Abstract
Studies examining the cellular mechanisms of inflammation and protease production in the lung tissue and airways of COPD patients have shed light on the important role of kinase-based signaling cascades. These pathways can be activated by environmental stimuli such as tobacco smoke, and by endogenous signals such as cytokines, growth factors, and inflammation-derived oxidants. The three most widely characterized cascades are those directed by the classical mitogen activated protein (MAP) kinase (ERK1/2), stress activated protein kinase/c-Jun N-terminal protein kinase, and p38 enzymes. These phosphorylation cascades transmit and amplify extracellular, receptor-mediated signals through the cytoplasm of the cell to activate nuclear transcription factors which bind and induce expression of target genes. The result is tight control of diverse cellular events, and rapid responses to external stimuli. However, recent research suggests that constitutive or aberrant activation of MAP kinases contributes to several COPD-associated phenotypes, including mucus overproduction and secretion, inflammation, cytokine expression, apoptosis, T cell activation, matrix metalloproteinase production, and fibrosis. This review explores the biological functions of the MAP kinase pathways in the pathogenesis of COPD, their activation by cigarette smoke, and discusses the potential role of MAP kinase inhibitors in COPD therapy.
Introduction
COPD is a debilitating lung disorder that kills over 110 000 individuals each year, making it the fourth largest cause of death in the US. Multiple initiating events including inflammation, protease–antiprotease imbalance, and oxidant–antioxidant imbalance damage the parenchyma and airways, leading to tissue remodeling. An emerging hypothesis in the field is that subsequent changes in epithelial gene expression and cellular function result in permanent airway injury and destruction of lung matrix. Many of these events are mediated in part by MAP kinase signal transduction pathways. Five distinct MAP kinase pathways have been identified in eukaryotes. These cascades are activated by distinct stimuli (including cigarette smoke) and direct a variety of biological events. While it is unlikely that an individual signaling cascade mediates a disease with such complex pathologies, recent research efforts have greatly improved our understanding of the activation of MAP kinase pathways and how these pathways direct cellular responses in the lung of COPD patients.
A remarkable feature of these pathways is that different stimuli (environmental toxins, oxidants, steroid hormones, mitogens, UV light, heat shock, or changes in pH, osmolarity, and nutrient supply) can activate the same pathway, which impressively induces diverse cell behaviors through reversible phosphorylation of transcription factor targets. For example, although interleukin (IL)-8 expression typically occurs through p38, ERK1/2 and SAPK/JNK have recently been found to mediate increased expression of this cytokine (Brand et al 2005). Therefore, the “specificity” of MAP kinase signaling depends not only on the cellular environment, but also on the cell type involved. The diversity of stimuli and biological responses associated with MAP kinase signaling is detailed in .
Table 1 Overview of the components of the ERK1/2, p38, and SAPK/JNK MAP kinase pathways
The classical MAP kinase cascade (Ras/ERK)
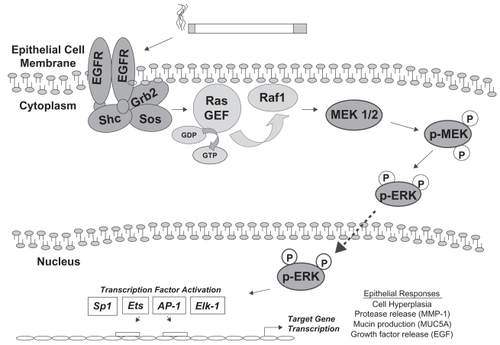
The best-characterized mammalian MAP kinase pathway is the Ras/ERK or classical MAP kinase pathway, composed of two genes with 90% sequence identity: ERK-1 (p44) and ERK-2 (p42) (CitationBoulton et al 1991). Additional ERK pathways include the less-often studied ERK-3 and ERK-5 cascades, which will not be reviewed here. Homologs for the ERK1/2 pathway are the pheromone-regulated kinases KSS1 and FUS3 in the yeast Saccharomyces cerevisiae, with similar modules in Drosophila melanogaster and Caenorhabditis elegans (CitationTreisman 1996). The phosphorylation substrate for ERK1/2 MAP kinases has a core motif with the short amino acid sequence serine/threonine-proline (S/T-P) (CitationCruzalegui et al 1999). The ERK1/2 MAP kinase pathway is typically activated, as the name suggests, by mitogenic stimuli, such as peptide growth factors EGF or PDGF (). Binding of growth factor to its cell surface receptor tyrosine kinase leads to receptor dimerization and autophosphorylation. Phosphorylation of the intracellular domain of the receptor activates GEFs, such as sos, which are attached to the cytoplasmic receptor tail by adaptor molecules grb-2 or shc. GEFs facilitate the activation of the monomeric GTPase Ras, via exchange of GDP to GTP. Ras-GTP recruits and activates the serine–threonine MAP kinase kinase kinase kinase (MKKK) c-Raf at the membrane, leading to Raf-mediated phosphorylation of the dual-specificity MAP kinase kinase-1 and -2 (MKKs or MEKs), MEK1/2. Next, MEK1/2 phosphorylates threonine and tyrosine amino acid residues on MAP kinases ERK1/2. Active transit of ERK1/2 through the nuclear membrane pore allows ERK1/2 to phosphorylate a variety of transcription factors such as the TCF member ELK-1, mediating DNA binding and gene transcription. As a result of these molecular events cell proliferation usually occurs. For this reason the Ras/ERK pathway is best studied for its direct role in tumorigenesis. In vitro (CitationVicent et al 2004), animal (CitationSebolt-Leopold et al 1999), and human studies (CitationHan et al 2005) have shown correlations between cancer incidence and increased Ras activation, ERK1/2 activity, or DNA binding by ERK1/2 transcription factor targets. Activation of ERK1/2 is shown in .
Figure 1 The ERK1/2 pathway in airway epithelial cell responses to cigarette smoke. Cigarette smoke exposure has been shown to activate the EGFR in lung epithelial cells. Following dimerization and autophosphorylation of EGFR, a cascade of adaptor molecules and GTPases leads to the recruitment of Raf1 to the plasma membrane and its activation. Raf1 is a MAP kinase kinase kinase, which phosphorylates the MAP kinase kinase MEK1/2. MEK1/2 activation leads to phosphorylation of ERK1/2 MAP kinase, which can translocate to the nucleus and phosphorylate transcription factors which bind to regulatory elements in the promoters of target genes, inducing their expression. Transcription factors that are phosphorylated by ERK1/2 include Sp1, Ets1, AP-1, and ELK-1. Cigarette smoke-mediated activation of this cascade in lung epithelial cells is associated with hyperplasia, MMP-1 expression, MUC5AC expression, and release of EGF ligand. The list of transcription factors and cell responses is not comprehensive.
