Abstract
Alzheimer’s disease (AD) is incredibly common. Increasing longevity ensures its prevalence will rise even further. Ongoing efforts to understand AD pathogenesis reveal numerous tantalizing leads. Formulating a comprehensive AD pathogenesis theory capable of incorporating these disparate leads, though, has proven difficult. This review discusses current attempts to formulate a comprehensive AD pathogenesis theory. In doing so, it focuses on clinical and molecular relationships between AD and aging. A better understanding of these relationships could inform and impact future development of AD-directed treatment strategies.
Introduction
Alzheimer’s disease (AD) is the most common disease of aging. Countless clinicians and scientists have dedicated careers to its investigation. For some time the field has been too large to comprehensively summarize. While this review broadly discusses both molecular and clinical AD-relevant themes, it considers these themes within the narrower context of aging. By focusing in this way, it hopes to provide a better understanding of aging-AD relationships. Considering AD as a disease of aging provides perspective on recent therapeutic development efforts, which are discussed later on. First, though, it is necessary to define AD and describe why it is so intimately related to aging. While doing so, it is necessary to consider which is more appropriate – to consider AD a condition of aging, or to consider it as a distinct disease entity. Although this argument is largely a semantic one, it does have important implications for future AD research.
What is Alzheimer’s disease?
This review will first consider the term “Alzheimer’s disease”. This is a worthwhile endeavor because AD perspectives abound. Affected patients and their family members may not distinguish AD from the generic dementia clinical syndrome. Physicians not specializing in AD often don’t go much further, using it as a default diagnosis for persons dementing despite negative diagnostic tests. To AD sub-specialists able to identify and categorize different patterns of cognitive decline, AD is less a diagnosis of exclusion and more a recognizable entity. AD sub-specialists can predict with reasonable accuracy the histopathologic features existing within the brains of those with an AD-consistent clinical syndrome (CitationGearing et al 1995). However, the pathogenic relevance of these histopathologic features is itself unclear.
The first defined histopathologic features of AD were extracellular amyloid plaques and intracellular neurofibrillary tangles. More recently recognized histopathologic features include synaptic degeneration, hippocampal neuronal loss, and aneuploidy. AD histopathologic criteria, though, currently take into account only plaques and tangles. Several AD histopathologic criteria are commonly used (CitationKhachaturian 1985; CitationMirra et al 1991; CitationConsensus Criteria 1997). Approximately 90% of the time, when pathologists evaluate brains from persons diagnosed with AD by trained clinicians, enough plaques and tangles are found to meet histopathologic diagnostic standards (CitationGearing et al 1995). Interestingly, for some time pathologists have recognized non-demented elderly individuals will also qualify for a histopathologic diagnosis of AD (reviewed in CitationSwerdlow 2006). This has caused fundamental problems for the AD field. If plaque-tangle quantity represents the AD “gold standard”, then clinical dementia becomes less relevant to the diagnosis.
De-emphasizing dementia as a required component raises several conceptual questions. First, how should one consider deceased individuals with plaque-laden brains but no dementia? The term “preclinical AD” is often used in these situations (CitationPrice et al 2001). Preclinical AD assumes histopathologic changes can precede clinical changes, and that if preclinical AD subjects lived longer dementia would manifest. Until recently this assumption has not been testable. The advent of in vivo plaque and plaque-tangle imaging may facilitate more critical assessment of this hypothesis. To date, such imaging has corroborated the pathology literature, in that non-demented elderly subjects often have plaque burdens equivalent to those with clinically diagnosed AD (CitationShoghi-Jadid et al 2002; CitationKlunk et al 2004; CitationFagan et al 2006; CitationRentz et al 2006).
The second conceptual question relates to how many people either have AD or the potential to get AD. Certainly, how to address this question depends heavily on the definition used. If histopathology represents the gold standard, then up to 95% of those making it over 100 years of age have AD (CitationJicha et al 2005). One large survey of brains from persons over 85 found at least some degree of AD histopathology in all brains examined (CitationPolvikoski et al 2005). A possible interpretation of this is that all individuals, should they live long enough, have the potential to develop AD. While this point is also not testable, it is minimally relevant. Rather, it is more important to consider whether in the most at-risk demographic groups it becomes more common to have AD than not to have AD. This does appear to be the case. Since approximately 75%–90% of centenarians have a dementia syndrome and 85%–95% meet at least minimal histopathologic criteria for AD (CitationMizutani and Shamada 1992; CitationThomassen et al 1998; CitationBlansjaar et al 2000; CitationPerls 2004; CitationJicha et al 2005), it seems reasonable to conclude centenarians are expected to have AD, and those who do not have it are the exception. Therefore, it is worth considering that at some point in the aging continuum AD ceases to become a disease because it becomes the norm.
The third conceptual question relates to the fact individuals commonly experience age-related revision of their cognitive skills. The neuropsychology literature suggests that upon reaching adulthood, insidious cognitive changes ensue (CitationSnowdon et al 1996; CitationSmyth et al 2004). These changes, which occur over decades, generally represent decline from previously higher levels of function as opposed to lateral shifts in cognitive strategies. While associated with at most subtle functional consequences not felt to be clinically relevant on an individual basis, it is worthwhile considering whether these changes do in fact represent part of an AD continuum. The relevance of this question is emphasized by the recently evolved concept of mild cognitive impairment (MCI) (CitationPetersen et al 1999). MCI was originally intended to define a transitional state between normal cognition and dementia. Retrospective perspectives now suggest the majority of those with MCI are actually manifesting AD in its earliest recognizable clinical form (CitationMorris 2006). The operational definition of MCI is similar to that of an older term, age-associated cognitive decline (AACD) (CitationLevy 1994). AACD was formulated to classify those with “benign” cognitive changes. The main difference between AACD and MCI is therefore one of perspective. AACD was intended as an optimistic diagnosis, whereas MCI is considerably more pessimistic. Whether the physiology underlying age-associated cognitive changes is fundamentally different from that underlying AD-associated cognitive change remains unknown.
It is now common to lump under the AD umbrella all dementia syndromes manifesting plaque and tangle accumulation. This in turn has driven classification of various AD subtypes. Dementia of the Alzheimer’s Type (DAT) corresponds to the classic presenile form identified by Alois Alzheimer. Senile dementia of the Alzheimer’s Type (SDAT) includes individuals that until the 1970’s may have been assigned a diagnosis of senility, hardening of the arteries, or just getting older. Late-onset AD (LOAD) generally applies to those developing signs and symptoms after the age of 65. Early-onset AD variably refers to those developing signs or symptoms before the age of 55, 60, or 65. The exact upper age limit is therefore arbitrarily defined. Familial AD (FAD) loosely refers to those AD subjects with a positive AD family history, but without additional clarification this designation is confusing. The reason is that if the simple presence of a single affected relative is enough to qualify a patient for FAD, then FAD is incredibly common. If more stringent criteria are used, such as the presence of a recognizable Mendelian inheritance pattern, then FAD comprises an exceedingly small percentage (less than 1%) of the total number of AD cases. Most Mendelian FAD cases are autosomal dominant and have a presenile onset. Nevertheless, even among early-onset cases clear-cut autosomal dominant inheritance patterns are rare. Despite their rarity, the autosomal dominant FAD cases demonstrate genetic heterogeneity, as mutations in at least three different genes cause the phenotype (). Therefore, when considering AD pathogenesis, it is perhaps expedient to consider the fact that what we now consider AD actually appears to consist of multiple pathogenically different Alzheimer’s diseases.
