, sensitizes human cells to UV- and BPDE-induced killing and mutagenesis through inhibition of nucleotide excision repairMutat Res200660112513616872641
- ChangCLMarraGChauhanDPOxidative stress inactivates the human DNA mismatch repair systemAm J Physiol Cell Physiol2002283C148C15412055083
- HillJWHuJJEvansMKOGG1 is degraded by calpain following oxidative stress and cisplatin exposureDNA Repair (Amst)2008764865418294929
- BravardAVacherMGougetBRedox regulation of human OGG1 activity in response to cellular oxidative stressMol Cell Biol2006267430743616923968
- GraziewiczMADayBJCopelandWCThe mitochondrial DNA polymerase as a target of oxidative damageNucleic Acids Res2002302817282412087165
- GatzSAWiesmüllerLp53 in recombination and repairCell Death Differ2006131003101616543940
- QiaoDGaitondeSVQiWMartinezJDDeoxycholic acid suppresses p53 by stimulating proteasome-mediated p53 protein degradationCarcinogenesis20012295796411375905
- RomagnoloDFChirnomasRBKuJDeoxycholate, an endogenous tumor promoter and DNA damaging agent, modulates BRCA-1 expression in apoptosis-sensitive epithelial cells: loss of BRCA-1 expression in colonic adenocarcinomasNutr Cancer200346829212925308
- BernsteinHPayneCMKunkeKA proteomic study of resistance to deoxycholate-induced apoptosisCarcinogenesis20042568169214729586
- KomichiDTazumaSNishiokaTHyogoHChayamaKGlyco-chenodeoxycholate plays a carcinogenic role in immortalized mouse cholangiocytes via oxidative DNA damageFree Rad Biol Med2005391418142716274877
- KulkarniMSYieldingKLDNA damage and repair in epithelial (mucous) cells and crypt cells from isolated colonChem Biol Interact1985523113183971470
- Pool-ZobelBLLeuchtUInduction of DNA damage by risk factors of colon cancer in human colon cells derived from biopsiesMutat Res19973751051159202721
- ScottDWMutambaSHopkinsRGLooGIncreased GADD gene expression in human colon epithelial cells exposed to deoxycholateJ Cell Physiol200520229530315316935
- RosignoliPFabianiRDe BartolomeoAFuccelliRPelliMAMorozziGGenotoxic effect of bile acids on human normal and tumour colon cells and protection by dietary antioxidants and butyrateEur J Nutr20084730130918685914
- MalangaMAlthausFRThe role of poly(ADP-ribose) in the DNA damage signaling networkBiochem Cell Biol20058335436415959561
- BurmaSChenBPMurphyMKurimasaAChenDJATM phosphorylates histone H2AX in response to DNA double-strand breaksJ Biol Chem2001276424624246711571274
- TheisenJPetersJHFeinMThe mutagenic potential of duodenoesophageal refluxAnn Surg2005241636815621992
- FeinMPetersJHDeMeesterTR 2007Carcinogenesis in reflux disease – in search for bile-specific effectsMicrosurgery20072764765017929260
- NaraharaHTatsutaMIishiHK-ras point mutation is associated with enhancement by deoxycholic acid of colon carcinogenesis induced by azoxymethane, but not with its attenuation by all-trans-retinoic acidInt J Cancer20008815716111004662
- WatabeJBernsteinHThe mutagenicity of bile acids using a fluctuation testMutat Res198515845512995801
- AssinderSJUpshallAMitotic aneuploidy induced by sodium deoxycholate in Aspergillus nidulansMutat Res1982931011087038466
- FergusonLRParryJMMitotic aneuploidy as a possible mechanism for tumour promoting activity in bile acidsCarcinogenesis198454474516368034
- AlbertiniSFriederichUWurglerFEInduction of mitotic chromosome loss in the diploid yeast Saccharomyces cerevisiae D61.M by genotoxic carcinogens and tumor promotersEnviron Mol Mutagen1988114975083286249
- FenechMCrottJTurnerJBrownSNecrosis, apoptosis, cytostasis and DNA damage in human lymphocytes measured simultaneously within the cytokinesis-block micronucleus assay: description of the method and results for hydrogen peroxideMutagenesis19991460561210567036
