Abstract
The molecular mechanisms involved in human aging are complicated. Two progeria syndromes, Werner’s syndrome (WS) and Hutchinson-Gilford progeria syndrome (HGPS), characterized by clinical features mimicking physiological aging at an early age, provide insights into the mechanisms of natural aging. Based on recent findings on WS and HGPS, we suggest a model of human aging. Human aging can be triggered by two main mechanisms, telomere shortening and DNA damage. In telomere-dependent aging, telomere shortening and dysfunction may lead to DNA damage responses which induce cellular senescence. In DNA damage-initiated aging, DNA damage accumulates, along with DNA repair deficiencies, resulting in genomic instability and accelerated cellular senescence. In addition, aging due to both mechanisms (DNA damage and telomere shortening) is strongly dependent on p53 status. These two mechanisms can also act cooperatively to increase the overall level of genomic instability, triggering the onset of human aging phenotypes.
Introduction
Aging is an extremely complicated process and is known to be driven by a variety of different mutually interacting mechanisms. Conventionally, it is seen as a process of progressive failure of homeostasis involving genes for maintenance and repair, environmental factors leading to molecular damage and molecular heterogeneity, and chance events with potentially significant consequences for death (CitationAlberts et al 2002). Since several human progeroid synqdromes (PSs) (human premature aging-like syndromes) are characterized by features resembling precocious aging, the identification of the genes involved in PSs has provided important clues to understanding the molecular mechanisms underlying normal human aging (CitationHutchinson 1886, CitationGilford 1904; CitationMartin 1978, Citation1985).
The classic example of PS is Werner’s syndrome (WS), a progeria of the adult, which is caused by a mutation in a gene coding for a member of the RecQ helicase family, WRN (CitationYu et al 1996) and is characterized by features resembling precocious aging, appearing as a variety of visible features associated with aging, such as graying of the hair and skeletal changes, which occur much earlier than normal (CitationGoto 1997). WRN acts as a “caretaker of the genome” and functions in both DNA repair and transcription, suggesting that breakdown of these processes is critical in promoting aging (CitationEpstein et al 1966; CitationGoto 1997; CitationBachrati et al 2003). Hutchinson-Gilford progeria syndrome (HGPS), a progeria of the child, is the other classic example, and is caused by mutations in the gene LMNA (1q21.2) (CitationEriksson et al 2003), encoding a nuclear envelope protein, lamin A, which has been shown to affect RNA polymerase II transcription, probably by alterations in chromatin organization (CitationMartin and Oshima 2000; CitationSpann et al 2002; CitationHickson 2003; CitationAndressoo and Hoeijmakers 2005). In addition, skin fibroblast biopsies from patients with HGPS fail to show evidence of normal DNA strand rejoining in vitro after exposure to irradiation, suggesting that DNA repair may be absent or greatly reduced in the cells (CitationEpstein et al 1973). Thus, these two PSs directly mimic the clinical and molecular features of natural aging, providing unique insights into the molecular mechanisms underlying human aging (CitationMartin 1978, Citation1985). In this review, we will focus on these two best characterized PSs, with particular emphasis on the functions of the mutated syndrome-causing genes, and their potential implications for resemble aging. Based on the current information on these two syndromes, we also propose a model to explain human aging.
Werner’s syndrome
Werner’s syndrome is a rare autosomal recessive disorder characterized by premature aging (CitationEpstein et al 1966; CitationMartin et al 1970; CitationGoto et al 1996; CitationYu et al 1996). The WRN gene responsible for WS encodes the 1432 amino acid WRN protein, which is a member of the RecQ DNA helicase family (CitationYu et al 1996). The C-terminal region of WRN contains the conserved RQC domain, which includes a nucleolar targeting sequence (NTS) (CitationMarciniak et al 1998; Citationvon Kobbe and Bohr 2002), and the helicase and RNaseD C-terminal (HRDC) domain (CitationLiu et al 1999). WRN has DNA-dependent ATPase, DNA strand annealing, DNA helicase, and exonuclease activities (CitationGray et al 1997; CitationSuzuki et al 1997, Citation1999; CitationHuang et al 1998). WRN, as a “caretaker of the genome”, is needed to prevent telomere dysfunction and consequent genomic instability (CitationOpresko et al 2003; CitationCrabbe et al 2004; CitationDu et al 2004). Since most of the mutations identified in WRN cause premature termination of translation (CitationGoto 1997; CitationYu et al 1997), resulting in impaired nuclear import of the protein (CitationMatsumoto et al 1997), the clinical features and cellular phenotypes of WS patients are major due to an absolute lack of normal WRN in the nucleus. WS cells have defects in DNA replication, resulting in dysfunction in multiple cellular DNA metabolic processes, such as DNA replication initiation, replication foci establishment, and the resolution of stalled replication forks during replication and DNA repair (CitationMartin et al 1970; CitationHasty et al 2003). In addition, a large number of reports have shown that many cellular events, including transcription and apoptosis, are affected in WS cells (CitationOgburn et al 1997; CitationBalajee et al 1999; CitationSpillare et al 1999, Citation2006; CitationSommers et al 2005). The loss of WRN function in WS appears as chromosome instability, a shorter life span in culture (CitationMartin 1977), and accelerated telomere shortening (CitationSchulz et al 1996), suggesting that breakdown of WRN function is important in promoting aging (CitationMartin et al 1970; CitationHasty et al 2003).
Multiple functions of the WRN protein
Biochemistry and catalytic activities
WRN, a multifunctional nuclear protein, interacts with a number of proteins to catalyze four major DNA-dependent enzymatic activities, acting (1) as a DNA-dependent ATPase, (2) as an intrinsic 3′→5′ helicase (CitationGray et al 1997), (3) as a 3′→5′ exonuclease (CitationHuang et al 1998, Citation2000), and (4) in DNA strand annealing (CitationMachwe et al 2005), and interacts with factors with established roles in DNA metabolic pathways (CitationBrosh and Bohr 2002). WRN contains three distinct structure-specific DNA-binding domains, one N-terminal domain and two different C-terminal fragments (the RecQ and HRDC domains), each of which plays roles in distinct DNA metabolic pathways (Citationvon Kobbe et al 2003a).
ATP-stimulated activities are required for the successive steps in the hydrolytic reaction, such as structure recognition, DNA binding, and 3′-terminal hydrolysis. In this context, WRN plays a DNA-dependent ATPase, using the energy from ATP hydrolysis to unwind double-stranded DNA, and its exonuclease and helicase activities act in concert to catalyze structure-dependent DNA degradation in resolving aberrant DNA structures (CitationShen and Loeb 2000). As a helicase, WRN is active on the forked end of a DNA duplex, while, as an exonuclease, it acts on the blunt end of the same duplex (CitationGray et al 1997; CitationHuang et al 1998; CitationOpresko et al 2001). WRN exonuclease can efficiently remove a mismatched nucleotide at a 3′ recessed terminus and can initiate DNA degradation from a 12-nucleotide gap or a nick (CitationHuang et al 2000). Furthermore, WRN forms a trimer, which interacts with the proliferating cell nuclear antigen (PCNA) to participate in the replication restart process (CitationHuang et al 2000; CitationRodríguez-López et al 2003; CitationJeziorny et al 2006). WRN exonuclease activity is suppressed by interaction with p53 (CitationBrosh et al 2001a) or BLM (Citationvon Kobbe et al 2002) and stimulated by interaction with Ku70/80 (CitationLi and Comai 2001) or phosphorylation (CitationKarmakar et al 2002). WRN helicase activity is stimulated by interaction with replication protein A (RPA) (CitationShen et al 1998) or telomere repeat binding factor 2 (TRF2) (CitationOpresko et al 2002) and phosphorylation (CitationKarmakar et al 2002). Moreover, WRN colocalizes and interacts with RAD52 and has strand annealing activity in addition to its DNA unwinding activity (CitationBaynton et al 2003; CitationMachwe et al 2005, Citation2006). WRN-dependent unwinding activity is significantly stronger than previously believed (CitationMachwe et al 2006).
Post-translational modifications
In response to DNA damage, WRN can be modulated by post-translational modifications, including phosphorylation, sumoylation, and acetylation. It is phosphorylated at serine/threonine and tyrosine residues in vivo after bleomycin treatment or after replication stress (CitationKarmakar et al 2002; CitationCheng et al 2003; CitationPichierri et al 2003). WRN phosphorylation at serine/threonine residues is primarily dependent on DNA-PKcs and c-Abl kinases (CitationKarmakar et al 2002; CitationCheng et al 2003; CitationPichierri et al 2003). Importantly, the serine/threonine phosphorylation of WRN by the DNA-PK complex results in inhibition of both WRN helicase and exonuclease activities, whereas dephosphorylation of WRN enhances both these activities. Thus, the serine/threonine phosphorylation status of WRN plays a role in the regulation of its catalytic activities (CitationKarmakar et al 2002; CitationCheng et al 2003). c-Abl was also found to phosphorylate WRN (CitationCheng et al 2003). c-Abl kinase, a regulator of the DNA damage response, mediates WRN nuclear localization and catalytic activities in response to DNA damage. WRN directly binds to c-Abl and this interaction is disrupted in the early cellular response to bleomycin (CitationCheng et al 2003). As with phosphorylation by the DNA-PK complex, c-Abl phosphorylation inhibits WRN helicase and exonuclease activities (CitationCheng et al 2003).