Table 1 Genes implicated in familial autosomal dominant AD
Disease of aging versus aging
Alois Alzheimer’s first “Alzheimer’s disease” patient, Auguste D., was reported in detail in 1907 (CitationAlzheimer 1907). Auguste D. presented early in her sixth decade, and so could officially be considered a case of presenile dementia. On autopsy, her brain manifested the plaques and tangles that have since been associated with the AD condition. As no prior descriptions of plaque and tangle presenile dementia existed, the door was open for naming a new disease, which was shortly thereafter accomplished by Emil Kraepelin, the chairman of Alzheimer’s academic department (CitationKraepelin 1910). Simultaneously, Oscar CitationFischer (1907) at a rival institution recognized brains of elderly demented individuals also contained plaques. In subsequent years the medical community assimilated these observations so that the presenile tangle-and-plaque dementia was considered a disease, AD. The other situation (senile tangle-and-plaque dementia) was considered a normal part of aging, and for many decades hence syndromically referred to as senile dementia (CitationAmaducci et al 1986; CitationBoller and Forbes 1998). In the decades following this, AD remained a rather uncommon entity.
While life expectancy increased in westernized countries during the course of the twentieth century, senile dementia became increasingly prevalent. Interest in treating the demented elderly therefore grew, and investigators increasingly noted the similarities between AD and senile dementia. As Katzman pointed out in 1976, “neither the clinician, the neuropathologist, nor the electron microscopist can distinguish between the two disorders, except by the age of the patient” (CitationKatzman 1976). It was further argued “Alzheimer disease and senile dementia are a single process and should therefore be considered a single disease” (CitationKatzman 1976). This view justified making dementia syndrome research a national health priority and invigorated research into the phenomenon. Expanding the definition of AD to include those with senile dementia swelled the ranks of those diagnosed to such an extent it inextricably linked AD to aging. The incidence of AD rises with increasing age, so that among centenarians the histopathologic and clinical changes needed to justify a diagnosis of AD probably coexist in greater than 75% (CitationMizutani and Shamada 1992; CitationThomassen et al 1998; CitationBlansjaar et al 2000; CitationPerls 2004; CitationJicha et al 2005). One oft-quoted study concluded almost half those over age 85 have AD or at least an AD syndrome (CitationEvans et al 1989).
Relevant to this debate are recent data suggesting AD may develop in individuals over the course of decades (CitationSnowdon et al 1996; CitationSmyth et al 2004). If this view is correct, then AD is a disease most people are in the process of developing throughout adulthood. From an epidemiologic standpoint, therefore, the debate about whether tangle-and-plaque dementia is a distinct disease or part of aging remains unresolved. An in-depth discussion of the aging-AD controversy has been reviewed elsewhere (CitationSwerdlow 2006).
Does the histopathology drive the disease, or does the disease drive the histopathology?
With AD, the cause-consequence debate goes back to Alzheimer’s time. Alzheimer personally believed the histopathologic features he observed were a marker of an upstream process and not the root cause of the disease (CitationDavis and Chisholm 1999). There are biochemical studies that support this view. For example, inhibition of the electron transport chain (ETC) enzyme cytochrome oxidase alters amyloid precursor protein (APP) processing to an amyloidogenic derivative (CitationGabuzda et al 1994). Oxidative stress activates beta secretase (BACE) activity, a requisite event in the processing of APP to Aβ (CitationTamagno et al 2002). Data such as these suggest amyloidosis in AD is a secondary event.
On the other hand, it is clear AD sometimes represents a primary amyloidosis. In 1991, APP gene mutation was shown to cause an early-onset, autosomal dominant AD variant (CitationGoate et al 1991). Mutation of two other genes, presenilin 1 and presenilin 2, were subsequently found to also cause early-onset, autosomal dominant AD (CitationLevy-Lahad et al 1995; CitationSherrington et al 1995). Functional studies revealed these mutations alter APP processing. In each case, Aβ42 to Aβ40 ratios increase (CitationScheuner et al 1996).
Whether most AD represents a primary or secondary amyloidosis goes to the heart of the AD pathogenesis debate. Two pathogenic AD hypotheses are discussed below, the amyloid cascade hypothesis (which assumes AD is always a primary amyloidosis), and the mitochondrial cascade hypothesis (which assumes most AD is a secondary amyloidosis).
The amyloid cascade hypothesis
The cortical plaques of AD brains largely consist of Aβ protein. Aβ is produced via processing of its parent protein, APP. The gene encoding APP resides on chromosome 21. Specific APP physiologic roles are not entirely clear, but in a general sense it is felt to contribute to proper neuronal function and perhaps cerebral development (CitationZheng et al 1995).
Because plaques contain Aβ and Down’s syndrome patients with trisomy 21 manifest both plaque pathology and presenile cognitive decline below their baseline, the role of the APP gene in AD was previously considered. In 1991, an APP gene mutation was uncovered in a family with autosomal dominant, early onset AD (CitationGoate et al 1991). Soon after this, the amyloid cascade hypothesis was formulated. In its original form, the amyloid cascade hypothesis proposed altered APP processing drove Aβ production, Aβ gave rise to plaques, plaques induced neurodegeneration, and this neuronal loss resulted in the clinical dementia syndrome typical of AD (CitationHardy and Allsop 1991; CitationHardy and Higgins 1992).
While subsequent research failed to show APP mutation was a common cause of AD, the findings of other genetic and molecular research also lent support to the amyloid cascade hypothesis. Specifically, mutations in two other genes, presenilin 1 on chromosome 14 and presenilin 2 on chromosome 1, were also shown to cause variants of early onset, autosomal dominant AD (CitationLevy-Lehad et al 1995; CitationSherrington et al 1995). While the functional role of presenilin proteins was unknown at the time of these discoveries, it was nevertheless found that as was the case with APP mutations, presenilin mutations enhanced production of the 42 amino acid APP C-terminal degradation product (Aβ42) at the expense of the 40 amino acid C-terminal APP C-terminal degradation product (Aβ40) (CitationScheuner et al 1996). Aβ42 is toxic to cells in culture, prone to aggregation, and found in plaques. More recent data suggest the presenilin gene products comprise part of the γ-secretase complex that is so intimately involved in APP processing (CitationWolfe et al 1999; CitationKimberly et al 2000).
The amyloid cascade hypothesis () has substantially evolved since its initial formulation. While Aβ sequestered in plaques was at first proposed to represent the critical toxic species, more recent versions of the hypothesis assume Aβ that is not sequestered in plaques actually drives the disease (CitationLesne and Kotilinek 2005; CitationWalsh et al 2005). Even so, the amyloid cascade hypothesis seems most applicable in cases of early onset, autosomal dominant AD. Certainly, these cases are the ones most likely to represent a primary amyloidosis. Such cases comprise far less than 1% of AD, though, and it is not clear whether it is reasonable to etiologically extrapolate to the late-onset form (which afflicts the vast majority of those affected). Whether individuals with late-onset AD also carry genetic variations that promote a primary Aβ amyloidosis remains to be shown. If this turns out not to be the case, the possibility that amyloidosis in late-onset AD is secondary to a more upstream event will require consideration.