Studies of mice with targeted deletion of ERK genes have shown that ERKs are essential for normal development and survival. Erk1 knockout mice (CitationPages et al 1999) develop normally and are fertile, likely due to the compensatory function of ERK2, but demonstrate behavioral hyperactivity (CitationSelcher et al 2001) and a defect in T cell proliferation and differentiation (CitationPages et al 1999). Erk2 null mice die at embryonic day 6.5, prior to lung formation, with significant apoptosis occurring in all tissues, and impaired angiogenesis (CitationYao et al 2003). Erk5 null animals die at embryonic day 9.5–10.5 from impaired heart and vessel development (the heterozygous animals grow to adulthood normally and are fertile) (CitationRegan et al 2002). These models demonstrate the role for ERKs during organogenesis, but conditional knockout animals are still needed to understand the role of ERKs in specific adult tissues and during adult-onset injury.
p38 MAP kinase cascades
The p38 MAP kinase family comprises four enzymes: p38α, p38β, p38γ, and p38δ. Early studies identified a 38 kDa protein that is tyrosine phosphorylated during lipopolysaccharide exposure or hyperosmolarity (CitationHan et al 1994). These enzymes have been studied for their ability to regulate TNF-α-induced inflammation (CitationLee et al 1994; CitationLee et al 2000). In particular, the p38 pathway is well characterized for its role in cytokine production in immune cells. This pathway can be activated not only by cellular stress such as osmotic shock, but also by growth factors, UV light, GPCR ligands, and hormones. Activation of p38 occurs through dual tyrosine phosphorylation on a motif (TGT) distinct from that of ERKs and SAPK/JNKs. The activation loops in which these tyrosines rest is 6 amino acids shorter than in the other MAP kinases. These differences suggest that the mechanism of phosphorylation of p38 is distinct from that for ERKs and JNKs. In addition, the upstream MKK3 is specific for p38 (). Knockout mice have been generated for the p38α, p38β, and p38γ MAP kinases. Similar to the ERK5 null mice, the p38α null mice are embryonic lethal due to severe cardiac malformations, resulting from impaired placental angiogenesis (CitationAdams et al 2000). There are no obvious phenotypes reported for the p38β and p38γ null mice (CitationKuida and Boucher 2004).
SAPK/JNK signaling
The SAPK/JNK pathway (p46, p54, and p55) is most notably involved in the control of apoptosis (CitationTournier et al 2000). This pathway is activated by UV light, cell stress, TNF-α, IL-1, and osmotic shock, and typically targets the transcription factor c-Jun. Additional stimuli include inhibition of protein translation (which stresses the cell), growth factors, and shifts in temperature. Activation of this pathway leads to phosphorylation of several additional substrates, including those of the early-response proto-oncogene family c-fos, which leads to the formation of Jun-fos heterodimers or Jun homodimers to create the AP-1 transcription factor (CitationAngel and Karin 1991). The binding activity of Jun is tightly controlled by phosphorylation of residues near the DNA-binding domain, but the role of the upstream SAPK/JNK molecules is not clear (CitationBinetruy et al 1991).
The JNK proteins phosphorylate a variety of substrates, including paxillin and mitochondrial Bcl-2. Much information on the biological roles of JNK signaling was obtained from JNK null mice. Individual JNK1, JNK2, or JNK3 null mice are generally normal, but the compound (JNK1/JNK2) knockout animals exhibit serious defects, dying in utero due to neural tube malformation (CitationKuan et al 1999). JNK3 is expressed in the nervous tissue and, interestingly, loss of JNK3 protects mice from kainic acid-induced neuronal death (CitationYang et al 1997; CitationKuan et al 2003). The JNK1/JNK3 and the JNK2/JNK3 compound null animals appear normal (CitationKuan et al 1999). JNK1 null mice have defective T cell proliferation and differentiation (CitationDong et al 1998), and CD4 T cells produce Th2 cytokines (IL-2, IL-4) in the absence of a Th2 stimulus (CitationDong et al 1998). Overall, targeted disruption of the JNK MAP kinase enzymes has revealed a great deal about their function during development. However, little is known about the specific role of these proteins in the adult lung.
Cigarette smoke and kinase activation in the lung
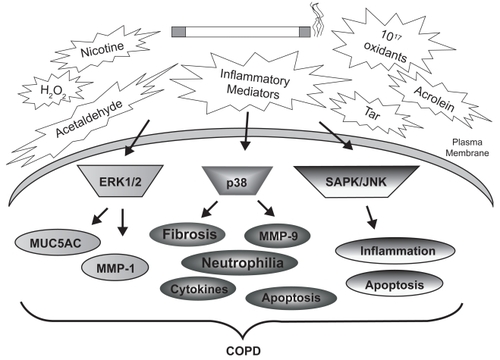
Signal transduction cascades are generally initiated by diverse stimuli which activate transmembrane receptors such as receptor tyrosine kinases and G-protein coupled receptors (). It is unclear whether cigarette smoke alters ligand-receptor interactions, or whether smoke can activate membrane-bound receptors directly. It is known, however, that smoke exposure is associated with rapid and persistent induction of several kinase pathways, typically within minutes. For example, our laboratory demonstrated rapid and lasting cigarette smoke-induced activation of ERK-1/2 MAP kinase in cultured SAECs (CitationMercer et al 2004). We have also detected elevated pulmonary ERK-1/2 (p44/42) phosphorylation in mice exposed to cigarette smoke for 10 days (CitationMercer et al 2004), which is in agreement with previous studies conducted in rats (CitationChang et al 2001). Most importantly, the relevance of these events to COPD was established by the discovery of significantly elevated ERK1/2 activity in airway and alveolar epithelial cells of patients with emphysema, compared with nonemphysematous controls (CitationMercer et al 2004). demonstrates the involvement of MAP kinase signaling in epithelial and inflammatory cell responses to cigarette smoke.
Figure 2 Cigarette smoke-induced MAP kinase activation and lung injury in COPD. The many chemicals, oxidants, and metabolites of cigarette smoke stimulate MAP kinase cascades within resident and inflammatory cells of the airways and parenchyma. Comparison of MAP kinase activities in the lung tissue of smokers, nonsmokers, and COPD patients has identified significant differences in these cascades. These signaling modules are linked to the indicated cellular processes, many of which are associated with COPD pathogenesis.
A central role for ERK1/2 in emphysema pathogenesis was established when our laboratory discovered that induction of MMP-1 by cigarette smoke in SAECs requires ERK1/2 MAP kinase signaling (CitationMercer et al 2004). MMP-1 is an interstitial collagenase upregulated in emphysema (CitationSelman et al 1996; CitationImai et al 2001), and generates structural and functional emphysema in transgenic mice (CitationD’Armiento et al 1992; CitationForonjy et al 2003). Indeed, polymorphisms in MMP-1 may play a role in lung function and COPD susceptibility (CitationJoos et al 2002). Future work is needed to determine how ERK1/2 and MMP signaling persists even after the patient has stopped smoking (CitationMercer et al 2004).