Phosphorylation of WRN may influence other forms of WRN post-translational modification, such as sumoylation and/or acetylation. Modification of proteins by the small ubiquitin-related modifier 1 (SUMO-1) conjugating system requires a set of enzymes, including SUMO-activating (E1), conjugating (Ubc9), and ligating enzymes (CitationMuller et al 2004). WRN interacts with Ubc9, which is required for conjugation of SUMO-1 to the N-terminal fragment (amino acids 272–514) of WRN (CitationKawabe et al 2000). In addition, binding of p14 Arf to WRN is multivalent and resembles the binding of p14 Arf to Mdm2, promoting sumoylation of WRN in a synergistic manner with the SUMO-conjugating enzyme Ubc9 (CitationWoods et al 2004). p14 Arf causes redistribution of WRN within the nucleus, and this effect is reversed by expression of a SUMO-specific protease, thus implicating the SUMO conjugation pathway in controlling WRN relocalization (CitationWoods et al 2004). Besides phosphorylation and sumoylation, WRN can be acetylated after mitomycin C or methyl methane-sulfonate treatment (CitationSharma et al 2005). On exposure to UV or ionizing radiation, WRN is acetylated by acetyltransferase p300, a transcriptional coactivator, which has acetylation activity (CitationBlander et al 2002).
DNA replication
WRN helicase unwinds replication fork structures very efficiently (CitationMohaghegh et al 2001), acting to resolve the block and/or in the replication restart process (CitationRodríguez-López et al 2003). Compared to normal cells, cells from WS patients undergo premature replicative senescence (CitationMartin et al 1970; CitationSalk et al 1985), display an extended S-phase (CitationPoot et al 1992), and show a reduced frequency of replication initiation sites (CitationTakeuchi et al 1982; CitationHanaoka et al 1985), thus exhibiting defects in DNA replication consistent with the inability to properly recover from DNA replication fork demise (CitationHickson 2003).
WRN is able to interact with PCNA, DNA topoisomerase I (topo I), polδ, and RPA (CitationWold 1997; CitationLebel and Leder 1998; CitationBrosh et al 1999; CitationLebel et al 1999; CitationConstantinou et al 2000; CitationHuang et al 2000; CitationLaine et al 2003) and catalyze DNA unwinding in vitro for DNA replication, recombination, and repair (CitationBrosh and Bohr 2002).
WRN and PCNA colocalize at replication foci, suggesting a physiological interaction between them in cycling primary cells (CitationRodríguez-López et al 2003). In addition, WRN is as part of the 17S multiprotein DNA replication complex, and establishes PCNA and topoisomerase I as the two WRN-interacting components (CitationWarbrick 1998, Citation2000; CitationLebel et al 1999; CitationRodríguez-López et al 2003).
Polδ participates in DNA replication and repair of DNA damage. WRN interacts specifically with the p50 subunit of polδ, and WRN directly modifies DNA replication via its interaction with p50 and is involved in the dynamic relocalization of polδ complexes within the nucleus (CitationKamath-Loeb et al 2000; CitationSzekely et al 2000). Moreover, WRN enhances the rate of nucleotide incorporation in polδ-mediated replication in the absence of PCNA and its helicase activity enables polδ to overcome hairpin and G-quadruplex DNA structures (CitationFry and Loeb 1999; CitationSzekely et al 2000; CitationKamath-Loeb et al 2000, Citation2001; CitationBrosh et al 2001b; CitationMohaghegh et al 2001). Thus, WRN may facilitate polδ-mediated DNA replication, and disruption of the WRN-polδ interaction in WS cells may contribute to the S-phase defects.
Human RPA is a heterotrimeric, single stranded DNA-binding protein required for DNA replication, recombination, and repair (CitationWold 1997; CitationBrosh et al 1999, Citation2002). RPA directly interacts with to WRN, markedly increasing the DNA helicase activity of WRN (CitationBrosh et al 1999; CitationShen et al 2003) and its ability to unwind forked telomeric DNA structures (CitationOhsugi et al 2000; CitationOpresko et al 2001). After DNA damage, WRN can colocalize with BLM and RPA (CitationConstantinou et al 2000; CitationBischof et al 2001; CitationSakamoto et al 2001). RPA binds to WRN and BLM to stimulate their unwinding of long DNA duplexes (CitationBachrati and Hickson 2003).
Another interesting replication protein, FEN-1, interacts with the 144-amino acid RQC domain on the C-terminal region of WRN (CitationBrosh et al 2001c). FEN-1, a DNA structure-specific nuclease, participates in pathways of DNA metabolism that are important for genomic stability (CitationBrosh et al 2002) and is involved in the maturation of Okazaki fragments during lagging strand DNA replication (CitationBambara et al 1997; CitationMerrill and Holm 1998). WRN stimulates FEN-1-mediated cleavage activity of displaced flaps that occur during lagging strand DNA synthesis at Okazaki fragments (CitationBrosh et al 2001c; CitationSharma et al 2004). WRN-FEN-1 complex colocalizes in foci associated with arrested replication forks, and this complex plays a role in the unwinding and degradation of Holliday junction structures associated with regressed replication forks (CitationSharma et al 2004). Defective Okazaki fragment processing causes DSBs, which may lead to the genomic instability in WS (CitationBrosh et al 2002).
WRN colocalizes with, and directly interacts with, human topo I (CitationLaine et al 2003). WRN stimulates the ability of topo I to relax negatively supercoiled DNA and specifically stimulates the religation step of the relaxation reaction, and cell extracts from WS fibroblasts exhibit a decreased ability to unwind negatively supercoiled DNA (CitationLaine et al 2003). These findings provide the interrelationship between WRN helicases and topoisomerases in the maintenance of genomic integrity.
DNA repair
WS cells display sensitivity to 4-nitroquinoline 1-oxide (CitationOgburn et al 1997; CitationPoot et al 2002), a carcinogen which causes the formation of DNA strand breaks and bulky DNA adducts (CitationNagao and Sugimura 1976), and are also hypersensitive to O6-methylguanine, a site-specific alkylating agent that can block DNA replication (CitationBlank et al 2004). WS cells are sensitive to DNA crosslinking drugs (CitationPoot et al 2001, Citation2002) and, since DNA crosslinks are normally repaired by homologous repair (HR), this suggests a defect in this repair pathway. WS cells display extensive deletions at nonhomologous joined ends (CitationOshima et al 2002; CitationChen et al 2003), while expression of wild-type WRN prevents excessive DNA deletions (CitationOshima et al 2002; CitationChen et al 2003). These findings suggest that lack of WRN may increase DNA damage and disrupt the regulatory processes controlling DNA repair, for example, nonhomologous end joining (NHEJ), HR, and base excision repair (BER).
Double-strand break repair
The first evidence for a link between WRN and double-strand break repair was the discovery that WRN interacts with both Ku and DNA-dependent protein kinase catalytic subunits (DNA-PKcs) (CitationCooper et al 2000; CitationLi and Comai 2000; CitationCheng et al 2003; CitationOpresko et al 2003) to participate in NHEJ (CitationLi and Comai 2002). Assembly of DNA-PK and WRN at DNA ends allows DNA-PKcs to phosphorylate WRN, thus stimulating WRN enzymatic activity and facilitating efficient processing of double-strand breaks (DSBs) prior to ligation (CitationYannone et al 2001). Deficiencies in either Ku or DNA-PKcs result in sensitivity to ionizing radiation due to defects in DSB repair. Complementation with the exonuclease/helicase double mutant or wild-type WRN restores NHEJ activity, suggesting that WRN is necessary for normal repair of DSBs (CitationChen et al 2003).
Chromatographic studies showed that WRN is bound to PARP-1 in a complex that contains Ku70/80 (CitationLi et al 2004). PARP-1 can induce apoptosis or necrosis in cells with extensive DNA damage. Absence of functional WRN prevents activation of PARP-1 in response to DNA damage caused by oxidative stress and alkylating agents (Citationvon Kobbe et al 2003b).
Some proteins that participate in the recombinational repair pathway have been found to functionally interact with WRN. WRN is involved in resolving recombination intermediates in RAD51-dependent HR (CitationPrince et al 2001; CitationSaintigny et al 2002) and forms distinct nuclear foci that partially overlap with the RAD51 nuclear foci formed in response to DNA damage (including DSBs) (CitationSakamoto et al 2001). In vivo data show that WRN interacts functionally with NBS1 (CitationCheng et al 2004), which is thought to act downstream of RAD51 (CitationSaintigny et al 2002; CitationTauchi et al 2002; CitationMonnat and Saintigny 2004). WRN colocalizes with, and interacts with, RAD52 (CitationBaynton et al 2003), NBS1 in the MRN complex (CitationCheng et al 2004), and flap endonuclease 1 (FEN-1) (CitationBrosh et al 2001c; CitationSharma et al 2004). In addition, it interacts physically with the Mre11-RAD50-NBS1 complex, which also functions in HR for DSB processing (CitationCheng et al 2004).