- FenechMBonassiSTurnerJIntra- and inter-laboratory variation in the scoring of micronuclei and nucleoplasmic bridges in binu-cleated human lymphocytes. Results of an international slide-scoring exercise by the HUMN projectMutat Res2003534456412504754
- HuntCRSimJESullivanSJGenomic instability and catalase gene amplification induced by chronic exposure to oxidative stressCancer Res199858398639929731512
- LimoliCLGiedzinskiEInduction of chromosomal instability by chronic oxidative stressNeoplasia2003533934614511405
- SamperENicholisDGMelovSMitochondrial oxidative stress causes chromosomal instability of mouse embryonic fibroblastsAging Cell2003227728514570235
- D’AngiolellaVSantarpiaCGriecoDOxidative stress overrides the spindle checkpointCell Cycle2007657657917351333
- FangLFangGCentromere cohesion: regulating the guardianCell Res20071766466517694095
- BarberTDMcManusKYuenKWChromatid cohesion defects may underlie chromosome instability in human colorectal cancersProc Natl Acad Sci U S A20081053443344818299561
- GreenRAKaplanKBChromosome instability in colorectal tumor cells is associated with defects in microtubule plus-end attachments caused by a dominant mutation in APCJ Cell Biol200316394996114662741
- TomonagaTNomuraFChromosome instability and kinetochore dysfunctionHistol Histopathol20072219119717149692
- DashBCEl-DeiryWSCell cycle checkpoint control mechanisms that can be disrupted in cancerMethods Mol Biol20042809916115187251
- KopsGJPLWeaverBAAClevelandDWOn the road to cancer:aneuploidy and the mitotic checkpointNature Rev20055773785
- Perez de CastroIde CarcerGMalumbresMA census of mitotic cancer genes:new insights into tumor cell biology and cancer therapyCarcinogenesis20072889991217259655
- WangXCheungHWChunACJinDYWongYCMitotic checkpoint defects in human cancers and their implications to chemotherapyFront Biosci2008132103211417981695
- RudolphKLMillardMBosenbergMWDePinhoRATelomere dysfunction and evolution of intestinal carcinoma in mice and humansNat Genet20012815515911381263
- PlentzRRWiemannSUFlemmingPTelomere shortening of epithelial cells characterises the adenomacarcinoma transition of human colorectal cancerGut2003521304130712912862
- NiggEAOrigins and consequences of centrosome aberrations in human cancersInt J Cancer20061192717272317016823
- BourkeEDodsonHMerdesADNA damage induces Chk1-dependent centrosome amplificationEMBO Rep2007860360917468739
- Burum-AuensenEDeAngelisPMSchjolbergARRoislienJAndersenSNClausenOPFSpindle proteins Aurora A and BUB1B, but not Mad2, are aberrantly expressed in dysplastic mucosa of patients with longstanding ulcerative colitisJ Clin Pathol2007601403140817322345
- Burum-AuensenEDe AngelisPMSchjolbergARKravikKLAureMClausenOPFSubcellular localization of the spindle proteins aurora A, Mad2, and BUBR1 assessed by immunohistochemistryJ Histochem Cytochem20075547748617242465
- Burum-AuensenEDe AngelisPMSchjolbergARRoislienJMjalandOClausenOPFReduced level of the spindle checkpoint protein BUB1B is associated with aneuploidy in colorectal cancersCell Prolif20084164565918699967
- BarberTDMcManusKYuenKWYChromatid cohesion defects may underlie chromosome instability in human colorectal cancersProc Natl Acad Sci U S A20081053443344818299561
- KangHCKimI-JJangS-GCoding region polymorphisms in the CHFR mitotic stress checkpoint gene are associated with colorectal cancer riskCancer Lett200826017017918079053
- SpitzDRMackeyMALiGCElwellJHMcCormickMLOberleyLWRelationship between changes in ploidy and stable cellular resistance to hydrogen peroxideJ Cell Physiol19891395925982738104