Figure 1 The amyloid cascade hypothesis. A black box is shown in the middle of the figure, since mechanisms through which Aβ42 drives downstream pathology are not well defined.
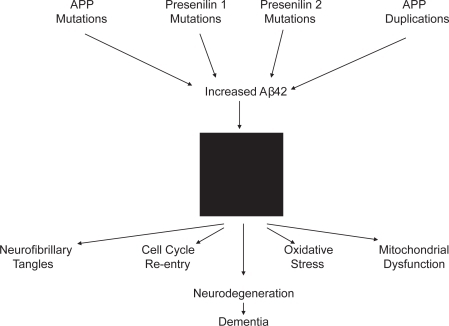
Pursuit of the amyloid cascade hypothesis has yielded in depth insight into how APP processing actually occurs. Enzymes are capable of cutting APP in several places near its C-terminal end. One group of enzymes called α-secretases cut APP 83 amino acids from its carboxyl terminus. Enzymes with α-secretase activity include the ADAM (“A Disintegrin and Metalloproteinase”) protease family. The β-secretase enzymes consist of at least two different complexes named BACE1 and BACE2. The BACE enzymes cut APP 99 amino acids from the carboxyl terminus. The third type of APP cut is rendered by the γ-secretase enzyme complex. The catalytic function of γ-secretase is mediated by either presenilin 1 or presenilin 2. The γ-secretase cuts APP twice. One of these cuts occurs 50 amino acids from the APP carboxyl terminus. This generates a 50 amino acid peptide consisting of the extreme APP C-terminal end, which is titled the amyloid intracellular domain (AICD). The other γ-secretase cut is somewhat variable, but tends to occur 57, 59, or 61 amino acids from the APP C-terminus.
The APP secretases work in combination. Sequential processing by the α and γ-secretases results in a large N-terminal peptide called soluble APPα (sAPPα) and a smaller 3 kD peptide called P3. Proteolysis by enzymes with α-secretase activity precludes sequential β-γ secretase activity. When both the β and γ secretases process APP, the β-secretase cut produces a large N-terminal peptide called soluble APPβ (sAPPβ), as well as a smaller C-terminal fragment called CTFβ. The γ-secretase cuts CTFβ, and the more upstream (from the APP carboxyl terminus) 4 kD peptide defined by the γ and β secretases is the Aβ peptide. With sequential β-γ secretase activity the exact γ secretase cut site varies, yielding an Aβ peptide which is typically 38–43 amino acids.
Aβ degradation is also enzymatically mediated. Enzymes that degrade Aβ include neprilysin and insulin degrading enzyme (IDE). Interestingly, in the common non-autosomal dominant forms of AD Aβ overproduction is accompanied by IDE downregulation (CitationCook et al 2003). This suggests amyloidosis in AD is not a toxic accident, but rather part of a coordinated cell response to an upstream event. Defining this event could potentially drive further evolution of the amyloid cascade hypothesis. As it currently stands, the amyloid cascade hypothesis assumes all AD is a primary amyloidosis. This assumption is extrapolated from a handful of AD cases which almost certainly are primary amyloidoses. If an upstream event is shown to initiate AD amyloidosis, though, it would suggest amyloidosis in the vast majority of AD cases is a secondary event. Taking this argument one step further, some have even argued amyloidosis in AD not only represents a secondary event, but also a compensatory one (CitationCastellani et al 2006).
Other clues to AD pathogenesis
Plaque-oriented research dominates the AD field. Much investigation, though, has focused on neurofibrillary tangles. These intracellular aggregations contain abnormally configured, excessively phosphorylated tau protein. In differentiated cells tau is normally unphosphorylated. It associates with microtubule cytoskeleton elements. This differs from undifferentiated cells, in which microtubules and tau do not form a permanent cytoskeleton and tau is phosphorylated. Mutations in the tau gene are associated with familial frontotemporal dementia, especially in cases with concomitant parkinsonism and tangle histopathology (CitationNeary et al 2005). Thus, while primary tauopathy can drive neurodegeneration, it is not recognized to cause an AD phenotype.
Interestingly, AD brains exhibit neuronal cell cycle re-entry (CitationVincent et al 1996; CitationMcShea et al 1999; CitationArendt et al 2000; CitationYang et al 2001; CitationBowser and Smith 2002; CitationZhu et al 2004). Cell cycle re-entry refers to a phenomenon in which differentiated, non-dividing cells manifest molecular changes typically associated with cell division. These manifestations include an increase in cyclin-dependent kinase (CDK) activities and DNA content. Cell cycle re-entry produces aneuploid neuronal nuclei with replicated chromosomes. AD neurons can actually reach G2, the penultimate stage of the cell cycle that immediately precedes mitosis (M). Nevertheless, these neurons are unable to complete mitosis, which leaves them in a state called G2-M arrest.
CDK proteins also phoshorylate tau. It is tempting to speculate tau phosphorylation and cell cycle re-entry pathology are related. A mechanistically critical relationship between cell-cycle re-entry, oxidative stress, and neuronal demise was recently postulated (CitationZhu et al 2004).
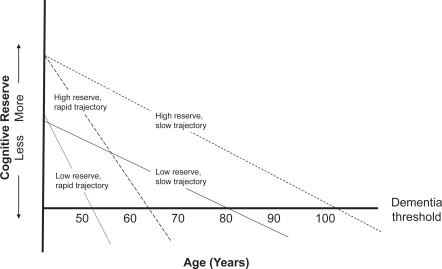
As discussed above, AD and aging are epidemiologically intertwined. The strength of this relationship suggests these processes share mechanistic commonalities. Clinically, AD is not an all-or-nothing entity. It is a continuum. Mild, moderate, and severe stages are arbitrarily defined. Preceding AD proper is a recognizable transition phase, MCI. Neuropsychological testing now suggests MCI is itself preceded by a period of cognitive change (reviewed in CitationSwerdlow 2006). Some call this period pre-MCI (CitationSwerdlow 2006). Pre-MCI may last decades; some data suggest it starts early in adulthood (CitationSnowdon et al 1996; CitationSmyth et al 2004). Pre-MCI may provide a conceptual framework for “cognitive reserve” phenomena in AD (CitationStern 2006). Those with higher education levels, a cognitive reserve surrogate, are said to have reduced AD risk. It is heuristically reasoned high cognitive reserve individuals begin their decades-long cognitive decline further from the dementia finish line than low cognitive reserve individuals. Given equal pre-MCI cognitive decline trajectories, low cognitive reserve individuals will cross dementia thresholds before high cognitive reserve individuals. Pre-MCI decline trajectories, though, probably vary between individuals. A combination of genetic and environmental factors may influence decline trajectories. By this reasoning, those with the most cognitive reserve and slowest decline trajectories are more likely to develop clinical AD at older ages than those with less cognitive reserve and faster decline trajectories ().