Base excision repair
Oxidative DNA lesions are repaired primarily by base excision repair (BER), and the accumulation of oxidative products and the resulting phenotypic changes have been implicated in the aging process (CitationBeckman et al 1998). In general, BER can be divided into short patch repair and long patch repair. WRN has been shown to physically interacts with polβ and participate in short patch BER (CitationHarrigan et al 2003). The active WRN helicase domain stimulates polβ strand displacement DNA synthesis at a nick on a BER substrate (CitationHarrigan et al 2003). In addition, it has been shown to interact physically and/or functionally with several replication proteins which participate in long patch BER, including PCNA, RPA, polδ, and FEN-1 (CitationShen et al 1998; CitationBrosh et al 1999, Citation2002; CitationKamath-Loeb et al 2001). PCNA is part of a sliding clamp which forms a ring which maintains the connection between polymerase and its DNA template, allowing uninterrupted synthesis. WRN has been shown to directly interact with PCNA in vitro, suggesting a unique role for WRN in DNA synthesis (CitationHuang et al 2000). WRN stimulates FEN-1 flap cleavage (CitationBrosh et al 2001c, Citation2002) and nucleotide incorporation by polδ (CitationKamath-Loeb et al 2000, Citation2001). RPA stimulates WRN helicase unwinding of long substrates (CitationShen et al 1998; CitationBrosh et al 1999) and the poly(ADP-ribosyl)ation state of PARP-1 regulates WRN helicase and exonuclease activities (Citationvon Kobbe et al 2004). PARP-1 binds strongly to strand breaks and acts in the DNA damage surveillance network, partly by ribosylating a variety of nuclear proteins in response to DNA damage.
DNA recombination
Cellular DNA recombination can occur physiologically during meiotic DNA replication and V(D)J recombination, or can be induced by DNA damaging agents. Some reports define a physiological role for WRN RecQ helicase in recombination via RAD51-dependent HR (CitationPrince et al 2001; CitationSaintigny et al 2002). WRN and BLM colocalize to DNA damage-induced RAD51 foci, implicated in HR (CitationBischof et al 2001; CitationSakamoto et al 2001; CitationWu et al 2001; CitationSaintigny et al 2002; CitationVon Kobbe et al 2002; CitationWu and Hickson 2003; CitationSpillare et al 2006). The WRN and BLM helicase activities are possible in a synergistic manner to intermediate DNA recombination, since a coimmunoprecipitation and colocalization study showed that the exonuclease domain of WRN interacts with BLM (Citationvon Kobbe and Bohr 2002). In addition, a biochemical study showed that WRN interacts with the homologous recombination mediator protein RAD52, and that WRN and RAD52 form a complex, should be a general response to replication forks arrested by DNA damage (CitationBaynton et al 2003).
WRN interacts physically and functionally with the MRN complex via NBS1 and they colocalize in response to ionizing radiation or mitomycin C treatment (CitationCheng et al 2004). WS cells display a deficiency in resolving DNA recombination intermediates which contributes to DNA damage hypersensitivity, limited cell growth, and genomic instability (CitationPrince et al 2001). The generation of viable mitotic recombinant progeny was rescued by the expression of WRN, which also improved WS cell survival after DNA damage (CitationSaintigny et al 2002). These results define a physiological role for the WRN RecQ helicase protein in RAD51-dependent HR and identify a mechanistic link between defective recombination resolution and limited cell division potential, DNA damage hypersensitivity, and genetic instability in human somatic cells (CitationSaintigny et al 2002). WRN also colocalizes with RAD51 and RPA in response to DNA damaging agents (CitationConstantinou et al 2000; CitationSakamoto et al 2001), and both WRN and BLM interact with RPA (CitationBrosh et al 1999, Citation2000).
Telomere maintenance
Telomeres are specialized nucleoprotein structures consisting of G-rich repetitive sequences that cap the ends of eukaryotic chromosomes and are crucial for the maintenance of chromosomal integrity and cell viability (CitationMcClintock et al 1941). They are maintained by the enzyme telomerase, which consists of an essential telomerase RNA component (TERC), which serves as a template for the addition of telomere repeats, and a protein component, the telomerase reverse transcriptase catalytic subunit (TERT). Telomere shortening eventually results in diverse pathophysiological consequences, primarily through accelerated telomere erosion, and triggers entry into premature senescence (CitationWright and Shay 1992; CitationBlasco 2002; CitationChang et al 2004; CitationDu et al 2004). Defects in telomere structure can initiate a DNA damage response and may lead to telomeric end fusions and chromosome breakage if not properly repaired (CitationDe Lange 2002).
Biochemical and cellular evidence suggest that WRN may dissociate secondary structures at the telomere to allow replication, repair, and telomerase activity at the telomere end (CitationOpresko et al 2003). Under normal conditions, WRN associates with telomeres in S phase to prevent loss of individual telomeres (CitationCrabbe et al 2004). Accelerated loss of telomere reserves and activation of cellular checkpoints appear integral to the decreased replicative potential seen in WS, as evidenced by the capacity of enforced TERT expression to impart unlimited replicative potential (CitationWyllie et al 2000).
WRN functionally interacts with a number of proteins involved in telomere length maintenance, including Ku 70/86 (CitationOrren et al 2001), RPA (CitationBrosh et al 1999, Citation2000; CitationSanz et al 2000), TRF1, and TRF2 (CitationOpresko et al 2002, Citation2004; CitationMachwe et al 2004). RPA is a single strand DNA-binding protein that is required for all aspects of DNA metabolism (CitationBrosh et al 2000), while TRF1 and TRF2 are homodimeric proteins that have been shown to bind exclusively to double stranded telomeric DNA throughout the cell cycle and are thought to be involved in the regulation of telomeric length (CitationFairall et al 2001). TRF2 as a promoter for the helicase activity of WRN, the interaction between WRN and TRF2 may serve to stabilize TRF2 in its active form or to improve TRF2’s interaction with DNA (CitationOpresko et al 2002). Due to the absence of WRN in WS patients, TRF2 is unable to perform its duty and telomeric-specific structures that need to be degraded are not (CitationGriffith 1999; CitationOpresko 2002; CitationJeziorny 2006). These structures then act as barriers against various transcription factors and telomeres are left without being fully transcribed.
Taken together, these results suggest that WRN is necessary for the efficient replication of G-rich telomeric DNA and that WRN deficiency and dysfunctional telomeres appear to act cooperatively to increase the overall level of genomic instability, triggering the onset of premature aging phenotypes (CitationOrren et al 2001; CitationOpresko et al 2002, Citation2004).
DNA apoptosis
A link between WRN and apoptosis was first proposed by studies demonstrating that WS fibroblasts exhibit a decreased p53- mediated apoptotic response, and this deficiency can be rescued by expression of wild-type WRN (CitationSpillare et al 1999). p53 is a key cellular component in maintaining genomic stability either by regulating the cell cycle to allow DNA repair or by inducing apoptosis (CitationHaupt et al 2003; CitationHofseth et al 2004; CitationLane 2004). The C-terminus of WRN binds to the C-terminal domain of p53 to induce p53-mediated apoptosis (CitationClarke et al 1993; CitationSymonds et al 1994; CitationWang et al 1996, Citation2001; CitationSpillare et al 1999; CitationBrosh et al 2001a).
Fibroblasts from WS patients have a decreased ability to undergo p53-mediated apoptosis (CitationSpillare et al 1999), so the absence of WRN-p53 direct interaction could serve as a signal for programmed cell death. Moreover, the expression of wild-type WRN is sufficient to rescue WS−/− cells from the attenuation of p53-mediated apoptosis (CitationSpillare et al 1999; Blander et al 2001). p53 may exert its effect on WRN by its interaction with RPA. Overexpression of p53 results in a decrease in Sp-1-mediated transcription of the WRN gene, suggesting that p53 regulates WRN expression (CitationYamabe et al 1998). Thus, the interaction of WRN with p53 and/or the WRN-RPA complex may be critical in preventing entry into S phase or in directing S phase cells towards apoptosis. The absence of a p53-WRN helicase interaction may disrupt the signal to direct S-phase cells into apoptosis for programmed cell death and contribute to the pronounced genomic instability and cancer predisposition seen in WS cells (CitationSommers et al 2005). In addition, epigenetic inactivation (promoter hypermethylation) of WRN can lead to the loss of WRN-associated exonuclease activity and increased chromosomal instability and apoptosis induced by topoisomerase inhibitors (CitationAgrelo et al 2006).