Analyses of baseline MAP kinase activity in AM of smokers and nonsmokers reveal, perhaps surprisingly, that smokers exhibit significantly lower active p38 levels than nonsmokers, and that there is no difference in the activity of ERK1/2 and SAPK/JNK kinases in AM between these two groups (CitationMochida-Nishimura et al 2001). These data suggest that different lung cell types (epithelial and inflammatory cell) can activate different MAP kinase signaling pathways in response to smoke exposure. However, one must be cautious when interpreting studies of molecular signaling in lung tissue from smokers, since not all smokers will develop COPD.
In vitro studies of A549 cells and in vivo studies with rats demonstrate tobacco smoke-mediated induction of c-Fos, MEK1, and ERK2 MAP kinase activities (CitationChang et al 2001; CitationHellermann et al 2002). Activation of these signal transduction molecules can trigger pro-proliferative or pro-inflammatory transcription factors such as c-fos, c-myc, AP-1, and Elk-1 (CitationPuddicombe and Davies 2000), which translocate to the nucleus and enhance gene expression (). One study reported increased mRNA and protein expression of Egr-1, an ERK1/2 substrate, in the lung tissue of patients with emphysema, suggesting that targets of ERK1/2 signaling are involved in pathogenesis (CitationZhang et al 2000). It is also likely that activation of different MAP kinases induces the same transcription factor. For example, ERK1/2 (CitationChen et al 2004) and JNK pathways have each been shown to activate NFκB signaling in human monocytes and airway epithelial cells (CitationTuyt et al 1999), demonstrating the potential for crosstalk between these pathways.
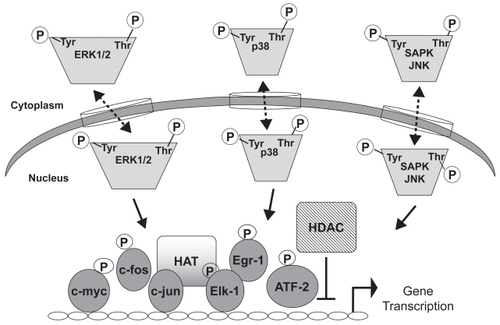
Figure 3 Transcriptional regulation by MAP kinases. Activated MAP kinases enter the nucleus and phosphorylate transcription factors such as Ets, AP-1, or ATF2. Transcription factor binding to cis-elements in the promoters of genes enhances transcription. Transcriptional control is also mediated via histone acetyltransferase (HAT) or histone deactylase (HDAC), which generally mediate activation or repression, respectively. Dashed lines indicate that kinase movement across the nuclear membrane occurs in both directions. Phosphorylation of transcription factors can also take place in the cytoplasm.
Research examining the interactions between cigarette smoke and the plasma membrane of lung epithelial cells has identified a role for the EGF receptor in the initial activation of MAP kinase signaling. EGFR is activated by several ligands: EGF, TGF-α, epiregulin, and amphiregulin. Several groups have demonstrated direct phosphorylation of EGFR by tobacco smoke. CitationTakeyama et al (2001) identified rapid transcriptional and post-translational activation of EGFR by tobacco smoke in NCI-H292 bronchial epithelial cells (a pulmonary mucoepidermoid carcinoma cell line which constitutively expresses EGFR). CitationLemjabbar et al (2003) later showed that EGFR activity was required for MUC5AC induction by smoke and elucidated the cytosolic events (CitationGensch et al 2004). These investigations showed that smoke and reactive oxygen species induce MUC5AC gene expression through both 1) an EGFR-independent mechanism, in which reactive oxygen species traverse the membrane to activate JNK and 2) an EGFR-dependent pathway which activates ERK1/2. The two pathways converge in the nucleus, where respective transcription factor targets JunD and Fra-2 bind AP-1 sites in the distal region of the MUC5AC gene to increase transcription (CitationGensch et al 2004). The role of EGFR in the activation of other genes in COPD has not been fully examined, and little is known about this process in primary epithelial cells of the small airways. models the steps in the activation of ERK1/2 via EGFR activation by cigarette smoke.
Lung cell apoptosis and MAP kinase signaling
Apoptosis of several lung cell types has been detected in the lung tissue in COPD (CitationSegura-Valdez et al 2000; CitationAoshibai et al 2003; CitationTuder et al 2003; CitationYokohori et al 2004), and positively correlates with disease severity (CitationImai et al 2005). In general, apoptosis is associated with changes in p38 or JNK signaling. In lung fibroblasts apoptosis can occur as a result of an increased oxidant burden; however, the mechanism is unclear (CitationIshii et al 2003). Recent data reveal that ceramide production in alveolar cells contributes to apoptosis and emphysema in mice (CitationPetrache et al 2005). No data on MAP kinase signaling exists in this model, but ceramide-induced apoptosis of A549 cells was recently shown to be mediated by JNK (CitationKurinna et al 2004), and ceramide-induced apoptosis resulting from cellular stress has been shown to require SAPK/JNK signaling (CitationVerheij et al 1996). Interestingly, ceramide can induce MMP-1 expression in dermal fibroblasts through ERK1/2- and p38-dependent mechanisms (CitationReunanen et al 1998). Data on primary lung epithelial cells are lacking.
Apoptosis in the lung tissue of animal models and patients with COPD may result from chronic exposure to irritants in smoke. Acrolein induces apoptosis of cultured bronchial epithelial cells (CitationNardini et al 2002), and acrolein-induced cytotoxicity occurs via ERK1/2 in vascular smooth muscle cells (CitationRanganna et al 2002). Nicotine also has been shown to suppress apoptosis of neutrophils (CitationAoshiba et al 1996), and is a potent inducer of ERK1/2 MAP kinase, with little effect on JNK or p38 pathways (CitationHeusch and Maneckjee 1998). The complexity of these apoptosis-inducing events merits further research.
Cytokine stimulation of and by MAP kinase cascades
One result of smoke-induced MAP kinase signaling is increased gene transcription due to altered chromatin architecture. For example, there is an increase in the intrinsic histone acetyltransferase (HAT) activity of ATF-2 through phosphorylation by either JNK or p38 (CitationKawasaki et al 2000). In addition, in vitro studies with A549 lung epithelial cells, for example, have demonstrated a cigarette smoke extract-mediated decrease in histone deactylase (HDAC) activity, contributing to unwinding of nuclear chromatin and cytokine gene expression (CitationMoodie et al 2004) (). Similarly, HDAC activity inversely correlates with COPD severity, and its reduced activity is thought to contribute to disease pathogenesis via enhanced inflammatory cytokine production (CitationIto et al 2005). These regulatory mechanisms are not fully elucidated, but it is hypothesized that control of cytokine gene expression is regulated in part through histone modification.