Figure 2 Alzheimer’s disease dementia develops over decades. Those with more cognitive reserve and slower decline trajectories dement at the oldest ages. Those with less cognitive reserve and more rapid decline trajectories dement at younger ages.
Apolipoprotein E (APOE) alleles influence AD related-markers in ways potentially relevant to pre-MCI cognitive decline trajectories (CitationSmall et al 1995; CitationReiman et al 1996; CitationCaselli et al 1999). The APOE gene on chromosome 19 encodes a protein, apolipoprotein E, which was previously recognized to play a role in cholesterol transport. Three APOE alleles are distributed throughout the population. Among these, the APOE4 allele associates with a younger age of AD onset than the APOE3 or APOE2 alleles (CitationCorder et al 1993; CitationLocke et al 1995; CitationBlacker and Tanzi 1998). For perhaps this reason APOE4 is also associated with a greater lifetime AD risk. The underlying biochemical basis for this association is unknown, but hypotheses abound (CitationSaunders 2000). The cysteine to arginine substitutions that define APOE4 may minimize any inherent ability of the protein to mitigate oxidative stress (CitationMiyata and Smith 1996). Apolipoprotein E protein variations may affect cholesterol transport, thereby indirectly affecting amyloidosis (CitationPoirier 2000). Direct effects on Aβ cerebrovascular transport might explain the association (CitationDeMattos et al 2002; CitationZlokovic et al 2005). Apoliprotein E fragments also accumulate in mitochondria and affect mitochondrial function (CitationChang et al 2005). Apolipoprotein E4 produces more of these toxic fragments than apolipoprotein E3.
Mitochondrial dysfunction clearly exists in AD subjects (CitationParker et al 1990; reviewed in CitationSwerdlow and Kish 2002). Structural and functional changes are evident. Mitochondrial pathology in AD is not limited to the brain. Mitochondria are implicated in aging, amyloidosis, oxidative stress, tau phosphorylation, and cell cycling. The protean manifestations of mitochondrial dysfunction and systemic nature of mitochondrial dysfunction in AD suggest to some a central pathogenic role for these organelles (CitationParker et al 1990; CitationSwerdlow and Khan 2004).
The mitochondrial cascade hypothesis
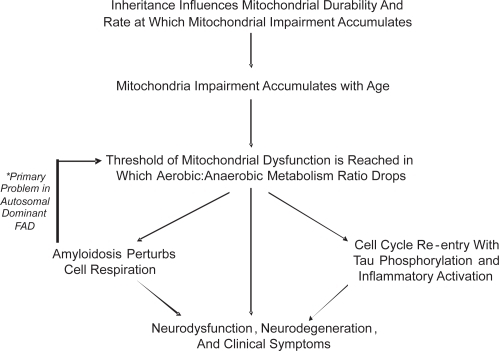
The mitochondrial cascade hypothesis attempts a unified explanation for the clinical, biochemical, and histologic features of AD (CitationSwerdlow and Khan 2004). The mitochondrial cascade hypothesis takes several conceptual liberties. It assumes similar physiologic mechanisms underlie AD and brain aging. It postulates because AD mitochondrial dysfunction is systemic, it cannot simply represent a consequence of neurodegeneration. The mitochondrial cascade hypothesis argues non-Mendelian genetic factors contribute to non-autosomal dominant AD. Finally, it posits AD brain mitochondrial dysfunction drives amyloidosis, tau phosphorylation, and cell cycle re-entry.
Certainly, mitochondria are indirectly featured in past theories of aging and directly in current aging theory. The rate of living hypothesis arose in the 1920’s from observations that animals with low metabolic rates tend to outlive those with high metabolic rates (CitationPearl 1928). CitationHarman refined this when he proposed the free radical theory of aging in 1956 (CitationHarman 1956). The free radical theory of aging specifically postulated over time cells accumulate structural damage from oxidative byproducts. By the 1970’s mitochondria were recognized sites of free radical production, and for many the free radical theory of aging morphed into the mitochondrial theory of aging (CitationHarman 1972; CitationMiquel et al 1980). The late 1980’s envisaged a role for somatic mitochondrial DNA (mtDNA) damage in aging (CitationLinnane et al 1989; CitationWallace 1992). Corroborating this possibility are recent studies showing mtDNA mutation acquisition accelerates aging in laboratory animals (CitationTrifunovic et al 2004; CitationKujoth et al 2005).
Mitochondrial dysfunction is observed in multiple AD tissues (reviewed in CitationSwerdlow and Kish 2002). At least brain, platelet, and fibroblast mitochondria are involved. Defects of three mitochondrial enzymes are reported. This includes reduced activities of pyruvate dehydrogenase complex, alpha ketoglutarate dehydrogenase complex, and cytochrome oxidase (CitationGibson et al 1998). Spectral analysis of cytochrome oxidase indicates AD brains contain normal amounts of cytochrome oxidase, but the enzyme itself is structurally altered (CitationParker and Parks 1995). Various mechanisms, such as oxidative stress and proteasome dysfunction, have been postulated to facilitate mitochondrial dysfunction in neurodegenerative diseases such as AD (CitationDing et al 2006). Also, cytoplasmic hybrid (cybrid) studies indicate mtDNA at least in part accounts for reduced cytochrome oxidase activity in AD, and through this perhaps oxidative stress (reviewed in CitationSwerdlow 2007).
Cybrid studies involve transfer of exogenous mtDNA to cultured cells depleted of endogenous mtDNA (reviewed in CitationKhan et al 2006). “ρ” DNA is an alternative term for mtDNA. These mtDNA-depleted cells are therefore called ρ0 cells. They do not produce mtDNA-encoded proteins and lack cytochrome oxidase activity. Mitochondrial DNA transferred to ρ0 cells replicates and accomplishes mtDNA replacement. This enables expression of mtDNA-encoded ETC subunits and restoration of cytochrome oxidase activity. When cybrid lines containing mtDNA from AD subject platelets are compared to cybrid lines containing mtDNA from age-matched controls without AD, cytochrome oxidase activity is lower in the cybrid lines containing AD subject mtDNA (CitationSwerdlow et al 1997; reviewed in CitationSwerdlow 1997). Because nuclear genetic and cell culture conditions are equivalent between all cybrid cell lines, differences between mtDNA from the donor subjects likely account for the observed differences in cytochrome oxidase activity.
The exact nature of the implied AD cybrid-control cybrid mtDNA difference is unclear. This uncertainty is partly due to mtDNA-related complexities. Mitochondrial genetics and nuclear genetics differ in several key ways. One difference is that cells contain multiple copies of mtDNA. The mtDNA molecules within a cell can be identical, a state referred to as homoplasmy. However, the nucleotide sequences of mtDNA molecules within a cell can also vary. This is called heteroplasmy. If mtDNA sequence variation is present within a cell, it is necessary to consider whether it represents a homoplasmic or heteroplasmic variation. If the variation is heteroplasmic, it is necessary to further consider whether it represents a majority (high abundance heteroplasmy) or minority (low abundance heteroplasmy) of that cell’s mtDNA copies.