Hutchinson-Gilford progeria syndrome
Hutchinson-Gilford progeria syndrome, a childhood progeroid disorder, is a rare, fatal genetic disorder characterized by segmental accelerated aging. Affected children appear normal at birth, but within a year develop characteristic features of failure to thrive, delayed dentition, alopecia, atherosclerosis, prominent scalp veins, a high pitched voice, and sclerodermatous skin changes, with death at approximately 13 years from atherosclerosis of the coronary and cerebrovascular arteries. CitationEriksson and colleagues (2003) identified the disease causing mutations in the LMNA gene (encoding lamin A/C). The vast majority of HGPS cases are caused by a single-base substitution (GGC > GGT), which does not cause an amino acid change (G608G), but results in deletion of 150 nucleotides in exon 11, causing an alternatively spliced truncated variant of lamin A mRNA and an in-frame deletion of 50 amino acids near the carboxy terminus, leading to changes in the nuclear architecture (CitationDe Sandre-Giovannoli et al 2003; CitationEriksson et al 2003). Other HGPS mutations that have been described in LMNA include E145K, R471C, R527C, G608S, T623S, and 1824C > T (CitationCao and Hegele 2003; CitationCsoka et al 2004a; CitationFukuchi et al 2004). Lamins form microfilaments in the nucleus and are important in maintaining the proper structure of the nuclei, but they also influence on chromatin structure, regulation of gene expression, localization and probably protein degradation (CitationLy et al 2000; CitationGoldman et al 2002; CitationScaffidi and Misteli 2005). Immunofluorescence of HGPS fibroblasts with antibodies directed against lamin A revealed that many cells show visible abnormalities of the nuclear membrane (CitationEriksson et al 2003). In addition, HGPS cells have also altered histone modification patterns, including reduced heterochromatin-specific trimethylation of Lys9 on histone H3 (Tri-Me-K9H3) (CitationScaffidi and Misteli 2005, Citation2006). A recent study implicated lamin A in physiological aging, showing that the molecular mechanism responsible for the premature aging (CitationHasty and Vijg 2004; CitationScaffidi and Misteli 2006). Thus, accelerated aging in HGPS might thus reflect an exaggerated lamin A-dependent mechanism, which contributes to physiological aging.
Defects in prelamin A processing
Lamin A has a conserved C-terminal CAAX motif, which is a potential target for subsequent processing steps. To generate mature lamin A, prelamin A undergoes substantial post-translational modification of its CAAX motif via four processing steps (CitationZhang and Casey 1996; CitationYoung et al 2006). First, a 15-carbon farnesyl lipid is added to the thiol group of the cysteine by a cytosolic enzyme, protein farnesyltransferase. Second, the last three amino acids of the protein (ie, the -AAX) are clipped off by the metalloprotease ZMP-STE24 and/or RCE1 (CitationBergo et al 2002; CitationCorrigan et al 2005). Third, the newly exposed farnesylcysteine is carboxylmethylated by ICMT, a prenylprotein-specific methyltransferase in the endoplasmic reticulum (CitationClarke et al 1988; CitationDai et al 1998). Fourth, the last 15 amino acids of the protein, including the farnesylcysteine methyl ester, are clipped off by ZMPSTE24 and degraded, releasing mature lamin A (CitationWeber et al 1989; CitationBeck et al 1990; CitationCorrigan et al 2005; CitationYoung et al 2006). In HGPS, a 50-amino acid deletion in the C-terminus of the protein (amino acids 607–656) lacks an important endoprotease cleavage site recognized by ZMPSTE24 during prelamin A post-translational processing, so no mature lamin A is formed and a farnesylated mutant prelamin A (progerin) accumulates in cells (CitationDe Sandre-Giovannoli et al 2003; CitationEriksson et al 2003; CitationNavarro et al 2006; CitationSun and Schatten 2006; CitationTsai et al 2006; CitationYoung et al 2006).
Progerin accumulation and nuclear morphology abnormalities
The mutant prelamin A is targeted to the nuclear rim, where it disrupts the integrity of the nuclear lamina, leading to premature cell death (CitationEriksson et al 2003; CitationGoldman et al 2004; CitationVarela et al 2005; CitationYang et al 2005; CitationFong et al 2006). The GGC > GGT (G608G) mutation of LMNA causes accumulation of farnesylprelamin A in the nucleus in a cellular age-dependent manner, and the cells display irregular nuclear shapes, including lobulation of the nuclear envelope, thickening of the nuclear lamina, loss of peripheral hetero-chromatin, and clustering of nuclear pores (CitationEriksson et al 2003; CitationGoldman et al 2004). Aberrant nuclear morphology is also reported with other LMNA mutations, which have been linked to other “laminopathies”, such as Emery–Dreifuss muscular dystrophy, dilated cardiomyopathy-1A, Dunnigan-type familial partial lipodystrophy, mandibuloacral dysplasia, and atypical WS (CitationGoldman et al 2002; CitationChen et al 2003). In HGPS cells, the nuclei and lamina appeared normal at early passages, but, at later passages, the nuclei are severely misshapen and contain an abnormally thick lamina (Citationde Sandre-Giovannoli et al 2003; CitationEriksson et al 2003). These structural defects worsen as HGPS cells age in culture, and their severity correlates with an obvious increase in mutant lamin A (CitationGoldman et al 2004). An abnormal distribution of nuclear pore complexes is seen in late passage HGPS cells. The changes in nuclear pore complexes gradually affect various aspects of the normal trafficking of protein and RNA across the nuclear envelope, having severe effects on the physiological state of HGPS cells (CitationYoshida and Blobel 2001). In addition, mutant lamin A induces decreased cellular proliferation, premature senescence, and altered motility (CitationGoldman et al 2004). The highly lobulated late passage HGPS cells primarily exhibit PCNA patterns resembling early S phase, suggesting that there is a block in the transition from the early chain-elongation phase of DNA replication to the mid- and later phases of replication. Mutant lamin A (progerin) progressively accumulates in the nucleus with cellular age, resulting in premature cessation of growth in the later passages of HGPS cells (CitationGoldman et al 2004).
Universal transcriptional alterations
Lamin A is a major constituent of the nuclear membrane, and an immunofluorescence study of HGPS fibroblast nuclei demonstrated abnormalities (CitationGoldman et al 2004). Given the prominent structural role of lamin A in the nuclear membrane, it is suggested that this protein has diverse roles in DNA metabolism, including DNA replication and transcription. Gene expression in HGPS was investigated by measuring mRNA levels in fibroblasts isolated from young, middle aged, and old humans with or without progeria (CitationLy et al 2000). Of the 152 genes studied, 47 (31%) were differentially transcribed in both old and HGPS compared to young. The direction of the change was the same in old and HGPS for all coregulated genes. Genes involved in mitosis were downregulated and the observed changes might result in increased rates of somatic mutation, leading to chromosome aberrations and mutations manifesting as an aging phenotype. CitationCsoka and colleagues (2004b) found that the genes differentially expressed in HGPS fibroblasts compared to age-matched control cell lines are involved in a variety of biological processes. Of the approximately 33,000 genes analyzed, 361 (1.1%) showed at least a 2 fold change in HGPS compared to the aged controls. The most prominent categories encoded transcription factors and extracellular matrix proteins, many of which are known to function in the tissues severely affected in HGPS. The most affected gene was MEOX/GAX, a homeobox gene that functions as a negative regulator of proliferation. Several genes involved in DNA replication and chromatin remodeling were downregulated. These changes were interpreted as contributing to depression of cellular proliferation. Some of the transcription changes suggested excess extracellular matrix deposition through the increased expression of extracellular matrix components and decreased expression of extracellular matrix remodeling enzymes. Of the 58 genes examined by both of these groups, the expression of 17 (29%) changed in the same direction, demonstrating a reasonable agreement between the two studies considering their different designs. The studies by CitationKyng and Bohr (2005) and CitationCsoka and colleagues (2004b) point to a shared mechanism of aging acceleration in PSs ie, misregulated transcription. The idea that aging is due to the loss of the proper transcriptional state of the cell followed by “dysdifferentiation” had been previously proposed by other investigators (CitationKator et al 1985; CitationZs-Nagy et al 1988; CitationFossel 2003; CitationProlla 2005). These findings that HGPS patients show inappropriate transcriptional patterns provides new evidence that transcriptional deregulation can contribute to the aging process in humans.