- TarinJJVendrellFJTenJBlanesRVan BlerkomJCanoAThe oxidizing agent tertiary butyl hydroperoxide induces disturbances in spindle organization, c-meiosis, and aneuploidy in mouse oocytesMol Human Reprod19962895901
- KarlhuberGMBauerHCEcklPMCytotoxic and genotoxic effects of 4-hydroxynonenal in cerebral endothelial cellsMutat Res19973812092169434877
- TarinJJVendrellFJTenJCanoAAntioxidant therapy counteracts the disturbing effects of diamide and maternal ageing on meiotic division and chromosomal segregation in mouse oocytesMol Human Reprod19984281288
- HagstromKAMeyerBJCondensin and cohesin: More than chromosome compactor and glueNature Reviews Genetics20034520534
- GertonJChromosome cohesion: A cycle of holding together and falling apartPloS Biol20053371374
- KimDSFranklynJASmithVESecurin induces genetic instability in colrectal cancer by inhibiting double-stranded DNA repair activityCarcinogenesis20072874975917071631
- EytanEBraunsteinIGanothDTwo different mitotic checkpoint inhibitors of the anaphase-promoting complex/cyclosome antagonize the action of the activator Cdc20Proc Natl Acad Sci U S A20081059181918518591651
- WangXCheungHWChunACJinDYWongYCMitotic checkpoint defects in human cancers and their implications to chemotherapyFront Biosci2008132103211417981695
- EmanueleMJLanWJwaMAurora B kinase and protein phos-phatase 1 have opposing roles in modulating kinetochore assemblyJ Cell Biol200818124125418426974
- NasmythKPetersJMUhlmannFSplitting the chromosome: cutting the ties that bind sister chromatidsScience20002881379138510827941
- ChangTSJeongWLeeDYChoCSRheeSGThe RING-H2-finger protein APC11 as a target of hydrogen peroxideFree Radic Biol Med20043752153015256223
- AriyoshiKSuzukiKGotoMWatanabeMKodamaSIncreased chromosome instability and accumulation of DNA double-strand breaks in Werner syndrome cellsJ Radiat Res20074821923117449919
- ZhengZYBernsteinHBernsteinCPayneCMMartinezJDGernerEWBile acid activation of the gadd153 promoter and of p53-independent apoptosis: relevance to colon cancerCell Death Differ1996340741417180111
- BernsteinHPayneCBernsteinCBeardSSchneiderJCrowleyCActivation of the promoters of genes associated with DNA damage, oxidative stress, ER stress and protein malfolding by the bile salt, deoxycholateToxicol Lett1999108374610472808
- McCulloughKDMartindaleJLKlotzLOAwTYHolbrookNJGadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox stateMol Cell Biol2001211249125911158311
- LiXDingXAdrianTEArsenic trioxide causes redistribution of cell cycle, caspase activation, and GADD expression in human colonic, breast, and pancreatic cancer cellsCancer Invest20042238940015493360
- LiJHolbrookNJElevated gadd153/chop expression and enhanced c-Jun N-terminal protein kinase activation sensitizes aged cells to ER stressExp Gerontol20043973574415130668
- OyadomariSMoriMRoles of CHOP/GADD153 in endoplasmic reticulum stressCell Death Differ20041138138914685163
- GotohTMoriMNitric oxide and endoplasmic reticulum stressArterioscler Thromb Vasc Biol2006261439144616645155
- TamakiNHatanoETauraKCHOP deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injuryAm J Physiol Gastrointest Liver Physiol2008294G498G50518174271
- TagawaYHiramatsuNKasaiAHayakawaKOkamuraMInduction of apoptosis by cigarette smoke via ROS-dependent endoplasmic reticulum stress and CCAAT/enhancer-binding protein-homologous protein (CHOP)Free Radic Biol Med200845505918394432
- WuYZhangHDongYParkYMIpCEndoplasmic reticulum stress signal mediators are targets of selenium actionCancer Res2005659073907916204082
- PayneCMCrowley-WeberCLDvorakovaKMitochondrial perturbation attenuates bile acid-induced cytotoxicityCell Biol Toxicol20052121523116323058