Distinct homoplasmic or high abundance heteroplasmic mutations of mtDNA cytochrome oxidase (CO) likely account for at most a very small percentage of AD (CitationHamblet et al 2006), and therefore should not account for AD-control cybrid differences. Mitochondrial DNA polymorphisms could potentially account for AD-control cybrid differences, as these are a common source of mtDNA inter-individual variability. Although AD-mtDNA polymorphism associations are reported (CitationChagnon et al 1999; reviewed in CitationSwerdlow and Kish 2002; Citationvan der Walt et al 2004), the effect of specific polymorphisms or linked polymorphisms (haplogroups) on cybrid cytochrome oxidase activity is unstudied. Low abundance heteroplasmy might also cause AD-control cybrid cytochrome oxidase differences. A recent study did in fact suggest unique low abundance mtDNA heteroplasmies occur in AD (CitationCoskun et al 2004). Other data indicate low abundance mtDNA heteroplasmies manifest with increasing age, and these heteroplasmies are associated with reduced cytochrome oxidase activity (CitationLin et al 2002). However, the presence and role of low abundance mtDNA heteroplasmies in AD cybrids has not been critically evaluated.
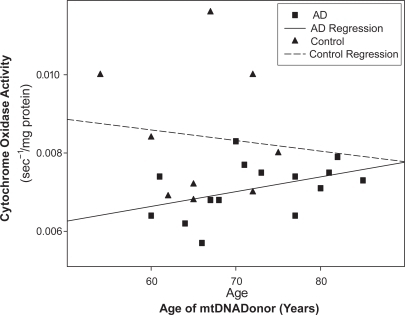
Whether somatic or inherited mtDNA features account for AD-control cybrid cytochrome oxidase differences also requires consideration. Data pertinent to this question permit speculation. First, complex I activity is normal in AD cybrids (CitationGhosh et al 1999). Complex I contains seven mtDNA-encoded subunits. Absence of complex I dysfunction suggests somatic mutation does not account for reduced cytochrome oxidase activity in AD cybrids, since acquired mtDNA somatic mutation should also reduce complex I activity. Second, reduction of cytochrome oxidase activity in multiple non-degenerating tissues is more consistent with mtDNA inheritance than somatic mutation. Third, plotting one AD cybrid study’s cytochrome oxidase data by mtDNA donor age actually reveals the youngest AD subjects show the biggest activity reductions (). These data suggest mtDNA signatures might affect intrinsic cognitive decline trajectories. One interpretation of this is the lower the cytochrome oxidase activity, the sooner the subject reaches the dementia threshold. Epidemiologic data showing maternal AD status correlates better with offspring AD status than paternal AD status is consistent with this possibility (CitationEdland et al 1996).
Figure 3 Cytochrome oxidase activity may influence cognitive decline rates. Cytochrome oxidase activity was assayed in cybrid lines expressing mtDNA from 15 different AD subjects. The youngest AD mtDNA donors tended towards lower activies. Nine cybrid lines expressing mtDNA from control subjects indicate at most subtle age-related activity reductions.
Any comprehensive theory of AD pathogenesis must explain the different pathologies observed in Alzheimer’s disease. AD cybrids with reduced cytochrome oxidase activity overproduce Aβ42 and Aβ40 (CitationKhan et al 2000). Under in vitro conditions sodium azide, a cytochrome oxidase inhibitor, alters APP processing towards amyloidogenic pathways (CitationGabuzda et al 1994). Administering sodium azide to mice also causes tau phosphorylation (CitationSzabados et al 2004). Fibroblasts from FAD subjects phosphorylate tau following exposure to CCCP, a mitochondrial uncoupler (CitationBlass et al 1990). Mitochondrial ETC dysfunction increases free radical production (reviewed in CitationSwerdlow 2002). Enhanced reliance on anaerobic metabolism is associated with cell cycling (reviewed in CitationSwerdlow and Khan 2004).
Interestingly, Aβ inhibits ETC activity in general and cytochrome oxidase activity specifically (CitationPeriera et al 1998; CitationCasley et al 2002; CitationCrouch et al 2005; CitationDevi et al 2006). Therefore, a reciprocal relationship exists between mitochondrial function and amyloidosis. Several independent observations reinforce this concept. Neuronal-like NT2 human teratocarcinoma cells exposed to Aβ show high rates of demise. NT2 ρ0 cells, though, are impervious to Aβ (CitationCardoso et al 2001). The main difference between native NT2 cells and NT2 ρ0 cells is the absence of a functional ETC in the ρ0 cells. This suggests under cell culture conditions the mitochondrial ETC mediates Aβ toxicity. Also, APP, Aβ, and the entire γ-secretase complex are found either within mitochondria or mitochondrial membranes (CitationAnandtheerthavarada et al 2003; CitationLustbader et al 2004; CitationHansson et al 2004; CitationCrouch et al 2005; CitationManczak et al 2006). The role APP, Aβ, and γ-secretase play in mitochondrial function is currently unknown. Recent work showing oligomeric β-sheet proteins permeabilize membranes (CitationGlabe and Kayed 2006) raises the possibility these proteins allow cells to disable mitochondria when certain conditions are met. If mitochondrial dysfunction is one of these conditions, one physiologic role of APP or Aβ may be to “shut down” abnormal mitochondria.
The mitochondrial cascade hypothesis takes aging phenomena into account, and applies to individuals long before they develop AD. The hypothesis postulates among individuals, inherited ETC differences influence mitochondrial durability. More durable mitochondria maintain adequate function longer than less durable mitochondria, and this in part determines aging success. Eventually, though, the balance between aerobic and anaerobic metabolism is not sustainable. At this point various cell responses are triggered. Cell cycle re-entry occurs. Tau phosphorylation ensues as neurons commit to de-differentiation. Amyloidosis facilitates this commitment by further altering aerobic metabolism. Ultimately, the neuronal de-differentiation response fails. Neurodegeneration results.
Just as the mitochondrial cascade hypothesis identifies with aging theory, links to the amyloid cascade hypothesis are postulated. In early onset, autosomal dominant AD altered APP or Aβ physiology initiate pathogenesis. If altered APP or Aβ physiology inadvertently induces mitochondrial dysfunction, it would trigger the same series of events as aging. Mitochondrial failure may therefore represent a final common pathway between autosomal dominant AD and non-autosomal dominant AD. Such would explain the clinical and histologic similarities observed between the different Alzheimer’s diseases ().
Figure 4 The mitochondrial cascade hypothesis.
Treatment development and clinical trials: Critical assessments of pathogenic hypotheses?
Current drug development strategies fall broadly into one of two categories. The first category includes treatments specifically designed to reduce the burden of brain Aβ. The second category includes all other strategies.
In order to reduce brain amyloid levels, approaches to both reducing Aβ production and enhancing its removal are undergoing evaluation. As discussed above, Aβ is produced through the β and γ-secretase mediated processing of APP. β-secretase inhibitors have gone to phase II human trials. Agents that specifically inhibit γ-secretase could prove problematic from a side effect perspective, as γ-secretase is also critical for processing Notch3, a protein of developmental importance and perhaps brain maintenance. Notch3 mutations cause another dementia syndrome, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL).