Genomic instability
DNA damage accumulation and the effects of repair defects can lead to genomic instability associated with premature aging and have causal roles in normal aging (CitationLombard et al 2005; Citationvon Zglinicki et al 2005). Indeed, defective recruitment of 53BP1 and RAD51 to sites of DNA lesion is seen in HGPS fibroblasts, resulting in a delayed checkpoint response and defective DNA repair (CitationLiu et al 2005). Wild-type mouse embryonic fibroblasts ectopically expressing unprocessible prelamin A show similar defects in checkpoint response and DNA repair (CitationLiu et al 2005). These results indicate that unprocessed prelamin A and truncated lamin A act dominant negatively to perturb the DNA damage response and repair, resulting in genomic instability, which might contribute to laminopathy-based premature aging (CitationLiu et al 2005). In HGPS cells, DNA damage checkpoints are persistently activated, and inactivation of checkpoint kinases ATM and ATR can partially restore cell cycle progression into S-phase (CitationCortez et al 2001; CitationLiu et al 2006), suggesting that senescence can be suppressed by inactivating DNA damage response pathways in HGPS cells. Organismal aging has been linked to activation of p53-dependent signaling pathways and initiation of the senescence program in a premature aging mouse model (CitationVarela et al 2005). Inhibition of aberrant splicing of lamin A results in significant downregulation of p21, IGFBP3, and GADD45B compared with mock-treated cells (CitationScaffidi and Misteli 2006). Consistent with the reduction in p53 activation, upon elimination of Δ50 lamin A from old cells, the fraction of 5-bromo-2′-deoxyuridine-positive proliferating cells increased by 30% and was similar to that in mock-treated young cells. Over the past few years, there have been reports that progerin leads to defective DNA repair and genome instability (CitationLiu et al 2005), overexpression of p53 target genes (CitationVarela et al 2005), and changes in histone methylation that affect heterochromatin organization (CitationShumaker et al 2006).
Farnesyltransferase inhibitor
Farnesyl-prelamin A is targeted to the nuclear envelope, where it interferes with the integrity of the nuclear envelope and causes misshapen cell nuclei. It has also been shown to affect the mechanical stability of the nucleus (CitationDahl et al 2006). Farnesyltransferase inhibitors (FTIs) can block prelamin A processing and reduce the percentage of cells with misshapen nuclei (CitationCapell et al 2005; CitationGlynn and Glover 2005; CitationMallampalli et al 2005; CitationToth et al 2005). Thus, the favorable effects of FTIs raise the question whether an FTI might improve disease phenotypes in HGPS (CitationYoung et al 2006). It is also important to define the extent to which these abnormalities are affected by an FTI and to determine whether FTIs will be a useful therapy in children with HGPS. A recent study found that treatment of patient’s cells with an FTI did not result in a reduction in DNA DSBs and damage checkpoint signaling, although it significantly reversed the aberrant shape of their nuclei (CitationLiu et al 2006), suggesting that DNA damage accumulation and aberrant nuclear morphology are independent phenotypes arising from prelamin A accumulation in these PSs. CitationYang and colleagues (2006) created gene-targeted mice with an HGPS mutation (LmnaHG/+) and examined the effect of an FTI on the disease phenotypes. The LmnaHG/+ mice exhibited phenotypes similar to those in human HGPS patients, including retarded growth, reduced amounts of adipose tissue, micrognathia, osteoporosis, and osteolytic lesions in bone. In addition, osteolytic lesions in the ribs led to spontaneous bone fractures. Treatment with an FTI ameliorated the disease phenotypes, resulting in an increased adipose tissue mass, improved body weight curves, a reduction in the number of rib fractures, and improved bone mineralization and bone cortical thickness, suggesting that FTIs could be useful for treating HGPS patients (CitationYang et al 2006). Though FTIs fall short of curing the disease (CitationYang et al 2006), these findings have established a paradigm for ameliorating the most obvious cellular pathology in HGPS and suggest a potential strategy for treating this disease.
A hypothetical model of aging
The molecular mechanisms involved in human senescence are complicated. Two canonical PSs, WS and HGPS, characterized by clinical features mimicking physiological aging at an early age, have provided insights into the mechanisms of natural aging. In these PSs, several cellular pathways are affected, resulting in the formation of endogenous and exogenous sources of oxidative stress, telomere attrition, and a decline in DNA repair, which can jointly contribute to genomic instability, and subsequently result in growth arrest and apoptosis, leading to the human aging phenotypes (CitationKaranjawala and Lieber 2004; CitationProlla 2005; CitationCollado 2007). The gene defective in WS, WRN, encodes a helicase of the RecQ family and possesses an exonuclease domain. WRN is involves in multiple DNA repair pathways and plays a significant role in the maintenance of overall genomic stability (CitationBachrati and Hickson 2003) (as seen in WS). The clinical features of HGPS show similarities to WS, but progress more rapidly (CitationHennekam 2006). LMNA, the gene defective in HGPS, affects the structure or post-translational maturation of lamin A, a major nuclear component (CitationEriksson et al 2003). Recently, several studies have established a functional link between DNA repair and A-type lamin-associated syndromes, which are associated with transcriptional alterations, abnormal DNA replication, changed organization of higher order chromatin structure, and genomic instability (CitationSerrano and Blasco 2007), suggesting a link between these syndromes and physiological aging (as seen in HGPS). DNA damage is generated throughout life and causes continuous damage to the macromolecular components of cells. Importantly, the rate of DNA damage production increases with ageing (CitationHasty and Vijg 2004; CitationSerrano and Blasco 2007).
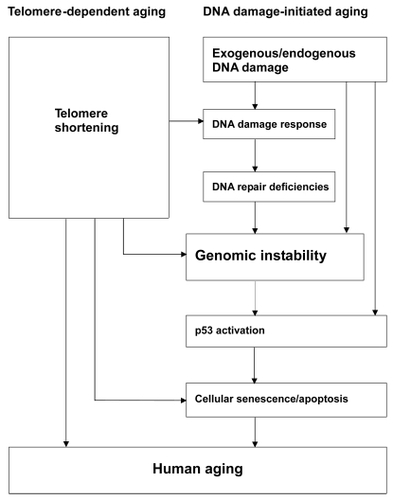
Thus, the causal relationship between the DNA damage response and cellular senescence suggests that DNA damage-initiated genomic instability can induce human aging phenotypes (CitationHarman 1956). p53-mediated senescence and apoptosis in response to DNA damage also probably contribute to aging. Indeed, p53, as a master integrator of cellular stress, is able to respond to a wide range of DNA damage (CitationHorn and Vousden 2007), then, if the stress persists, prevent propagation of the damaged cells (by apoptosis or senescence) (CitationVousden and Lane 2007). Following DNA damage, activation of p53 leads to transcription and upregulation of p53 target genes, among which P21 acts as the major effector of p53-induced cellular senescence. P21 levels increase gradually as cells pass into senescence (CitationAlcorta et al 1996). Collectively, these results suggest that p53 activation is at least partially responsible for the induction of cellular senescence in response to DNA damage. On the other hand, various lines of evidence have shown that telomere shortening and dysfunction can also trigger DNA damage responses and are sufficient to induce cellular senescence (d’Adda Citationdi Fagagna et al 2003; CitationTakai et al 2003; CitationChang et al 2004; CitationDu et al 2004). Some HGP fibroblasts also appear refractory to telomerase-mediated immortalization (CitationWallis et al 2004), and most cultures show elevated apoptosis and senescence (CitationBridger and Kill 2004). Numerous studies indicate that, in the setting of WRN deficiency, dysfunctional telomeres trigger the onset of premature aging phenotypes, suggesting a link between increased telomere dysfunction and the genomic instability associated with the aging process (d’Adda Citationdi Fagagna et al 2003; CitationTakai et al 2003; CitationSmogorzewska and de Lange 2004; CitationChang et al 2004; CitationDu et al 2004). On the basis of the information available for WS and HGPS, we suggest a model of human aging (). Human aging can be triggered by two main mechanisms, telomere shortening and DNA damage. In telomere-dependent aging, telomere shortening and dysfunction can lead to DNA damage responses, inducing cellular senescence. In DNA damage-initiated aging, DNA damage accumulates, along with DNA repair deficiencies, resulting in genomic instability and accelerated cellular senescence. Both aging mechanisms depend strongly on p53 status. These two mechanisms can act cooperatively to increase the overall level of genomic instability and trigger the onset of human aging phenotypes.
Figure 1 A hypothetical model: Genomic instability plays a central role during the aging process, triggered by two main stimuli, telomere shortening and DNA damage. (1) Telomere-dependent aging: Telomeres are essential for chromosomal stability. Telomere shortening and dysfunction can trigger DNA damage responses and are sufficient to induce cellular senescence. (2) DNA damage-initiated aging: DNA damage accumulates, along with DNA repair deficiencies, resulting in genomic instability and accelerated cellular senescence. Both mechanisms depend strongly on p53. These two mechanisms can act cooperatively to increase the overall level of genomic instability and trigger the onset of human aging phenotypes.
Conflict of interest
The authors report no conflicts of interest in this work.