Analyses of murine asthma models reveal that production of the Th2 cytokines IL-2, IL-13, and IFNγ in the airways (as represented by bronchoalveolar [BAL] fluid) is mediated via MAP kinase signaling, specifically ERK1/2 and JNK MAP kinases (CitationChialda et al 2005). MAP kinases themselves may be activated by cytokines (CitationCuenda et al 1995; CitationMeier et al 1996), but the effect depends upon the type of cytokine. For example, the proinflammatory Th1 cytokine IL-17 can induce ERK1/2, but not p38 or SAPK/JNK in A459 lung epithelial cells (CitationNing et al 2005). Mapping out these kinase-cytokine modules is complicated by the variety of cell types, signal crosstalk, and inflammatory mediators within the lung. Additional research into the role of MAP kinases in pulmonary cytokine expression and inflammation is needed.
Identifying the components of tobacco smoke that activate MAP kinases
The most common and preventable cause of COPD is cigarette smoking (CitationMarkewitz et al 1999). Therefore, determining which pathways are activated in lung cells during tobacco smoke exposure is an important step towards identifying cellular mechanisms involved in the pathogenesis of COPD. However, an additional objective of research is to determine which components or fractions of the smoke (or tobacco leaf) are mediating such an effect. For example, several well-conducted studies have examined the roles of nicotine (CitationZhang et al 1993; CitationArmstrong et al 1996; CitationCarty et al 1996; CitationZhang et al 2001), cotinine (CitationCarty et al 1996), acrolein (CitationBishop and Laurent 1995; CitationBorchers et al 1999; CitationNardini et al 2002), acetaldehyde (CitationAppelman et al 1982; CitationSaladino et al 1985; CitationSisson et al 1991; CitationMio et al 1997), and hydrogen peroxide (CitationSaladino et al. 1985) in animals or cultured cells. It has been estimated that there are over 4700 chemicals in tobacco smoke (CitationMoodie et al 2004), making it difficult to identify specific tobacco component(s) responsible for smoking-induced MAP kinase activation (). In addition, it is important to appreciate that in vivo ADME (absorption, distribution, metabolism, excretion) pharmacokinetics of combusted tobacco components cannot be predicted by in vitro studies of isolated components dissolved in dimethyl sulfoxide. Cell type-specific responses are also important, as demonstrated by studies showing that while oxidant-sensitive NFκB is activated by cigarette smoke extract in A549 cells (CitationMoodie et al 2004), no activation occurs in NHBE cells (CitationHellermann et al 2002).
Acrolein is a potent component of tobacco that has been shown to produce mucus metaplasia in the lungs of both mice (CitationBorchers et al 1999) and rats (CitationBorchers et al 1998). Acrolein also contributes to EGFR phosphorylation, MUC5AC transcription, and ERK1/2 activation in NCI-H292 airway epithelial cells (CitationDeshmukh et al 2005). In this cell culture system acrolein depleted glutathione (GSH) and increased the burden of oxidative stress, as is thought to occur in COPD. Acrolein also has been shown to inhibit apoptosis in neutrophils (CitationFinkelstein et al 2001), and activates all three MAP kinase pathways in smooth muscle cells (CitationRanganna et al 2002).
Nicotine is present naturally as an antimicrobial in tobacco leaves (CitationTomizawa and Casida 2003), and is required for the addictive properties of cigarettes. Nicotine has been shown to increase normal branching morphogenesis and gene expression in embryonic mouse lung buds. However, the mechanisms involved are not known, and the response varies in different genetic strains (CitationWuenschell et al 2004). In cultured human airway epithelial cells, nicotine has been shown to activate Ras, the MKKK in the classical ERK1/2 signaling pathway (CitationChu et al 2005; CitationGuo et al 2005). In lung cancer cells nicotine has no effect on the activities of JNK and p38 MAP kinases, but does activate ERK2 (CitationHeusch and Maneckjee 1998). It is thus reasonable that activation of the classical MAP kinase signaling pathway is one reason that nicotine is a potent anti-apoptotic agent in several different lung cell types (CitationAoshiba et al 1996; CitationHeusch and Maneckjee 1998).
Inhibitors of MAP kinase signaling
Cancer research stimulates broad interest in kinase inhibitors
MAP kinase inhibitors have been extensively studied in animal models of disease, particularly cancer. These efforts were driven in part by human studies revealing increased ERK phosphorylation or activity in tumor-derived lung tissue (CitationErman et al 2005; CitationHan et al 2005) or human lung cell lines (CitationVicent et al 2004). Mouse models of lung cancer also demonstrate significant increases in several MAP kinase pathways (CitationWilhelm et al 2004). Evidence from smoke-exposed mice, xenografted mice, or mice injected with the aggressive human A549 adenocarcinoma cells suggests that uncontrolled proliferation of lung cancer cells can be alleviated using kinase inhibitors, with subsequent improvements in lung structure (CitationKramer et al 2004). Although these compounds were initially designed as cancer therapies, recent preliminary studies suggest that they may also possess significant benefit in treating emphysematous lesions and associated COPD pathologies (see below).
Potential for MAP kinase inhibitors as therapeutics in COPD
Inhibition of transcription factor activity can occur at the level of activation, translocation, or DNA binding. However, inhibition of the upstream kinases trumps the need to block nuclear transcription factors, by preventing the phosphorylation of these cytosolic DNA-binding effectors. Several inhibitors exist which have selective affinities for specific kinases, and which are, importantly, orally available (). These drugs, which typically act in an ATP-competitive manner, have shown promise in their ability to block various COPD-relevant cellular behaviors in culture, such as protease production, mucus secretion, and cell proliferation. However, the side-effects of these drugs cannot be known until long-term animal studies are conducted. Impressively, the MAP kinase inhibitors currently used in humans demonstrate low incidences of side-effects. It is for this reason, perhaps, that protein kinases are postulated to be the primary drug targets of this century (CitationCohen 2002).
Table 2 MAP kinase inhibitors and their effect in various models of tissue injury
Inhibitors of the classical MAP kinase pathway
There are several points along the classical MAP kinase pathway that can be targeted for inhibition. At the membrane, inhibition can block receptor dimerization or the attachment of GEFs to the plasma membrane. Inhibitors of RTKs exist, such as EGFR inhibitors AG1478 and ZD1839 (gefitinib/ Iressa). ZD1839 has been shown to block lung injury in a mouse model of bleomycin-induced pulmonary fibrosis (CitationSuzuki et al 2003), and to slow breast cell tumorigenesis in transgenic mice (CitationLu et al 2003). This drug has shown promise in clinical trials for lung cancer but adverse effects are often an issue (CitationTsuboi et al 2005).
Compounds have also been designed to target the farnesylation of oncogenic Ras, thereby blocking its attachment to the plasma membrane (CitationCohen 2002; CitationDoll et al 2004). In addition, the cytoplasmic MEK inhibitors have been studied extensively for their ability to reduce or prevent the phosphorylation and activation of ERKs (CitationKramer et al 2004). Indeed, MAP kinase inhibitors have been shown to be effective in cancer therapy in mouse models (CitationSebolt-Leopold et al 1999).