Certain NSAIDS (ibuprofen and flurbiprofen) influence the γ-secretase but do not inhibit it outright. In general, these “selective amyloid lowering agents” (SALAs) alter where γ-secretase cuts the APP protein. Under in vitro conditions, ibuprofen and flurbiprofen reduce production of the Aβ42 APP derivative, with a secondary increase in the production of shorter Aβ fragments (CitationEricksen et al 2003). An enantiomer of flurbiprofen, R-flurbiprofen, recently completed a phase II human trial and is slated for a phase III efficacy trial.
Immunotherapy approaches have been studied as a way to enhance Aβ removal. The most extensive investigation involved AN1792, an Aβ-based vaccine (Aβ linked to an adjuvant). This vaccine first showed efficacy in transgenic mice that produce human Aβ. Such transgenic mice are engineered to express human APP transgenes containing mutations known to cause autosomal dominant, early-onset AD, and are widely used for preclinical drug screening in AD. In vaccinated mice, Aβ loads were reduced, there was preservation of cognitive abilities, and the vaccine was well tolerated (CitationSchenk et al 1999). When phase I human studies did not reveal obvious toxicity, a phase II trial was initiated. This trial was prematurely halted after a substantial percentage of those mounting a robust immune response to the vaccine experienced encephalitis, a potentially life-threatening brain inflammation. However, there were over 40 individuals in this study who were vaccinated, developed a robust immune response, and did not develop encephalitis. Most of these subjects, as well as the trial’s placebo group, received ongoing clinical follow-up. One-year post-vaccination neuropsychological data on these subjects have been published (CitationGilman et al 2005). On no single endpoint was a statistical benefit shown. Subjects in both treatment and placebo groups continued to decline. For most of the individual endpoints, trends towards lesser decline in the vaccination group were identified, and on a z score analysis these trends were significant, suggesting it was possible the vaccine group was progressing slightly more slowly than the placebo group. Other data pertinent to the AN1792 study derive from autopsies of vaccinated subjects that subsequently died for various reasons. Brain histopathology from these deceased subjects demonstrated clear-cut reductions in brain parenchyma Aβ (CitationNicoll et al 2003; CitationFerrer et al 2004; CitationMasliah et al 2005). To summarize the AN1792 experience from an efficacy perspective, available clinical data are inconclusive, but indicate over a one-year period activating the immune system to remove Aβ does not have a robust impact on cognition.
The next generation of amyloid clearance therapies are currently under development. These include modifications of the active immunization approach that will hopefully not trigger encephalitis. Passive immunization approaches via antibody infusions are also under investigation. The use of unique Aβ antibodies is being explored. The utility of treating AD subjects with intravenous immunoglobulin preparations, which naturally contain antibodies to Aβ, is being evaluated in an open label study (CitationRelkin et al 2005).
The second category of AD treatment development includes efforts not specifically intended to reduce brain Aβ levels. For example, neuroscientists are unraveling intracellular pathways involved in cell information processing, and drugs that can modulate these pathways are under consideration. Drugs that retard neurofibrillary tangle formation in mice expressing mutant human tau transgenes, such as valproic acid, are being tested in humans. Although standard antioxidants to date have shown no-to-minimal evidence of therapeutic efficacy, new antioxidants that target specific free radical production sites are in preclinical development (CitationReddy 2006). The thiazolidinedione drugs rosiglitazone and pioglitazone, which reduce insulin resistance and which may also have anti-inflammatory effects, are undergoing testing in humans with AD (CitationRisner et al 2006; CitationGeldmacher et al 2006). One small open label trial has involved intracranial implantation of fibroblasts engineered to produce neurotrophic factors (CitationTuszynski et al 2005).
Obviously, successful development of new AD treatments depends on elucidating AD’s true underlying pathophysiology. If AD is a primary amyloidosis, as is postulated by the amyloid cascade hypothesis, then reducing Aβ would seem a rationale way to proceed with drug development. If AD is not a primary amyloidosis, then the impact anti-amyloid therapies have on the disease will be limited at best. Moreover, if AD is not a primary amyloidosis, the usefulness of Aβ-overproducing transgenic animals for preclinical drug testing is called into question. Finally, at genetic and epidemiologic levels it is now possible to define several different Alzheimer’s diseases. It is reasonable to consider whether treatments useful in one type of AD may not benefit patients with another type. In any case, Aβ-oriented treatment development will likely provide a practical assessment of the amyloid cascade hypothesis. If treatments that efficiently reduce Aβ production or remove Aβ fail to arrest disease progression, it would argue amyloidosis is not the primary pathology in most of those with AD.
Conclusion
Unlike many recent AD reviews, this one assumes critical questions remain about AD pathogenesis. As AD now stands, there are at least several different Alzheimer’s diseases. The rare autosomal dominant forms are primary amyloidoses. Amyloidosis in the common age-related forms may prove secondary. Until the causes of Aβ accumulation in age-related AD are revealed, it seems wise not to assume Aβ aggregation drives the disease, or that Aβ removal will cure it.
AD is now associated with increasing numbers of clinical, biochemical, and histologic markers. The fidelity of these associations, though, does not necessarily address pathogenesis. After all, it would be wrong to conclude serum troponin causes myocardial infarcts simply because levels are consistently elevated in myocardial infarct states. On the other hand, the more AD phenomena we recognize, the more likely it is the pieces of the AD puzzle will one day come together.
Grant information
Supported by the National Institute of Aging (AG022407).