- GerasimenkoJVFlowerdewSEVoroninaSGBile acids induce Ca2+ release from both the endoplasmic reticulum and acidic intracellular calcium stores through activation of inositol triphosphate receptors and ryanodine receptorsJ Biol Chem2006281401544016317074764
- KawaharaKOyadomariSGotohTKohsakaSNakayamaHMoriMInduction of CHOP and apoptosis by nitric oxide in p53-deficient microglial cellsFEBS Lett200150613513911591387
- ArringtonDDSchnellmannRGTargeting of the molecular chaperone oxygen-regulated protein 150 (ORP150) to mitochondria and its induction by cellular stressAm J Physiol Cell Physiol2008294C641C65018094145
- KassGEPriceSCRole of mitochondria in drug-induced cholestatic injuryClin Liver Dis200812275118242496
- RoloAPOliveiraPJMorenoAJPalmeiraCMChenodeoxycholate induction of mitochondrial permeability transition pore is associated with increased membrane fluidity and cytochrome c release: protective role of carvedilolMitochondrion2003230531116120330
- WachsFPKriegRCRodriguesCMPBile salt-induced apoptosis in human colon cancer cell lines involves the mitochondrial transmembrane potential but not CD95 (Fas/Apo-1) receptorInt J Colorectal Dis20052010311315365741
- LemastersJJModulation of mitochondrial membrane permeability in pathogenesis, autophagy and control of metabolismJ Gastroenterol Hepatol2007Suppl 1S31S3717567461
- SchmuckerDLOhtaMKanaiSSatoYKitaniKHepatic injury induced by bile salts: correlation between biochemical and morphological eventsHepatology199012121612212227821
- GumprichtEDevereauxMWDahlRHSokolRJGlutathione status of isolated rat hepatocytes affects bile acid-induced cellular necrosis but not apoptosisToxicol Appl Pharmacol200016410211110739750
- RoloAPPalmeiraCMHolyJMWallaceKBRole of mitochondrial dysfunction in combined bile acid-induced cytotoxicity: the switch between apoptosis and necrosisToxicol Sci20047919620414976352
- BorgognoneMPerezLMBasiglioCLOchoaJERomaMGSignaling modulation of bile salt-induced necrosis in isolated rat hepatocytesToxicol Sci20058311412515496497
- KobakGEDahlRDevereauxMWIncreased susceptibility of fat-laden Zucker-rat hepatocytes to bile acid-induced oncotic necrosis: an in vitro model of steatocholestasisJ Lab Clin Med200514524726215902097
- GarewalHBernsteinHBernsteinCSamplinerRPayneCReduced bile acid-induced apoptosis in “normal” colorectal mucosa: a potential biological marker for cancer riskCancer Res199656148014838603388
- BernsteinHHolubecHWarnekeJAPatchy field defects of apoptosis resistance and dedifferentiation in flat mucosa of colon resections from colon cancer patientsAnn Surg Oncol2002950551712052764
- HagueAElderDJEHicksDJParaskevaCApoptosis in colorectal tumour cells: Induction by the short chain fatty acids butyrate, propionate and acetate and by the bile salt deoxycholateInt J Cancer1995604004067829251
- MarchettiMCMiglioratiGMoracaRPossible mechanisms involved in apoptosis of colon tumor cell lines induced by deoxycholic acid, short-chain fatty acids and their mixturesNutr Cancer19972874809200153
- MilovicVTellerICFaustDCasparyWFSteinJEffects of deoxycholate on human colon cancer cells: apoptosis or proliferationEur J Clin Invest200232293411851724
- MartinezJDStratagoulesEDLaRueJMDifferent bile acids exhibit distinct biological effects: The tumor promoter deoxycholic acid induces apoptosis and the chemopreventive agent ursodeoxycholic acid inhibits cell proliferationNutr Cancer1998311111189770722
- YuiSSaekiTKanamotoRIwamiKCharacteristics of apoptosis in HCT116 colon cancer cells induced by deoxycholic acidJ Biochem (Tokyo)200513815115716091589