PD98059 and UO126 are two of the few MAP kinase inhibitors that are not ATP competitive. While both have good specificity at inhibiting ERK1/2 activation (IC50 in the nm range), PD98059 has been shown to also inhibit cyclo-oxygenases 1 and 2, decreasing platelet aggregation (CitationBorsch-Haubold et al 1998). PD98059 blocks the activation of MKK1 by binding to it and preventing its activation by Raf-1, but it does not act on active MKK1. While PD98059 has outstanding activity in vitro, the potential for in vivo toxicity limits its long-term use in animal models or humans (CitationCohen 2002). One promising compound is CI-1040 (PD 184352), a specific small molecule inhibitor of MEK1/2. CI-1040 is well tolerated in humans and has shown efficacy as an anti-tumor agent (CitationKramer et al 2004). Use of ERK inhibitors in animal models of smoke-induced emphysema has not been reported, but it is conceivable that this treatment would block the activation of ERK1/2, preventing the subsequent changes in gene expression.
p38 inhibitors
Inhibitors of p38 MAP kinase signaling have shown efficacy in ameliorating several COPD pathologies in animal models (). These compounds were initially identified for their ability to prevent production of IL-1 and TNF from stimulated human monocytes (CitationLee et al 1994). The most popular p38 inhibitor used in basic science research applications is SB203580. Unfortunately, this compound has limited solubility and high toxicity and its use is generally restricted to cell culture assays (CitationCohen 2002). But use in the laboratory can identify roles for p38 in disease. For example, as was shown for the MEK1/2 inhibitor PD98059, SB203580 can inhibit cyclo-oxygenases-1 and -2 (CitationBorsch-Haubold et al 1998). These data demonstrate the importance of dose-response studies and careful analysis of the multiple pathways that may be affected by kinase inhibitors. SB203580 at 3–5 micromolar concentrations has been shown to block phosphorylation of phosphatidylinositol 3-kinase/protein kinase B (PKB)(Akt/Rac) kinase in IL-2 stimulated T cells (CitationLali et al 2000). The result is blockade of T cell proliferation via inhibition of cell cycle progression.
The efficacy of SB203580 on lung injury has also been tested in vivo (CitationArcaroli et al 2001), with mixed results. Although researchers demonstrated an increase in p38 activity within neutrophils during acute lung injury, SB203580 did not decrease lung neutrophil influx or pulmonary edema during hemorrhage or lipopolysaccharide endotoxemia (CitationArcaroli et al 2001). Similarly, the increased production of proinflammatory cytokines (MIP2, TNF-α) and activation of NF-κB in lung neutrophils induced in these models was not diminished by p38 inhibition.
Not all studies result in the same conclusions regarding the efficacy of p38 inhibitors. While the previous report did not demonstrate any benefit from p38 inhibition, others report positive outcomes. During synthesis of a different compound, SB239063, it was discovered that methylation of the nitrogen in the imidizole group greatly improved the drug’s bioavailability (CitationLiverton et al 1999). The resulting inhibitor was shown to reduce myocardial infarction in the mouse (CitationKaiser et al 2005). In addition, use in rats and guinea pigs demonstrates that SB239063 effectively blocks p38 signaling in lung tissue when delivered intragrastrically before and after lipopolysaccharide inhalation challenge (CitationUnderwood et al 2000), demonstrating that this compound has substrate efficacy in the lung. Most notably, this study showed that inhibition of p38 signaling with SB239063 could reduce pulmonary fibrosis, MMP-9 and IL-6 expression, and neutrophil influx in vivo (CitationUnderwood et al 2000) (). While the study by Arcaroli and colleagues demonstrated no benefit with SB203580, p38 inhibition with intratracheal SB239063 was much more effective. Taken together, these studies demonstrate that the benefit of p38 inhibition depends upon the type of inhibitor, delivery route, and disease model. The bioavailability of these compounds may be an important factor in their ability to control lung injury.
SAPK/JNK inhibitors
Inhibitors of SAPK/JNK signaling have only recently become commercially available to basic scientists, and thus it will be some time before their effects on COPD-relevant events become clear. It has been demonstrated in several studies that this pathway is an important regulator of gene transcription, in part for the ability of JNKs (JNKs1, 2, and 3) to bind and phosphorylate the transcriptional regulator c-Jun. Inhibitors of this pathway have shown promise in controlling rheumatoid arthritis (CitationHan et al 2001). There are 13 upstream MKKKs which regulate JNK signaling (Science STKE JNK Pathway Connections Map 2002; URL: http://stke.science-mag.org/cgi/cm/stkecm;CMP_10827), suggesting that there are many targets for SAPK/JNK signaling blockade. As was shown for inhibitors of the classical MAP kinase pathway, SAPK/JNK inhibitors can also slow the growth of tumor cells in vivo (CitationEnnis et al 2005). Using the SP600125 compound, researchers demonstrated that JNK blockade reduced tumor growth in a mouse model of Lewis lung carcinoma (CitationEnnis et al 2005). This drug demonstrated anti-angiogenic as well as anti-proliferative effects (CitationEnnis et al 2005). SP600125 also reduces arthritis symptoms (inflammation, joint swelling) (CitationHan et al 2001) and several parameters of lipopolysaccharide-induced lung injury in rats (CitationLee et al 2004) (). Other JNK inhibitors have shown promise in reducing neuronal apoptosis (CitationMaroney et al 1998) and pancreatic edema and inflammation (CitationWagner et al 2000). The SAPK/ JNK pathway is also involved in T cell differentiation and activation (CitationDong et al 2000). Several studies have shown activated T cells in lung tissue and peripheral circulation of patients with COPD (CitationBarnes and Cosio 2004), suggesting that inhibition of this MAP kinase cascade may impact on both central and peripheral inflammation in COPD. However, further studies are needed to define the precise roles of this pathway in the lung.
Pitfalls of kinase inhibition in COPD
A key objective in the design of therapeutics for COPD is to achieve significant functional and survival benefit for the patient. The first steps of kinase inhibition research involve a determination of the efficacy and toxicity of potential compounds. A major obstacle in the design of kinase inhibitors is obtaining high selectivity of the compound without side-effects. For example, MAP kinase blockade in embryonic mouse lung explants impairs branching morphogenesis and increases apoptosis (CitationKling et al 2002), which may translate into unwanted cellular and morphometric effects in adult lung tissue. Often, while a drug is able to inhibit the phosphorylation of its purified substrate in a test tube, it may not be biologically available, making it difficult to use for animal models of COPD, and ultimately humans. This has been shown to be the case for PD98059 and SB23580, two compounds that inhibit ERK1/2 and p38 kinases, respectively. Though cell permeable, these compounds demonstrate toxicity and solubility issues at higher concentrations which preclude their chronic use in animals, although short-term studies have been performed (CitationTamaoki et al 2004; Kase et al 2005). In addition, because COPD patients typically present with several comorbidities, such as right-sided heart failure, muscle weakness, cachexia, and pulmonary vascular disease, the effects of drug therapy must be cautiously assessed.