References
- AgreloRChengWHSetienF2006Epigenetic inactivation of the premature aging Werner syndrome gene in human cancerProc Natl Acad Sci U S A1038822716723399
- AlbertsBJohnsonALewisJAlbertsB2002Development of multicellular organismsMolecular Biology of the Cell4th editionNew YorkGarland Science
- AlcortaDAXiongYPhelpsD1996Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblastsProc Natl Acad Sci U S A931374278943005
- AndressooJOHoeijmakersJH2005Transcription-coupled repair and premature ageingMutat Res5771799416009385
- BachratiCZHicksonID2003RecQ helicases: Suppressors of tumorigenesis and premature agingBiochem J37457760612803543
- BalajeeASMachweAMayA1999The Werner syndrome protein is involved in RNA polymerase II transcriptionMol Biol Cell1026556810436020
- BambaraRAMuranteRSHendricksonLA1997Enzymes and reactions at the eukaryotic DNA replication forkJ Biol Chem2724647509081985
- BayntonKOtterleiMBjørasM2003WRN interacts physically and functionally with the recombination mediator protein RAD52J Biol Chem278364768612750383
- BeckLAHosickTJSinenskyM1990Isoprenylation is required for the processing of the lamin A precursorJ Cell Biol1101489992335559
- BeckmanKBAmesBN1998The free radical theory of aging maturesPhysiol Rev78547819562038
- BergoMOGavinoBRossJ2002Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defectProc Natl Acad Sci U S A99130495412235369
- BischofOKimSHIrvingJ2001Regulation and localization of the Bloom syndrome protein in response to DNA damageJ Cell Biol1533678011309417
- BlanderGZalleNDanielyY2002DNA damage-induced translocation of the Werner helicase is regulated by acetylationJ Biol Chem277509344012384494
- BlankABobolaMSGoldB2004The Werner syndrome protein confers resistance to the DNA lesions N3-methyladenine and O(6)-methyl-guanine: implications for WRN functionDNA Repair36293815135730
- BlascoMA2002Telomerase beyond telomeresNat Rev Cancer26273312154355
- BridgerJMKillIR2004Aging of Hutchinson-Gilford progeria syndrome fibroblasts is characterised by hyperproliferation and increased apoptosisExp Gerontol397172415130666
- BroshRMJrOrrenDKNehlinJO1999Functional and physical interaction between WRN helicase and human replication proteinA J Biol Chem2741834150
- BroshRMJrLiJLKennyMK2000Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its helicase activityJ Biol Chem27523500810825162
- BroshRMJrKarmakarPSommersJA2001ap53 modulates the exonuclease activity of Werner syndrome proteinJ Biol Chem2763509310211427532
- BroshRMJrMajumdarADesaiS2001bUnwinding of a DNA triple helix by the Werner and Bloom syndrome helicasesJ Biol Chem27630243011110789
- BroshRMJrvon KobbeCSommersJA2001cWerner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activityEMBO J20579180111598021
- BroshRMJrBohrVA2002Roles of the Werner syndrome protein in pathways required for maintenance of genome stabilityExp Gerontol3749150611830352
- BroshRMJrDriscollHCDianovGL2002Biochemical characterization of the WRN-FEN-1 functional interactionBiochemistry41122041612356323
- CaoHHegeleRA2003LMNA is mutated in Hutchinson-Gilford progeria (MIM 176670) but not in Wiedemann- Rautenstrauch progeroid syndrome (MIM 264090)J Hum Genet48271412768443
- CapellBCErdosMRMadiganJP2005Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndromeProc Natl Acad Sci USA102128798416129833
- ChangSMultaniASCabreraNG2004Essential role of limiting telomeres in the pathogenesis of Werner syndromeNature Genetics368778215235603
- ChenLHuangSLeeL2003WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repairAging Cell2191912934712
- ChengWHvon KobbeCOpreskoPL2003Werner syndrome protein phosphorylation by abl tyrosine kinase regulates its activity and distributionMol Cel Biol23638595
- ChengWHvon KobbeCOpreskoPL2004Linkage between Werner syndrome protein and the Mre11 complex via Nbs1J Bio Chem279211697615026416
- ClarkeSVogelJPDeschenesRJ1988Posttranslational modification of the Haras oncogene protein: evidence for a third class of protein carboxyl methyltransferasesProc Natl Acad Sci, USA85464373290900
- ClarkeARPurdieCAHarrisonDJ1993Thymocyte apoptosis induced by p53-dependent and independent pathwaysNature362849528479523
- ColladoMBlascoMASerranoM2007Cellular senescence in cancer and agingCell1302233317662938
- ConstantinouATarsounasMKarowJK2000Werner’s syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrestEMBO Rep180411256630
- CooperMPMachweAOrrenDK2000Ku complex interacts with and stimulates the Werner proteinGenes Dev149071210783163
- CorriganDPKuszczakDRusinolAE2005Prelamin A endoproteolytic processing in vitro by recombinant Zmpste24Biochem J3871293815479156
- CortezDGuntukuSQinJ2001ATR and ATRIP: partners in checkpoint signalingScience29417131611721054
- CrabbeLVerdunREHaggblomCI2004Defective telomere lagging strand synthesis in cells lacking WRN helicase activityScience3061951315591207
- CsokaABCaoHSammakPJ2004aNovel lamin A/C gene (LMNA) mutations in atypical progeroid syndromesJ Med Genet41304815060110
- CsokaABEnglishSBSimkevichCP2004bGenome-scale expression profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atherosclerosisAging Cell32354315268757
- d’Adda di FagagnaFReaperPMClay-FarraceL2003A DNA damage checkpoint response in telomere-initiated senescenceNature426194814608368
- DahlKNScaffidiPIslamMF2006Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndromeProc Natl Acad Sci USA10310271616801550
- DaiQChoyEChiuV1998Mammalian prenylcysteine carboxyl methyltransferase is in the endoplasmic reticulumJ Biol Chem2731503049614111
- De LangeT2002Protection of mammalian telomeresOncogene215324011850778
- De Sandre-GiovannoliABernardRCauP2003Lamin A truncation in Hutchinson–Gilford progeriaScience300205512702809
- DuXShenJKuganN2004Telomere shortening exposes functions for the Werner and Bloom syndrome genes in miceMol Cell Biol2484374615367665
- EpsteinCJMartinGMSchultzAL1966Werner’s syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging processMedicine451772215327241
- EpsteinJWilliamsJRLittleJB1973Deficient DNA repair in human progeroid cellsProc Natl Acad Sci U S A70977814515628
- ErikssonMBrownWTGordonLB2003Recurrent de novo point mutations in lamin a cause Hutchinson-Gilford progeria syndromeNature423293812714972
- FairallLChapmanLMossH2001Structure of the TRFH dimerization domain of the human telomeric proteins TRF1 and TRF2Mol Cell83516111545737
- FongLGNgJKLammerdingJ2006Prelamin A and lamin A appear to be dispensable in the nuclear laminaJ Clin Invest1167435216511604
- FosselM2003The progeriasJ Antiaging Med612338
- FryMLoebLA1999Human Werner syndrome DNA helicase unwinds tetrahelical structures of the fragile X syndrome repeat sequence d(CGG)nJ Biol Chem2741279780210212265
- FukuchiKKatsuyaTSugimotoK2004LMNA mutation in a 45 year old Japanese subject with Hutchinson-Gilford progeria syndromeJ Med Genet41e6715121795
- GilfordH1904Ateleiosis and progeria: continous youth and premature old ageBr Med J9148
- GlynnMWGloverTW2005Incomplete processing of mutant lamin A in Hutchinson-Gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibitionHum Mol Genet1429596916126733
- GoldmanRDGruenbaumYMoirRD2002Nuclear lamins: building blocks of nuclear architectureGenes Dev165334711877373
- GoldmanRDShumakerDKErdosMR2004Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndromeProc Natl Acad Sci, USA1018963815184648
- GotoMMillerRWIshikawaY1996Excess of rare cancers in Werner’s syndrome (adult progeria)Cancer Epidemiol Biomarkers Prev5239468722214
- GotoM1997Hierarchical deterioration of body systems in Werner’s syndrome: Implications for normal ageingMech Ageing Dev98239549352493
- GrayMDShenJCKamath-LoebAS1997The Werner syndrome protein is a DNA helicaseNat Genet1710039288107
- GriffithJDComeauLRosenfieldS1999Mammalian telomeres end in a large duplex loopCell975031410338214
- Halaschek-WienerJBrooks-WilsonA2007Progeria of stem cells: stem cell exhaustion in hutchinson-gilford progeria syndromeJ Gerontol A Biol Sci Med Sci623817301031
- HanaokaFYamadaMTakeuchiF1985Autoradiographic studies of DNA replication in Werner’s syndrome cellsAdv Exp Med Biol190439574083159
- HarmanD1956Aging: a theory based on free radical and radiation chemistryJ Gerontol1129830013332224
- HarriganJAOpreskoPLvon KobbeC2003The Werner syndrome protein stimulates DNA polymerase beta strand displacement synthesis via its helicase activityJ Biol Chem278226869512665521
- HastyPCampisiJHoeijmakersJ2003Aging and genome maintenance: lessons from the mouse?Science2991355912610296