- CrowleyCLPayneCMBernsteinHBernsteinCRoeDThe NAD+ precursors, nicotinic acid and nicotinamide protect cells against apoptosis induced by a multiple stress inducer, deoxycholateCell Death Diff20007314326
- Washo-StultzDCrowleyCPayneCMIncreased susceptibility of cells to inducible apoptosis during growth from early to late log phase: an important caveat for in vitro apoptosis researchToxicol Lett200011619920710996481
- HazaAIGlinghammarBGrandienARafterJEffect of colonic luminal components on induction of apoptosis in human colonic cell linesNutr Cancer200036798910798220
- SchlottmannKWachsFPKriegRCKullmannFScholmerichJRogierG 2000Characterization of bile salt-induced apoptosis in colon cancer cell linesCancer Res2000604270427610945641
- PowolnyAXuJLooG 2001Deoxycholate induces DNA damage and apoptosis in human colon epithelial cells expressing either mutant or wild-type p53Int J Biochem Cell Biol20013319320311240376
- PayneCMWaltmireCNCrowleyCCaspase-6 mediated cleavage of guanylate cyclase alpha 1 during deoxycholate-induced apoptosis: Protective role of nitric oxide signaling moduleCell Biol Toxicol20031937339215015762
- KrahenbuhlSTalosCFischerSReichenJ 1994Toxicity of bile acids on the electron transport chain of isolated rat liver mitochondriaHepatology1994194714797904981
- RoloAPOliveiraPJMorenoAJPalmeiraCMBile acids affect liver mitochondrial bioenergetics: possible relevance for cholestasis therapyToxicol Sci20005717718510966524
- PalmeiraCMRoloAPMitochondrially-mediated toxicity of bile acidsToxicology200420311515363577
- SolaSBritoMABritesDMouraJJRodriguesCMMembrane structural changes support the involvement of mitochondria in the bile salt-induced apoptosis of rat hepatocytesClin Sci (Lond)200210347548512401120
- SokolRJDahlRDevereauxMWYerushalmiBKobakGEGumprichtEHuman hepatic mitochondria generate reactive oxygen species and undergo the permeability transition in response to hydrophobic bile acidsJ Pediatr Gastroenterol Nutr20054123524316056106
- TanKPYangMItoSActivation of nuclear factor (erythroid-2 like) factor 2, by toxic bile acids provokes adaptive defense responses to enhance cell survival at the emergence of oxidative stressMol Pharmacol2007721380139017724089
- PayneCMBernsteinCBernsteinHApoptosis overview emphasizing the role of oxidative stress, DNA damage and signal-transduction pathwaysLeuk Lymphoma19951943938574171
- LongpreJMLooGParadoxical effect of diphenyleneiodonium in inducing DNA damage and apoptosisFree Radic Res20084253354318569011
- RokutanKKawaharaTKuwanoYTominagaKSekiyamaATeshima-KondoSNADPH oxidases in the gastrointestinal tract: a potential role of Nox1 in innate immune response and carcinogenesisAntioxid Redox Signal2006815738216987012
- KomatsuDKatoMNakayamaJMiyagawaSKamataTNADPH oxidase 1 plays a critical mediating role in oncogenic Ras-induced vascular endothelial growth factor expressionOncogene2008274724473218454179
- LaurentEMcCoyJW3rdMacinaRANox1 is over-expressed in human colon cancers and correlates with activating mutations in K-RasInt J Cancer200812310010718398843
- NisimotoYTsubouchiRDieboldBAActivation of NADPH oxidase 1 in tumor colon epithelial cellBiochem J2008415576518518859
- CoccoTDi PaolaMPapaSLorussoMArachidonic acid interaction with the mitochondrial electron transport chain promotes reactive oxygen species generationFree Radic Biol Med199927515910443919
- BeckerSReinehrRGrether-BeckSEberleAHaussingerDHydrophobic bile salts trigger ceramide formation through endosomal acidificationBiol Chem200738818519617261082
- ZhangXFLiBXDongCYRenRApoptosis of human colon carcinoma HT-29 cells induced by ceramideWorld J Gastroenterol2006123581358416773715