Conclusions
Transitioning COPD research
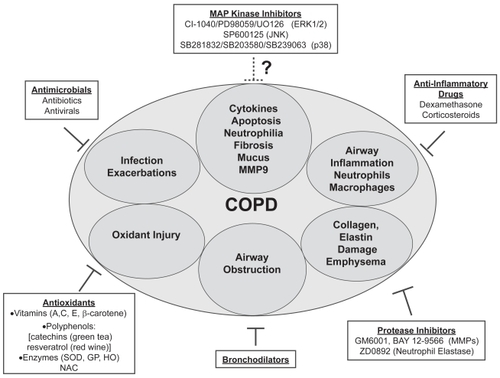
The molecular pathogenesis of COPD involves repeated smoke-induced injury to cells of the airways and parenchyma. These insults alter gene expression and epithelial cell function. Although diverse stimuli activate MAP kinase pathways in the lung, it is clear that tobacco smoke is an essential modifier of gene expression in COPD. Many cell processes are controlled by reversible phosphorylation of proteins. Analysis of the human genome revealed that the “kinome” (collection of all putative protein kinases) contains 518 genes, many with unknown function (CitationManning et al 2002). In vitro, animal, and human tissue studies of ERK1/2, SAPK/JNK, or p38 kinases have identified important roles for these enzymes in lung cell biology. Although individual MAP kinases have been shown to mediate specific COPD-relevant cellular events (CitationJohnson and Lapadat 2002), the crosstalk among these pathways within the lung milieu is complex (CitationPrice et al 1996). Thus it is conceivable that even small reductions in kinase activity will result in robust reductions in gene transcription. Indeed, the use of MAP kinase inhibitors to reduce inflammation, apoptosis, cytokine production, and tissue injury has already been demonstrated. Currently, drug treatment strategies for COPD can be organized into several branches of target pathology (). Although further studies are needed, the MAP kinase inhibitors, along with protease inhibitors and antioxidants, are beginning to emerge as promising therapeutic strategies for COPD.
Figure 4 Model of the potential for MAP kinase inhibitors as COPD treatment strategies. COPD treatment branches are numerous, targeting the most destructive and debilitating processes, including airway inflammation, infection, exacerbations, and airway obstruction. Ongoing research to design drugs that reduce protease activity and oxidant injury will be critical in future COPD treatment. The potential for MAP kinase inhibitors is another promising area of research. These drugs have already demonstrated efficacy in reducing apoptosis, inflammation, cytokine production, fibrosis, and MMP expression. Future studies may result in the inclusion of MAP kinase inhibitors to COPD therapeutic strategies.
Abbreviations
| AM | = | alveolar macrophages |
| AP-1 | = | activating protein 1 |
| BCR | = | B cell receptor |
| EGF | = | epidermal growth factor |
| EGFR | = | epidermal growth factor receptor |
| ERK | = | extracellular signal-regulated kinases |
| FGF | = | fibroblast growth factor |
| GEF | = | guanine exchange factors |
| GPCR | = | G-protein coupled receptor |
| MAP | = | mitogen activated protein |
| MMP-1 | = | matrix proteinase-1 |
| NHBE | = | normal human bronchial epithelial cells |
| PDGF | = | platelet-derived growth factor |
| RTK | = | receptor tyrosine kinase |
| SAEC | = | small airway epithelial cells |
| SAPK/JNK | = | stress activated protein kinase/c-Jun N-terminal protein kinase |
| TCF | = | ternary complex factor |
| TCR | = | T cell receptor |
| TNF | = | tumor necrosis factor |
References
- AdamsRHPorrasAAlonsoG2000Essential role of p38alpha MAP kinase in placental but not embryonic cardiovascular developmentMol Cell61091610949032
- AngelPKarinM1991The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformationBiochim Biophys Acta1072129571751545
- AoshibaKNagaiAYasuiS1996Nicotine prolongs neutrophil survival by suppressing apoptosisJ Lab Clin Med127186948636647
- AoshibaKYokohoriNNagaiA2003Alveolar wall apoptosis causes lung destruction and emphysematous changesAm J Respir Cell Mol Biol285556212707011
- AppelmanLMWoutersenRAFeronVJ1982Inhalation toxicity of acetaldehyde in rats. I. Acute and subacute studiesToxicology232933077123564
- ArcaroliJYumHKKupfnerJ2001Role of p38 MAP kinase in the development of acute lung injuryClin Immunol1012111911683580
- ArmstrongLRomWMartinuikFT1996Nicotine enhances expression of the neutrophil elastase gene and protein in a human myeloblast/promyelocyte cell lineAm J Respir Crit Care Med154152048912774
- BarnesPJCosioMG2004Characterization of T lymphocytes in chronic obstructive pulmonary diseasePLoS Med1e2015526047
- BinetruyBSmealTKarinM1991Ha-Ras augments c-Jun activity and stimulates phosphorylation of its activation domainNature35112271903181
- BishopJELaurentGJ1995Collagen turnover and its regulation in the normal and hypertrophying heartEur Heart J1638447556271
- BorchersMTCartyMPLeikaufGD1999Regulation of human airway mucins by acrolein and inflammatory mediatorsAm J Physiol276L5495510198352
- BorchersMTWertSELeikaufGD1998Acrolein-induced MUC5ac expression in rat airwaysAm J Physiol274L573819575876
- BorchersMTWesselkamperSWertSE1999Monocyte inflammation augments acrolein-induced Muc5ac expression in mouse lungAm J Physiol277L4899710484456
- Borsch-HauboldAGPasquetSWertSE1998Direct inhibition of cyclooxygenase-1 and -2 by the kinase inhibitors SB 203580 and PD 98059. SB 203580 also inhibits thromboxane synthaseJ Biol Chem27328766729786874
- BoultonTGNyeSHRobbinsDJ1991ERKs:a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGFCell65663752032290
- BrandSOlszakTBeilgelF2006Cell differentiation dependent expressed CCR6 mediates ERK-1/2, SAPK/JNK, and Akt signaling resulting in proliferation and migration of colorectal cancer cellsJ Cell Biochem977092316215992
- CartyCSSolowayPD1996Nicotine and cotinine stimulate secretion of basic fibroblast growth factor and affect expression of matrix metalloproteinases in cultured human smooth muscle cellsJ Vasc Surg2492734 discussion 934–58976346
- ChangWCLeeYCLiuCL2001Increased expression of iNOS and c-fos via regulation of protein tyrosine phosphorylation and MEK1/ERK2 proteins in terminal bronchiole lesions in the lungs of rats exposed to cigarette smokeArch Toxicol75283511357518
- ChenBCYuCCLeiHC2004Bradykinin B2 receptor mediates NF-kappaB activation and cyclooxygenase-2 expression via the Ras/Raf-1/ERK pathway in human airway epithelial cellsJ Immunol17352192815470067
- ChialdaLZhangMBruneK2005Inhibitors of mitogen-activated protein kinases differentially regulate costimulated T cell cytokine production and mouse airway eosinophiliaRespir Res63615833106