References
- AlzheimerA1907Uber eine eigenartige Erkrankung der HirnrindeAllg Z Psychiat Psych-Gerichtl Med641468
- AmaducciLARoccaWASchoenbergBS1986Origin of the distinction between Alzheimer’s disease and senile dementia: How history can clarify nosologyNeurology36149793531918
- AnandtheerthavaradaHKBiswasGRobinMAAvadhaniMG2003Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s precursor protein impairs mitochondrial function in neuronal cellsJ Cell Biol164154
- ArendtT2000Alzheimer’s disease as a loss of differentiation control in a subset of neurons that retain immature features in the adult brainNeurobiol Aging217839611124422
- BlackerDTanziRE1998The genetics of Alzheimer disease: Current status and future prospectsArch Neurol5529469520001
- BlansjaarBAThomassenRVan SchaickHW2000Prevalence of dementia in centenariansInt J Geriatr Psychiatry152192510713579
- BlassJPBakerACKoLBlackRS1990Induction of Alzheimer antigens by an uncoupler of oxidative phosphorylationArch Neurol4786492375692
- BollerFForbesMM1998History of dementia and dementia in history: An overviewJ Neurol Sci158125339702682
- BowserRSmithMA2002Cell cycle proteins in Alzheimer’s disease: Plenty of wheels but no cycleJ Alzheimers Dis42495412226545
- CardosoSMSantosSSwerdlowRHOliveiraCR2001Functional mitochondria are required for amyloid beta-mediated neurotoxicityFASEB J1514394111387250
- CaselliRJGraff-RadfordNRReimanEM1999Preclinical memory decline in cognitively normal apolipoprotein E-ɛ4 homozygotesNeurology53201710408560
- CasleyCSCanevariLLandJMClarkJBSharpeMA2002Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activitiesJ Neurochem809110011796747
- CastellaniRJLeeHGZhuXNunomuraAPerryGSmithMA2006Neuropathology of Alzheimer disease: Pathognomonic but not pathogenicActa Neuropathol111503916718346
- ChagnonPGeeMFilionM1999Phylogenetic analysis of the mitochondrial genome indicates significant differences between patients with Alzheimer disease and controls in a French-Canadian founder populationAm J Med Genet85203010377009
- ChangSran MaTMirandaRD2005Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicityProc Natl Acad Sci USA10218694916344479
- Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease1997The national institute on aging, and reagan institute working group on diagnostic criteria for the neuropathological assessment of Alzheimer’s diseaseNeurobiol Aging184 SupplS129330978
- CookDGLeverenzJBMcMillanPJ2003Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer’s disease is associated with the apolipoprotein E-epsilon4 alleleAm J Pathol162313912507914
- CorderEHSaundersAMStrittmatterWJ1993Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset familiesScience26192138346443
- CoskunPEBealMFWallaceDC2004Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replicationProc Natl Acad Sci USA101107263115247418
- CrouchPJBlakeRDuceJA2005Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1–42J Neurosci25672915659604
- DavisJN2ndChisholmJC1999Alois Alzheimer and the amyloid debateNature2681010476951
- DeMattosRBBalesKRParsadanianM2002Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer’s diseaseJ Neurochem812293612064470
- DeviLPrabhuBMGalatiDFAvadhaniNGAnandatheerthavaradaHK2006Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunctionJ Neurosci2690576816943564
- DingQDimayugaEKellerJN2006Proteasome regulation of oxidative stress in aging and age-related diseases of the CNSAntioxid Redox Signal81637216487050
- EdlandSDSilvermanJMPeskindER1996Increased risk of dementia in mothers of Alzheimer’s disease cases: Evidence for maternal inheritanceNeurology4725468710088
- EriksenJLSagiSASmithTE2003NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivoJ Clin Invest112440912897211
- EvansDAFunkensteinHHAlbertMS1989Prevalence of Alzheimer’s disease in a community population of older persons. Higher than previously reportedJAMA262255162810583
- FaganAMMintunMAMachRH2006Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta(42) in humansAnn Neurol59512916372280
- FerrerIBoada RoviraMSanchez GuerraMLReyMJCosta-JussaF2004Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s diseaseBrain Pathol14112014997933
- FischerO1907Miliare Nekrosen mit drusigen Wucherungen der Neurofibrillen, eine regelmabige Veranderung der Hirnrinde bei seniler DemenzMonatsschr Psychiatr Neurol2236172
- GabuzdaDBusciglioJChenLBMatsudairaPYanknerBA1994Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivativeJ Biol Chem2691362388175797
- GearingMMirraSSHedreenJC1995The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part X. Neuropathology confirmation of the clinical diagnosis of Alzheimer’s diseaseNeurology4546167898697
- GeldmacherDSFritschTMcClendonMJLernerAJLandrethGE2006A double-blind, placebo controlled, 18 momth pilot study of the PPAR-gamma agonist pioglitazone in Alzheimer’s diseaseProgram and abstracts of the 10th International Conference on Alzheimer’s Disease and Related Disorders, Abstract P2408
- GhoshSSSwerdlowRHMillerSW1999Use of cytoplasmic hybrid cell lines for elucidating the role of mitochondrial dysfunction in Alzheimer’s disease and Parkinson’s diseaseAnn N Y Acad Sci8931769110672237
- GibsonGESheuKFBlassJP1998Abnormalities of mitochondrial enzymes in Alzheimer’s diseaseJ Neural Transm105855709869323
- GilmanSKollerMBlackRS2005Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trialNeurology6415536215883316
- GlabeCGKayedR2006Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesisNeurology662 Suppl 1S74816432151
- GoateAChartier-HarlinMCMullanM1991Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s diseaseNature34970461671712
- HambletNSRaglandBAliMMutations in mitochondrial-encoded cytochrome c-oxidase subunits I, II, and III genes detected in Alzheimer’s disease using single-strand conformation polymorphismElectrophoresis2739840816358358
- HanssonCAFrykmanSFarmeryMR2004Nicastrin, presenilin, APH-1, and PEN-2 form active gamma-secretase complexes in mitochondriaJ Biol Chem279516546015456764
- HardyJAllsopD1991Amyloid deposition as the central event in the aetiology of Alzheimer’s diseaseTrends Pharmacol Sci1238381763432
- HardyJAHigginsGA1992Alzheimer’s disease: the amyloid cascade hypothesisScience25618451566067
- HarmanD1956Aging: a theory based on free radical and radiation chemistryJ Gerontol1129830013332224
- HarmanD1972A biological clock: The mitochondria?J Am Ger Soc201457
- JichaGAParisiJEDicksonDW2005Alzheimer and Lewy body pathology in centenarian case seriesNeurology6Suppl 1A275
- KatzmanR1976The prevalence and malignancy of Alzheimer’s disease: a major killerArch Neurol3321781259639
- KhachaturianZS1985Diagnosis of Alzheimer’s diseaseArch Neurol4210971062864910
- KhanSMCassarinoDSAbramovaNN2000Alzheimer’s disease cybrids replicate beta-amyloid abnormalities through cell death pathwaysAnn Neurol481485510939564
- KhanSMSmigrodzkiRMSwerdlowR2006Cell and animal models of mtDNA biology: Progress and prospectsAm J Physiol Cell Physiol89[Epub ahead of print]. PMID 16899549.
- KimberlyWTXiaWRahmaticJWolfeMSSelkoeDJ2000The transmembrane aspartates in presenilin 1 and 2 are obligatory for gamma-wcretase activity and amyloid beta-protein generationJ Biol Chem2753173810652302
- KlunkWEEnglerHNordbergA2004Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-BAnn Neurol553061914991808
- KraepelinEPsychiatrie. 1910 Ein Lehrbuch fur Studierende und Artze. II. Band, Klinische Psychiatrie. Verlag Johann Ambrosius Barth, Leipzig.