- MagnusonBAShirtliffNBirdRPResistance of aberrant crypt foci to apoptosis induced by azoxymethane in rats chronically fed cholic acidCarcinogenesis199415145914628033325
- BernsteinHPrasadAHolubecHReduced Pms2 expression in non-neoplastic flat mucosa from patients with colon cancer correlates with reduced apoptosis competenceAppl Immunohistochem Mol Morphol20061416617216785784
- HillMJDrasarBSAriesVCCrowtherJSHawksworthGSWilliamsREOBacteria and etiology of cancer of large bowelLancet19711951004099643
- HillMJTaylorAJThompsonMHWaitRFaecal steroids and urinary volatile phenols in four Scandinavian populationsNutr Cancer1982467737155919
- HillMJBile flow and colon cancerMutat Res19902383133202188127
- ReddyBSWynderELLarge bowel carcinogenesis: faecal constituents of populations with diverse incidence ratio of colon cancerJ Natl Cancer Inst197350143714424717561
- ReddyBSWynderELMetabolic epidemiology of colon cancer: fecal bile acids and neutral sterols in colon cancer patients and patients with adenomatous polypsCancer19773925332539872053
- ReddyBSHedgesARLaaksoKWyndeerELMetabolic epidemiology of large bowel cancer: fecal bulk and constituents of high-risk North American and low-risk Finnish populationsCancer19784228322838728877
- CrowtherJSDrasarBSHillMJFaecal steroids and bacteria and large bowel cancer in Hong Kong by socioeconomic groupsBr J Cancer197634191198962996
- JensenOMMacLennanRWahrendorfJDiet, bowel function, fecal characteristics, and large bowel cancer in Denmark and FinlandNutr Cancer198245197155918
- DomellofLDarbyLHansonDMathewsLSimiBReddyBSFecal sterols and bacterial β-glucuronidase activity: A preliminary metabolic epidemiology study of healthy volunteers from Umea, Sweden, and metropolitan New YorkNutr Cancer198241201276298751
- CheahPYHypotheses for the etiology of colorectal cancer – An overviewNutr Cancer1990145132195469
- MunchAStromMSoderholmJDDihydroxy bile acids increase mucosal permeability and bacterial uptake in human colon biopsiesScand J Gastroenterol2007421167117417852874
- BernsteinCBernsteinHPayneCMDvorakKGarewalHField defects in progression to gastrointestinal tract cancersCancer Lett200826011018164807
- BaylinSBOhmJEEpigenetic gene silencing in cancer – a mechanism for early oncogenic pathway addictionNature Rev Cancer2006610711616491070
- PolleyACJMulhollandFPinCProteomic analysis reveals field-wide changes in protein expression in the morphologically normal mucosa of patients with colorectal neoplasiaCancer Res2006666553656216818627
- ChenLCHaoCYChiuYSYAlteration of gene expression in normal-appearing colon mucosa of APCmin mice and human cancer patientsCancer Res2004643694370015150130
- ShenLKondoYRosnerGLMGMT promoter methylation and field defect in sporadic colorectal cancerJ Natl Cancer Inst2005971330133816174854
- HutchinsJTReadingCLGiavazziRHoaglundJJessupJMDistribution of mono-, di, and tri-O-acetylated sialic acids in normal and neoplastic colonCancer Res1988484834893335016
- ShenYTiralongoJKohlaGSchauerRRegulation of sialic acid O-acetylation in human colon mucosaBiol Chem200438514515215101557
- ShenYKohlaGLrhorfiALO-acetylation and de-O-acetylation of sialic acids in human colorectal carcinomaEur J Biochem200427128129014717696
- AntiMArmuzziAMoriniSSevere imbalance of cell proliferation and apoptosis in the left colon and in the rectosigmoid tract in subjects with a history of large adenomasGut20014823824611156647
- BadvieSHanna-MorrisAAndreyevHJNCohenPSainiSAllen-MershTGA “field change” of inhibited apoptosis occurs in colorectal mucosa adjacent to colorectal adenocarcinomaJ Clin Pathol20065994294616679352
- BronnerMPCulinCReedJCFurthEEThe bcl-2 proto-oncogene and the gastrointestinal epithelial tumor progression modelAm J Pathol199514620267856728