- ChuMGuoJChenCY2005Long-term exposure to nicotine, via ras pathway, induces cyclin D1 to stimulate G1 cell cycle transitionJ Biol Chem28063697915574422
- CohenP2002Protein kinases – the major drug targets of the twenty-first century?Nat Rev Drug Discov13091512120282
- CruzaleguiFHCanoETreismanR1999ERK activation induces phosphorylation of Elk-1 at multiple S/T-P motifs to high stoichiometryOncogene1879485710637505
- CuendaARouseJDozaYN1995SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1FEBS Lett364229337750577
- D’ArmientoJDalalSSOkadaY1992Collagenase expression in the lungs of transgenic mice causes pulmonary emphysemaCell71955611458541
- DeshmukhHSCaseLMWesselkamperSC2005Metalloproteinases mediate mucin 5AC expression by epidermal growth factor receptor activationAm J Respir Crit Care Med1713051415531749
- DollRJKirschmeierPBishopWR2004Farnesyltransferase inhibitors as anticancer agents:critical crossroadsCurr Opin Drug Discov Devel747886
- DongCYangDDTournierC2000JNK is required for effector T-cell function but not for T-cell activationNature40591410811224
- DongCYangDDWyskM1998Defective T cell differentiation in the absence of Jnk1Science282209259851932
- EnnisBWFultzKESmithKA2005Inhibition of tumor growth, angiogenesis, and tumor cell proliferation by a small molecule inhibitor of c-Jun N-terminal kinaseJ Pharmacol Exp Ther3133253215626722
- ErmanMGrunenwaldDPenault-LlorcaF2005Epidermal growth factor receptor, HER-2/neu and related pathways in lung adenocarcinomas with bronchioloalveolar featuresLung Cancer473152315713515
- FinkelsteinENardiniMVan der VlietA2001Inhibition of neutrophil apoptosis by acrolein; a mechanism of tobacco-related lung disease?Am J Physiol Lung Cell Mol Physiol281L732911504702
- ForonjyRFOkadaYColeR2003Progressive adult-onset emphysema in transgenic mice expressing human MMP-1 in the lungAm J Physiol Lung Cell Mol Physiol284L7273712676763
- GenschEGallupMSucherA2004Tobacco smoke control of mucin production in lung cells requires oxygen radicals AP-1 and JNKJ Biol Chem279390859315262961
- GuoJChuMAbbeyquayeT2005Persistent nicotine treatment potentiates amplification of the dihydrofolate reductase gene in rat lung epithelial cells as a consequence of Ras activationJ Biol Chem280304223115983034
- HanJLeeJDBibbsL1994A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cellsScience265808117914033
- HanSWHwangPGChungDH2005Epidermal growth factor receptor (EGFR) downstream molecules as response predictive markers for gefitinib (Iressa, ZD1839) in chemotherapy-resistant non-small cell lung cancerInt J Cancer1131091515386420
- HanZBoyleDLChangL2001c-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritisJ Clin Invest108738111435459
- HellermannGRNagySBKongX2002Mechanism of cigarette smoke condensate-induced acute inflammatory response in human bronchial epithelial cellsRespir Res32212204101
- HeuschWLManeckjeeR1998Signalling pathways involved in nicotine regulation of apoptosis of human lung cancer cellsCarcinogenesis1955169600337
- ImaiKDalalSChenE2001Human collagenase (matrix metal-loproteinase-1) expression in the lungs of patients with emphysemaAm J Respir Crit Care Med16378679111254539
- ImaiKMercerBShulmanLL2005Correlation of lung surface area to apoptosis and proliferation in human emphysemaEur Respir J2519
- IshiiTFujishiroMMasudaM2003Depletion of glutathione S-transferase P1 induces apoptosis in human lung fibroblastsExp Lung Res295233614710442
- ItoKItoMElliottWM2005Decreased histone deacetylase activity in chronic obstructive pulmonary diseaseN Engl J Med35219677615888697
- JohnsonGLLapadatR2002Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinasesScience2981911212471242
- JoosLHeJQSheperdsonMB2002The role of matrix metalloproteinase polymorphisms in the rate of decline in lung functionHum Mol Genet115697611875051
- KaiserRALyonsJMDuffyJY2005Inhibition of p38 reduces myocardial infarction injury in the mouse but not pig following ischemia-reperfusionAm J Physiol Heart Circ Physiol289H27475116143643
- KaseSYoshidaKHaradaT2006Phosphorylation of extracellular signal-regulated kinase and p27(KIP1) after retinal detachmentGraefes Arch Clin Exp Ophthalmol244352816075224
- KawasakiHSchiltzLChiuR2000ATF-2 has intrinsic histone acetyltransferase activity which is modulated by phosphorylationNature40519520010821277
- KlingDLorenzoHTrbovichA2002MEK-1/2 inhibition reduces branching morphogenesis and causes mesenchymal cell apoptosis in fetal rat lungsAm J Physiol Lung Cell Mol Physiol282L370811839529
- KramerBWGotzRRappUR2004Use of mitogenic cascade blockers for treatment of C-Raf induced lung adenoma in vivo: CI-1040 strongly reduces growth and improves lung structureBMC Cancer42415171791
- KuanCYWhitmarshAJYangDD2003A critical role of neural-specific JNK3 for ischemic apoptosisProc Natl Acad Sci U S A10015184914657393
- KuanCYYangDDSamanta RoyDR1999The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain developmentNeuron226677610230788
- KuidaKBoucherDM2004Functions of MAP kinases:insights from gene-targeting studiesJ Biochem (Tokyo)135653615213239
- KurinnaSMTsaoCCNicaAF2004Ceramide promotes apoptosis in lung cancer-derived A549 cells by a mechanism involving c-Jun NH2-terminal kinaseCancer Res647852615520191
- LaliFVHuntAETurnerSJ2000The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation, and retinoblastoma hyperphosphorylation in interleukin-2-stimulated T cells independently of p38 mitogen-activated protein kinaseJ Biol Chem275739540210702313
- LeeHSKimHJMoonCS2004Inhibition of c-Jun NH2-terminal kinase or extracellular signal-regulated kinase improves lung injuryRespir Res52315566575
- LeeJCKumarSGriswoldDE2000Inhibition of p38 MAP kinase as a therapeutic strategyImmunopharmacology4718520110878289
- LeeJCLaydonJTMcDonnellPC1994A protein kinase involved in the regulation of inflammatory cytokine biosynthesisNature372739467997261
- LemjabbarHLiDGallupM2003Tobacco smoke-induced lung cell proliferation mediated by tumor necrosis factor alpha-converting enzyme and amphiregulinJ Biol Chem27826202712711607