- HastyPVijgJ2004Accelerating aging by mouse reverse genetics: a rational approach to understand longevityAging Cell3556515038819
- HauptSBergerMGoldbergZ2003Apoptosis - the p53 networkJ Cell Sci11640778512972501
- HennekamRC2006Hutchinson-Gilford progeria syndrome: Review of the phenotypeAm J Med Genet A14026032416838330
- HicksonID2003RecQ helicases: caretakers of the genomeNat Rev Cancer31697812612652
- HofsethLJHussainSPHarrisCC2004p53: 25 years after its discoveryTrends Pharmacol Sci251778115116721
- HornHFVousdenKH2007Coping with stress: multiple ways to activate p53Oncogene2613061617322916
- HuangSLiBGrayMD1998The premature ageing syndrome protein, WRN, is a 3′→5′ exonucleaseNature Genet2011469771700
- HuangSBerestenSLiB2000Characterization of the human and mouse WRN 3′→5′ exonucleaseNucleic Acids Res28239640510871373
- HutchinsonJ1886Case of congenital absence of hair, with atrophic condition of the skin and its apendagesLancet923
- JeziornyLMcCurdyLMichaelK2006The Heli-CASE of the missing WRN geneEukaryon2349
- Kamath-LoebASJohanssonEBurgersPM2000Functional interaction between the Werner syndrome protein and DNA polymerase deltaProc Natl Acad Sci USA974603810781066
- Kamath-LoebASLoebLAJohanssonE2001Interactions between the Werner syndrome helicase and DNA polymerase d specifically facilitate copying of tetraplex and hairpin structures of the d(CGG)n trinucleotide repeat sequenceJ Biol Chem276164394611279038
- KaranjawalaZELieberMR2004DNA damage and agingMech Ageing Dev1254051615272504
- KarmakarPSnowdenCMRamsdenDA2002Ku heterodimer binds to both ends of the Werner protein and functional interaction occurs at the Werner N-terminusNucleic Acids Res3035839112177300
- KatorKCristofaloVCharpentierR1985Dysdifferentiative nature of aging: passage number dependency of globin gene expression in normal human diploid cells grown in tissue cultureGerontology31355613840764
- KawabeYSekiMSekiT2000Covalent modification of the Werner’s syndrome gene product with the ubiquitin-related protein, SUMO-1J Biol Chem27520963610806190
- KyngKJBohrVA2005Gene expression and DNA repair in progeroid syndromes and human agingAgeing Res Rev457960216246641
- LaineJPOpreskoPLIndigFE2003Werner protein stimulates topoisomerase I DNA relaxation activityCancer Res6371364614612507
- LaneD2004Anthony Dipple Carcinogenesis Award. p53 from pathway to therapyCarcinogenesis2510778115205365
- LebelMLederP1998A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacityProc Natl Acad Sci USA95130971029789047
- LebelMSpillareEAHarrisCC1999The Werner syndrome gene product co-purifies with the DNA replication complex and interacts with PCNA and topoisomerase IJ Biol Chem27437795910608841
- LiBComaiL2000Functional interaction between Ku and the Werner syndrome protein in DNA end processingJ Biol Chem275283495210880505
- LiBComaiL2001Requirements for the nucleoytic processing of DNA ends by the Werner syndrome protein: Ku70/80 complexJ Biol Chem276989690211152456
- LiBComaiL2002Displacement of DNA-PKcs from DNA ends by the Werner syndrome proteinNucleic Acids Res3036536112202749
- LiBNavarroSKasaharaN2004Identification and biochemical characterization of a Werner’s syndrome protein complex with Ku70/80 and poly(ADP-ribose) polymerase-1J Biol Chem279136596714734561
- LiuBWangJChanKM2005Genomic instability in laminopathy-based premature agingNat Med11780515980864
- LiuYRusinolASinenskyM2006DNA damage responses in progeroid syndromes arise from defective maturation of prelamin AJ Cell Sci1194644917062639
- LiuZMaciasMJBottomleyMJ1999The three-dimensional structure of the HRDC domain and implications for the Werner and Bloom syndrome proteinsStruct Fold Des7155766
- LombardDBChuaKFMostoslavskyR2005DNA repair, genome stability, and agingCell12049751215734682
- LyDHLockhartDJLernerRA2000Mitotic misregulation and human agingScience28724869210741968
- MachweAXiaoLOrrenDK2004TRF2 recruits the Werner syndrome (WRN) exonuclease for processing of telomeric DNAOncogene231495614712220
- MachweAXiaoLGrodenJ2005RecQ family members combine strand pairing and unwinding activities to catalyze strand exchangeJ Biol Chem2802339740715845538
- MachweALozadaEMXiaoL2006Competition between the DNA unwinding and strand pairing activities of the Werner and Bloom syndrome proteinsBMC Mol Biol7116412221
- MaierBGlubaWBernierB2004Modulation of mammalian life span by the short isoform of p53Genes Dev183061914871929
- MallampalliMPHuyerGBendaleP2005Inhibiting farnesylation reverses the nuclear morphology defect in a HeLa cell model for Hutchinson-Gilford progeria syndromeProc Natl Acad Sci, USA102144162116186497
- MarciniakRALombardDBJohnsonFB1998Nucleolar localization of the Werner syndrome protein in human cellsProc Natl Acad Sci USA956887929618508
- MartinGMSpragueCAEpsteinCJ1970Replicative life-span of cultivated human cells. Effects of donor’s age, tissue, and genotypeLab Inves238692
- MartinGM1977Cellular aging-clonal senescence. A review (Part I)Am J Pathol89484511335895
- MartinGM1978Genetic syndromes in man with potential relevance to the pathobiology of agingBirth Defects Orig Artic Ser14539147113
- MartinGM1985Genetics and aging: the Werner syndrome as a segmental progeroid syndromeAdv Exp Med Biol190161703909765
- MartinGMOshimaJ2000Lessons from human progeroid syndromesNature408263611089984
- MatsumotoTShimamotoAGotoM1997Impaired nuclear localization of defective DNA helicases in Werner’s syndromeNat Genet1633569241267
- McClintockB1941The stability of broken ends of chromosomes in Zea maysGenetics262348217247004
- MerrillBJHolmC1998The RAD52 recombinational repair pathway is essential in pol30 (PCNA) mutants that accumulate small single-stranded DNA fragments during DNA synthesisGenetics148611249504910
- MohagheghPKarowJKBroshJRJr2001The Bloom’s and Werner’s syndrome proteins are DNA structurespecific helicasesNucleic Acids Res2928434911433031
- MonnatRJJrSaintignyY2004Werner syndrome protein-unwinding function to explain diseaseSci Aging Knowledge Environ20043
- MüllerSLedlASchmidtD2004SUMO: a regulator of gene expression and genome integrityOncogene231998200815021887
- NagaoMSugimuraT1976Molecular biology of the carcinogen, 4- nitroquinoline 1-oxideAdv Cancer Res2313169818888
- NavarroCLCauPLevyN2006Molecular bases of progeroid syndromesHuman Molecular Genetics15R1516116987878
- OgburnCEOshimaJPootM1997An apoptosis-inducing genotoxin differentiates heterozygotic carriers for Werner helicase mutations from wild-type and homozygous mutantsHum Genet10112159402954
- OhsugiITokutakeYSuzukiN2000Telomere repeat DNA forms a large non-covalent complex with unique cohesive properties which is dissociated by Werner syndrome DNA helicase in the presence of replication protein ANucleic Acids Res283642810982887
- OpreskoPLLaineJ-PBroshRMJr2001Coordinate action of the helicase and 3′ to 5′ exonuclease of Werner syndrome proteinJ Biol Chem276446778711572872
- OpreskoPLvon KobbeCLaineJP2002Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicaseJ Biol Chem277411101912181313
- OpreskoPLChengWHvon KobbeC2003Werner syndrome and the function of the Werner protein: what they can teach us about the molecular aging processCarcinogenesis2479180212771022
- OpreskoPLOtterleiMGraakjaerJ2004The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2Mol Cell147637415200954
- OrrenDKMachweAKarmakarP2001A functional interaction of Ku with Werner exonuclease facilitates digestion of damaged DNANucleic Acids Res2919263411328876
- OshimaJHuangSPaeC2002Lack of WRN results in extensive deletion at nonhomologous joining endsCancer Res625475111809708
- PichierriPRosselliFFranchittoA2003Werner’s syndrome protein is phosphorylated in an ATR/ATM-dependent manner following replication arrest and DNA damage induced during the S phase of the cell cycleOncogene22149150012629512
- PootMHoehnHRungerTM1992Impaired S phase transit of Werner syndrome cells expressed in lymphoblastoid cellsExp Cell Res202267731327851
- PootMYomJSWhangSH2001Werner syndrome cells are sensitive to DNA cross-linking drugsFASEB J151224611344095
- PootMGollahonKAEmondMJ2002Werner syndrome diploid fibroblasts are sensitive to 4-nitroquinoline-N-oxide and 8-methoxypsoralen: implications for the disease phenotypeFASEB J16757811978740
- PrincePREmondMJMonnatRJJr2001Loss of Werner syndrome protein function promotes aberrant mitotic recombinationGenes Dev15933811316787