- SarelaAIMacadamRCAFarmerySMMarkhamAFGuillouPJExpression of the antiapoptosis gene survivin predicts death from recurrent colorectal carcinomaGut20004664565010764707
- GreavesLCPrestonSLTadrousPJMitochondrial DNA mutations are established in human clonic stem cells, and mutated clones expand by crypt fissionProc Natl Acad Sci U S A200610371471916407113
- PayneCMHolubecHBernsteinCCrypt-restricted loss and decreased protein expression of cytochrome C oxidase subunit I as potential hypothesis-driven biomarkers of colon cancer riskCancer Epidemiol Biomarkers Prev2005142066207516172211
- ScalmatiALipkinMProliferation and differentiation biomarkers in colorectal mucosa and their application to chemoprevention studiesEnviron Health Perspect1993991691738319616
- MeyerhardtJANiedzwieckiDHollisDAssociation of dietary patterns with cancer recurrence and survival in patients with stage III colon cancerJAMA200729875476417699009
- BartramHPScheppachWSchmidHProliferation of human colonic mucosa as an intermediate biomarker of carcinogenesis: effects of butyrate, deoxycholate, calcium, ammonia, and pHCancer Res199353328332888324739
- BartramHPEnglertSScheppachWAntagonistic effects of deoxycholic acid and butyrate on epithelial cell proliferation in the proximal and distal human colonZ Gastroenterol1994323893927975774
- VelazquezOCZhouDSetoRWIn vivo crypt surface hyper-proliferation is decreased by butyrate and increased by deoxycholate in normal rat colon: associated in vivo effects on c-Fos and c-Jun expressionJPEN J Parenter Enteral Nutr1996202432508865104
- VelazquezOCSetoRWChoiJButyrate inhibits deoxycho-late-induced increase in colonic mucosal DNA and protein synthesis in vivoDis Colon Rectum199740136813759369115
- RosignoliPFabianiRDe BartolomeoAProtective ativity of butyrate on hydrogen peroxide-induced DNA damage in isolated human colonocytes and HT29 tumour cellsCarcinogenesis2001221675168011577008
- AbrahamseSLPool-ZobelBLRechkemmerGPotential of short chain fatty acids to modulate the induction of DNA damage and changes in the intracellular calcium concentration by oxidative stress in isolated rat distal colon cellsCarcinogenesis19992062963410223191
- HeerdtBGHalseyHKLipkinMAugenlichtLHExpression of mitochondrial cytochrome c oxidase in human colonic cell differentiation, transformation, and risk for colonic cancerCancer Res199050159616002154329
- HeerdtBGAugenlichtLHEffects of fatty acids on expression of genes encoding subunits of cytochrome c oxidase and cytochrome c oxidase activity in HT29 human colonic adenocarcinoma cellsJ Biol Chem199126619120191261655774
- ScheppachWMullerJGBoxbergerFHistological changes in the colonic mucosa following irrigation with short-chain fatty acidsEur J Gastroenterol Hepatol199791631689058627
- MariadasonJMCornerGAAugenlichtLHGenetic reprogramming in pathways of colonic cell maturation induced by short chain fatty acids: comparison with trichostatin A, sulindac, and curcumin and implications for chemoprevention of colon cancerCancer Res2000604561457210969808
- BordonaroMLazarovaDLAugenlichtLHSartorelliACCell type- and promoter-dependent modulation of the Wnt signaling pathway by sodium butyrateInt J Cancer200297425111774242
- WilliamsEACoxheadJMMathersJCAnti-cancer effects of butyrate: use of micro-array technology to investigate mechanismsProc Nutr Soc20036210711512740065
- WongJMWde SouzaRKendallCWCEmamAJenkinsDJAColonic health: fermentation and short chain fatty acidsJ Clin Gastroenterol20064023524316633129
- Pool-ZobelBLSauerJOverview of experimental data on reduction of colorectal cancer risk by inulin-type fructansJ Nutr200713711 Suppl2580S2584S17951507