- KujothGCHionaAPughTD 309. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian agingScience309481416020738
- LesneSKotilinekL2005Amyloid plaques and amyloid-beta oligomers: An ongoing debateJ Neurosci2593192016221839
- LevyR1994Aging-associated cognitive decline. Working Party of the International Psychogeriatric Association in collaboration with the World Health OrganizationInt Psychogeriatr66388054494
- Levy-LahadEWascoWPoorkajP1995Candidate gene for the chromosome 1 familial Alzheimer’s disease locusScience26997377638622
- LinMTSimonDKAhnCHKimLMBealMF2002High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brainHum Mol Genet111334511809722
- LinnaneAWMarzukiSOzawaTTanakaM1989Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseasesLancet164252564461
- LockePAConneallyPMTanziREGusellaJFHainesJL1995Apolipoprotein E, survival in Alzheimer’s disease patients, and the competing risks of death and Alzheimer’s diseaseNeurology45132387617191
- LustbaderJWCirilliMLinC2004ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s diseaseScience3044485215087549
- ManczakMAnekondaTSHensonE2006Mitochondria are a direct site of A beta accumulation in Alzheimer‘s disease neurons: implications for free radical generation and oxidative damage in disease progressionHum Mol Genet1514374916551656
- MasliahEHansenLAdameA2005Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer diseaseNeurology641293115642916
- McSheaAWahlAFSmithMA1999Re-entry into the cell cycle: A mechanism for neurodegeneration in Alzheimer diseaseMed Hypotheses52525710459833
- MiquelJEconomosACFlemingJJohnsonJEJr1980Mitochondrial role in cell agingExp Gerontol15575917009178
- MirraSSHeymanAMcKeelD1991The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). II. Standardization of the neuropathological assessment of Alzheimer’s diseaseNeurology41479862011243
- MiyataMSmithJD1996Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and beta-amyloid peptidesNat Genet1455618782820
- MizutaniTShimadaH1992Neuropathological background of twenty-seven centenarian brainsJ Neurol Sci108168771517748
- MorrisJC2006Mild cognitive impairment is early-stage Alzheimer disease: Time to revise diagnostic criteriaArch Neurol6315616401731
- NearyDSnowdenJMannD2005Frontotemporal dementiaLancet Neurol47718016239184
- NicollJAWilkinsonDHolmesC2003Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: A case reportNat Med94485212640446
- ParkerWDJrFilleyCMParksJK1990Cytochrome oxidase deficiency in Alzheimer’s diseaseNeurology40130232166249
- ParkerWDJrParksJK1995Cytochrome c oxidase in Alzheimer’s disease brain: purification and characterizationNeurology4548267898701
- PearlR1928The rate of living, being an account of some experimental studies on the biology of life durationNew YorkAlfred A. Knopf
- PereiraCSantosMSOliveiraC1998Mitochondrial function impairment induced by amyloid beta-peptide on PC12 cellsNeuroreport91749559665595
- PerlsT2004Dementia-free centenariansExp Gerontol3915879315582273
- PetersenRCSmithGEWaringSC1999Mild cognitive impairment: Clinical characterization and outcomeArch Neurol56303810190820
- PoirierJ2000Apoliproptein E and Alzheimer’s disease. A role in amyloid catabolismAnn NY Acad Sci924819011193807
- PolvikoskiTSulkavaRRastasS2005Incidence of dementia in very elderly individuals: A clinical, neuropathological and molecular genetic studyNeuroepidemiology26768216352910
- PriceJLKoAIWadeMJNeuron number in the entorhinal cortex and CA1 in preclinical Alzheimer diseaseArch Neurol58139540211559310
- ReddyPH2006Mitochondrial oxidative damage in aging and Alzheimer’s disease: Implications for mitochondrially targeted antoxidant therapeuticsJ Biomed Biotechnol20063137217047303
- ReimanEMCaselliRJYunLS1996Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apoliprotein ENEJM33475288592548
- RelkinNSzaboPAdamiakB2005Intravenous immunoglobulin (IVIg) treatment causes dose-dependent alterations in β-amyloid (Aβ) levels and anti-Aβ antibody titers in plasma and cerebrospinal fluid (CSF) of Alzheimer’s disease (AD) patientsNeurology64A144
- RentzDMBeckerJAMoranEK2006Amyloid imaging with Pittsburgh Compound-B (PIB) in AD, MCI, and highly intelligent older adultsNeurology66suppl 2A66
- RisnerMESaundersAMAltmanJF2006Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s diseasePharmacogenomics J62464516446752
- SaundersAM2000Apolipoprotein E and Alzheimer disease: An update on genetic and functional analysesJ Neuropath Exp Neurol59751811005255
- SchenkDBarbourRDunnW1999Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouseNature400173710408445
- ScheunerDEckmanCJensenM1996Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s diseaseNat Med2864708705854
- SherringtonRRogaevaEILiangY1995Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s diseaseNature375754607596406
- Shoghi-JadidKSmallGWAgdeppaED2002Localization of neurofibrillary tangles and beta amyloid plaques in the brains of living patients with Alzheimer diseaseAm J Geriatr Psychiatry10243511790632
- SmallGWMazziottaJCCollinsMT1995Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer diseaseJAMA27394277884953
- SmythKAFritschTCookTB2004Worker functions and traits associated with occupations and the development of ADNeurology6349850315304581
- SnowdonDAKemperSJMortimerJA1996Linguistic ability in early life and cognitive function and Alzheimer’s disease in late life. Findings from the Nun StudyJAMA275528328606473
- SternY2006Cognitive reserve and Alzheimer diseaseAlzheimer Dis Assoc Disord203 suppl 2S697416917199
- SwerdlowRHParksJKCassarinoDS1997Cybrids in Alzheimer’s disease: A cellular model of the disease?Neurology49918259339668
- SwerdlowRH2002Mitochondrial DNA – related mitochondrial dysfunction in neurodegenerative diseasesArch Pathol Lab Med1262718011860299
- SwerdlowRHKishSJ2002Mitochondria in Alzheimer’s diseaseInt Rev Neurobiol533418512512346
- SwerdlowRHKhanSM2004A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s diseaseMed Hypotheses6382015193340
- SwerdlowRH2006Is aging part of Alzheimer’s disease, or is Alzheimer’s disease part of aging?Neurobiol Aging728[Epub ahead of print] PMID: 16876913.
- SwerdlowRH2007Mitochondria in cybrids containing mtDNA from persons with mitochondriopathiesJ Neurosci Res122[Epub ahead of print] PMID: 17243174.
- SzabadosTDulCMajtenyiK2004A chronic Alzheimer’s model evoked by mitochondrial poison sodium azide for pharmacological investigationsBehav Brain Res154314015302108
- TamagnoEBardiniPObbiliA2002Oxidative stress increases expression and activity of BACE in NT2 neuronsNeurobiol Dis102798812270690
- ThomassenRvan SchaickHWBlansjaarBA1998Prevalence of dementia over age 100Neurology5028369443495
- TrifunovicAWredenbergAFalkenbergM2004Premature ageing in mice expressing defective mitochondrial DNA polymeraseNature42941742315164064
- TuszynskiMHThalLPayM2005A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer diseaseNat Med11551515852017
- van der WaltJMDementievaYAMartinER2004Analysis of European mitochondrial haplogroups with Alzheimer disease riskNeurosci Lett365283215234467
- VincentIRosadoMDaviesP1996Mitotic mechanisms in Alzheimer’s disease?J Cell Biol132413258636218
- WallaceDC1992Mitochondrial genetics: a paradigm for aging and degenerative diseases?Science256628321533953
- WalshDMKlyubinIShankarGM2005The role of cell-derived oligomers of Abeta in Alzheimer’s disease and avenues for therapeutic interventionBiochem Soc Trans3310879016246051
- WolfeWTXiaWOstaszewskiBL1999Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activityNature398513710206644
- YangYGeldmacherDSHerrupK2001DNA replication precedes neuronal cell death in Alzheimer’s diseaseJ Neurosci2126628
- ZhengHJiangMTrumbauerME1995Beta-amyloid precursor protein-deficient mice show reactive gliosis and decreased locomoter activityCell81525317758106
- ZhuXRainaAKPerryGSmithMA2004Alzheimer’s disease: The two-hit hypothesisLancet Neurol32192615039034
- ZlokovicBV2005Neurovascular mechanisms of Alzheimer’s neurodegenerationTrends Neurosci28202815808355