- ProllaTA2005Multiple roads to the aging phenotype: insights from the molecular dissection of progerias through DNA microarray analysisMech Ageing Dev126461515722104
- Rodríguez-LópezAMJacksonDANehlinJO2003Characterisation of the interaction between WRN, the helicase/exonuclease defective in progeroid Werner’s syndrome, and an essential replication factor, PCNAMech Ageing Dev1241677412633936
- SaintignyYMakienkoKSwansonC2002Homologous recombination resolution defect in Werner syndromeMol Cell Biol226971812242278
- SakamotoSNishikawaKHeoSJ2001Werner helicase relocates into nuclear foci in response to DNA damaging agents and co-localizes with RPA and Rad51Genes Cells64213011380620
- SalkDBryantEHoehnH1985Growth characteristics of Werner syndrome cells in vitroAdv Exp Med Biol190305114083155
- SanzMMProytchevaMEllisNA2000BLM, the Bloom’s syndrome protein, varies during the cell cycle in its amount, distribution, and co-localization with other nuclear proteinsCytogenet Cell Genet912172311173860
- ScaffidiPMisteliT2005Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndromeNat Med11440515750600
- ScaffidiPMisteliT2006Lamin A-dependent nuclear defects in human agingScience31210596316645051
- SchulzVPZakianV AOgburnCE1996Accelerated loss of telomeric repeats may not explain accelerated replicative decline of Werner syndrome cellsHum Genet9775048641691
- SerranoMBlascoMA2007Cancer and ageing: convergent and divergent mechanismsNat Rev Mol Cell Biol87152217717516
- SharmaSOtterleiMSommersJA2004WRN helicase and FEN-1 form a complex upon replication arrest and together process branch-migrating DNA structures associated with the replication forkMol Biol Cell157345014657243
- SharmaSSommersJAGaryRK2005The interaction site of Flap Endonuclease-1 with WRN helicase suggests a coordination of WRN and PCNANucleic Acids Res3367698116326861
- ShenJCGrayMDOshimaJ1998Characterization of Werner syndrome protein DNA helicase activity: directionality, substrate dependence and stimulation by replication protein ANucleic Acids Res262879859611231
- ShenJCLoebLA2000Werner syndrome exonuclease catalyzes structure-dependent degradation of DNANucleic Acids Res283260810954593
- ShenJCLaoYKamath-LoebA2003The N-terminal domain of the large subunit of human replication protein A binds to Werner syndrome protein and stimulates helicase activityMech Ageing Dev1249213014499497
- ShumakerDKDechatTKohlmaierA2006Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature agingProc Natl Acad Sci U S A1038703816738054
- SmogorzewskaAde LangeT2004Regulation of telomerase by telomeric proteinsAnnu Rev Biochem7317720815189140
- SommersJASharmaSDohertyKM2005p53 modulates RPA-dependent and RPA-independent WRN helicase activityCancer Res6512233315735006
- SpannTPGoldmanAEWangC2002Alteration of nuclear lamin organization inhibits RNA polymerase II-dependent transcriptionJ Cell Biol156603811854306
- SpillareEARoblesAIWangXW1999p53-mediated apoptosis is attenuated in Werner syndrome cellsGenes Dev1313556010364153
- SpillareEAWangXWvon KobbeC2006Redundancy of DNA helicases in p53-mediated apoptosisOncogene2521192316288211
- SunQYSchattenH2006Role of NuMA in vertebrate cells: review of an intriguing multifunctional proteinFront Biosci1111374616146802
- SuzukiNShimamotoAImamuraO1997DNA helicase activity in Werner’s syndrome gene product synthesized in a baculovirus systemNucleic Acids Res25297389224595
- SuzukiNShiratoriMGotoM1999Werner syndrome helicase contains a 5′→3′ exonuclease activity that digests DNA and RNA strands in DNA/DNA and RNA/DNA duplexes dependent on unwindingNucleic Acids Res272361810325426
- SymondsHKrallLRemingtonL1994p53-dependent apoptosis suppresses tumor growth and progression in vivoCell78703118069917
- SzekelyAMChenYHZhangC2000Werner protein recruits DNA polymerase δ to the nucleolusProc Natl Acad Sci, USA97113657011027336
- TakaiHSmogorzewskaAde LangeT2003DNA damage foci at dysfunctional telomeresCurr Biol1315495612956959
- TakeuchiFHanaokaFGotoM1982Altered frequency of initiation sites of DNA replication in Werner’s syndrome cellsHum Genet6036587106772
- TauchiHKobayashiJMorishimaK2002Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cellsNature42093812422221
- TothJIYangSHQiaoX2005Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromesProc Natl Acad Sci U S A10212873816129834
- TsaiMYWangSHeidingerJM2006A mitotic lamin B matrix induced by RanGTP required for spindle assemblyScience31118879316543417
- VarelaICadinanosJPendasAM2005Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activationNature437564816079796
- von KobbeCBohrVA2002A nucleolar targeting sequence in the Werner syndrome protein resides within residues 949–1092J Cell Sci1153901712244128
- von KobbeCKarmakarPDawutL2002Colocalization, physical and functional interaction between Werner and Bloom syndrome proteinsJ Biol Chem277220354411919194
- von KobbeCThomaNHCzyzewskiBK2003aWerner syndrome protein contains three structure-specific DNA binding domainsJ Biol Chem27852997300614534320
- von KobbeCHarriganJAMayA2003bCentral role for the Werner syndrome protein/poly(ADP-ribose) polymerase 1 complex in the poly(ADP-ribosyl)ation pathway after DNA damageMol Cell Biol2386011314612404
- von KobbeCHarriganJASchreiberV2004Poly(ADP-ribose) polymerase 1 regulates both the exonuclease and helicase activities of the Werner syndrome proteinNucleic Acids Res3240031415292449
- von ZglinickiTSaretzkiGLadhoffJ2005Human cell senescence as a DNA damage responseMech Ageing Dev1261111715610769
- VousdenKHLaneDP2007p53 in health and diseaseNature Rev Mol Cell Biol82758317380161
- WallisCVSheerinANGreenMH2004Fibroblast clones from patients with Hutchinson-Gilford progeria can senesce despite the presence of telomeraseExp Gerontol39461715050279
- WangXWVermeulenWCoursenJD1996The XPB and XPD DNA helicases are components of the p53-mediated apoptosis pathwayGenes Dev101219328675009
- WangXWTsengAEllisNA2001Functional interaction of p53 and BLM DNA helicase in apoptosisJ Biol Chem276329485511399766
- WarbrickE1998PCNA binding through a conserved motifBioEssays2019599631646
- WarbrickE2000The puzzle of PCNA’s many partnersBioEssays22997100611056476
- WeberKPlessmannUTraubP1989Maturation of nuclear lamin A involves a specific carboxy-terminal trimming, which removes the polyisoprenylation site from the precursor: implications for the structure of the nuclear laminaFEBS Lett25741142583287
- WoldMS1997Replication protein A: a heterotrimeric, single stranded DNA binding protein required for eukaryotic DNA metabolismAnnu Rev Biochem6661929242902
- WoodsYLXirodimasDPPrescottAR2004p14 Arf promotes small ubiquitin-like modifier conjugation of Werners helicaseJ Biol Chem279501576615355988
- WrightWEShayJW1992The two-stage mechanism controlling cellular senescence and immortalizationExp Gerontol2738391333985
- WuLDaviesSLLevittNC2001Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51J Biol Chem276193758111278509
- WuLHicksonID2003The Bloom’s syndrome helicase suppresses crossing over during homologous recombinationNature426870414685245
- WyllieFSJonesCJSkinnerJW2000Telomerase prevents the accelerated cell ageing of Werner syndrome fibroblastsNat Genet2416710615119
- YamabeYShimamotoAGotoM1998Sp1-mediated transcription of the Werner helicase gene is modulated by Rb and p53Mol Cell Biol1861912009774636
- YangSHBergoMOTothJI2005Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome mutationProc Natl Acad Sci U S A10210291616014412
- YangSHMetaMQiaoX2006A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutationJ Clin Invest11621152116862216
- YannoneSMRoySChanDW2001Werner syndrome protein is regulated and phosphorylated by DNA-dependent protein kinaseJ Biol Chem27638242811477099
- YoshidaKBlobelG2001The karyopherin Kap142p/Msn5p mediates nuclear import and nuclear export of different cargo proteinsJ Cell Biol1527294011266464
- YoungSGMetaMYangSH2006Prelamin A farnesylation and progeroid syndromesJ Biol Chem28139741517090536
- YuCEOshimaJFuYH1996Positional cloning of the Werner’s syndrome geneScience272258628602509
- YuCEOshimaJWijsmanEM1997Mutations in the consensus helicase domains of the Werner syndrome gene. Werner’s Syndrome Collaborative GroupAm J Hum Genet60330419012406
- ZhangFLCaseyPJ1996Protein prenylation: molecular mechanisms and functional consequencesAnnu Rev Biochem65241698811180
- Zs-NagyICutlerRGSemseiI1988Dysdifferentiation hypothesis of aging and cancer: a comparison with the membrane hypothesis of agingAnn NY Acad Sci521215253288042