- KimYSMilnerJADietary modulation of colon cancer riskJ Nutr200713711 Suppl2576S2579S17951506
- StempeljMKedingerMAugenlichtLKlampferLEssential role of the JAK/STAT1 signaling pathway in the expression of inducible nitric-oxide synthase in intestinal epithelial cells and its regulation by butyrateJ Biol Chem20072829797980417251186
- BordonaroMLazarovaDLSartorelliACButyrate and Wnt signaling: a possible solution to the puzzle of dietary fiber and colon cancer risk?Cell Cycle200871178118318418037
- HamerHMJonkersDVenemaKVanhoutvinSTroostFJBrummerRJReview article: the role of butyrate on colonic functionAliment Pharmacol Ther20082710411917973645
- CummingsJHPomareEWBranchWJNaylorCPMacfarlaneGTShort chain fatty acids in human large intestine, portal, hepatic and venous bloodGut28122112273678950
- PrydeSEDuncanSHHoldGLStewartCSFlintHJThe microbiology of butyrate formation in the human colonFEMS Microbiol Lett200221713313912480096
- LouisPScottKPDuncanSHFlintHJUnderstanding the effects of diet on bacterial metabolism in the large intestineJ Appl Microbiol20071021197120817448155
- BalamuruganRRajendiranEGeorgeSSamuelGVRamakrishnaBSReal-time polymerase chain reaction quantification of specific butyrate-producing bacteria, Desulfovibrio and Enterococcus faecalis in the feces of patients with colorectal cancerJ Gastroenterol Hepatol2008231298130318624900
- WangXHuyckeMMExtracellular superoxide production by Enterococcus faecalis promotes chromosomal instability in mammalian cellsGastroenterol2007132551561
- HuyckeMMAbramsVMooreDREnterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNACarcinogenesis20022352953611895869
- Attene-RamosMAWagnerEDPlewaMJGaskinsHREvidence that hydrogen sulfide is a genotoxic agentMol Cancer Res2006491416446402
- Attene-RamosMAWagnerEDGaskinsHRPlewaMJHydrogen sulfide induces direct radical-associated DNA damageMol Cancer Res2007545545917475672
- BaskarRLiLMoorePKHydrogen sulfide-induces DNA damage and changes in apoptotic gene expression in human lung fibroblast cellsFASEB J20072124725517116745
- ChristlSUEisnerHDDuselGKasperHScheppachWAntagonistic effects of sulfide and butyrate on proliferation of colonic mucosa: a potential role for these agents in the pathogenesis of ulcerative colitisDig Dis Sci199641247724819011461
- FlintHJDuncanSHScottKPLouisPInteractions and competition within the microbial community of the human colon: links between diet and healthEnviron Microbiol200791101111117472627
- ChristlSUBartramHPPaulAKelberEScheppachWKasperHBile acid metabolism by colonic bacteria in continuous culture: effects of starch and pHAnn Nutr Metab19974145519195000
- BlottiereHMBuecherBGalmicheJPCherbutCMolecular analysis of the effect of short-chain fatty acids on intestinal cell proliferationProc Nutr Soc20036210110612740064
- YinLLaevskyGGiardinaCButyrate suppression of colonocyte NF-kappa B activation and cellular proteasome activityJ Biol Chem2001276446414464611572859
- WollowskiIRechkemmerGPool-ZobelBLProtective role of probiotics and prebiotics in colon cancerAmer J Clin Nutr2001732 Suppl451S455S11157356
- Pool-ZobelBVeeriahSBohmerFDModulation of xenobiotic metabolizing enzymes by anticarcinogens – focus on glutathione S-transferases and their role as targets of dietary chemoprevention in colorectal carcinogenesisMutat Res2005591749216083918
- YoungGPHuYLe LeuRKNyskohusLReview. Dietary fibre and colorectal cancer: A model for environment – gene interactionsMol Nutr Food Res20054957158415864783
- CoxheadJMWilliamsESMathersJCDNA mismatch repair status may influence anti-neoplastic effects of butyrateBiochem Soc Trans20053372872916042586