- LivertonNJButcherJWClaiborneCF1999Design and synthesis of potent, selective, and orally bioavailable tetrasubstituted imidazole inhibitors of p38 mitogen-activated protein kinaseJ Med Chem4221809010377223
- LuCSpeersCZhangY2003Effect of epidermal growth factor receptor inhibitor on development of estrogen receptor-negative mammary tumorsJ Natl Cancer Inst9518253314679152
- ManningGWhyteDBMartinezR2002The protein kinase complement of the human genomeScience29819123412471243
- MarkewitzBOwensMWPayneDK1999The pathogenesis of chronic obstructive pulmonary diseaseAm J Med Sci31874810452563
- MaroneyACGlicksmanMABasmaAN1998Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathwayJ Neurosci18104119412490
- MeierRRouseJCuendaA1996Cellular stresses and cytokines activate multiple mitogen-activated-protein kinase kinase homologues in PC12 and KB cellsEur J Biochem2367968058665897
- MercerBAKolesnikovaNSonettJ2004Extracellular regulated kinase/mitogen activated protein kinase is up-regulated in pulmonary emphysema and mediates matrix metalloproteinase-1 induction by cigarette smokeJ Biol Chem27917690614764579
- MioTRombergerDJThompsonAB1997Cigarette smoke induces interleukin-8 release from human bronchial epithelial cellsAm J Respir Crit Care Med155177069154890
- Mochida-NishimuraKSurewiczKCrossJ2001Differential activation of MAP kinase signaling pathways and nuclear factor-kB in bronchoalveolar cells of smokers and nonsmokersMolecular Medicine71778511471554
- MoodieFMMarwickJAAndersonCS2004Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-kappaB activation and proinflammatory cytokine release in alveolar epithelial cellsFaseb J181897915456740
- NardiniMFinkelsteinEIReddyS2002Acrolein-induced cyto-toxicity in cultured human bronchial epithelial cells. Modulation by alpha-tocopherol and ascorbic acidToxicology1701738511788155
- NingWChoiAMLiC2005Carbon monoxide inhibits IL-17-induced IL-6 production through the MAPK pathway in human pulmonary epithelial cellsAm J Physiol Lung Cell Mol Physiol289L2687316003000
- PagesGGuerinSGrallD1999Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout miceScience2861374710558995
- PetracheINatarajanVZhenL2005Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in miceNat Med11491815852018
- PriceMACruzaleguiFHTreismanR1996The p38 and ERK MAP kinase pathways cooperate to activate Ternary Complex Factors and c-fos transcription in response to UV lightEmbo J156552638978682
- PuddicombeSMDaviesDE2000The role of MAP kinases in intracellular signal transduction in bronchial epitheliumClin Exp Allergy3071110606925
- RangannaKYousefipourZNasifR2002Acrolein activates mitogen-activated protein kinase signal transduction pathways in rat vascular smooth muscle cellsMol Cell Biochem240839812487375
- ReganCPLiWBoucherDM2002Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defectsProc Natl Acad Sci U S A9992485312093914
- ReunanenNWestermarckJHäkkinenL1998Enhancement of fibroblast collagenase (matrix metalloproteinase-1) gene expression by ceramide is mediated by extracellular signal-regulated and stress-activated protein kinase pathwaysJ Biol Chem2735137459478967
- SaladinoAJWilleyJCLechnerJF1985Effects of formaldehyde, acetaldehyde, benzoyl peroxide, and hydrogen peroxide on cultured normal human bronchial epithelial cellsCancer Res456252263986791
- Sebolt-LeopoldJSDudleyDTHerreraR1999Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivoNat Med5810610395327
- Segura-ValdezLPardoAGaxiolaM2000Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPDChest1176849410712992
- SelcherJCNekrasovaTPaylorR2001Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learningLearn Mem811911160759
- SelmanMMontanoMRamosC1996Tobacco smoke-induced lung emphysema in guinea pigs is associated with increased interstitial collagenaseAm J Physiol271L734438944716
- SissonJHTumaDJRennardSI1991Acetaldehyde-mediated cilia dysfunction in bovine bronchial epithelial cellsAm J Physiol260L29361825452
- SuzukiHAoshibaKYokohoriN2003Epidermal growth factor receptor tyrosine kinase inhibition augments a murine model of pulmonary fibrosisCancer Res635054912941834
- TakeyamaKJungBShimJJ2001Activation of epidermal growth factor receptors is responsible for mucin synthesis induced by cigarette smokeAm J Physiol Lung Cell Mol Physiol280L1657211133506
- TamaokiJTagayaEKawataniK2004Airway mucosal thickening and bronchial hyperresponsiveness induced by inhaled beta 2-agonist in miceChest1262051215249464
- TomizawaMCasidaJE2003Selective toxicity of neonicotinoids attributable to specificity of insect and mammalian nicotinic receptorsAnnu Rev Entomol483396412208819
- TournierCHessPYangDD2000Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathwayScience288870410797012
- TreismanR1996Regulation of transcription by MAP kinase cascadesCurr Opin Cell Biol8205158791420
- TsuboiMKatoHNagaiK2005Gefitinib in the adjuvant setting: safety results from a phase III study in patients with completely resected non-small cell lung cancerAnticancer Drugs161123816222155
- TuderRMZhenLChoCY2003Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockadeAm J Respir Cell Mol Biol29889712600822
- TuytLMDokterWHBirkenkampK1999Extracellular-regulated kinase 1/2, Jun N-terminal kinase, and c-Jun are involved in NF-kappa B-dependent IL-6 expression in human monocytesJ Immunol162489390210202034
- UnderwoodDCOsbornRRBochnowiczS2000SB 239063, a p38 MAPK inhibitor, reduces neutrophilia, inflammatory cytokines, MMP-9, and fibrosis in lungAm J Physiol Lung Cell Mol Physiol279L89590211053025
- VerheijMBoseRLinXH1996Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosisNature3807598598911
- VicentSGarayoaMLopez-PicazoJM2004Mitogen-activated protein kinase phosphatase-1 is overexpressed in non-small cell lung cancer and is an independent predictor of outcome in patientsClin Cancer Res1036394915173070
- WagnerACMazzucchelliLMillerM2000CEP-1347 inhibits caerulein-induced rat pancreatic JNK activation and ameliorates caerulein pancreatitisAm J Physiol Gastrointest Liver Physiol278G1657210644575
- WilhelmSMCarterCTangL2004BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesisCancer Res64709910915466206
- WuenschellCKunimiMCatilloC2004Nicotine-responsive genes in cultured embryonic mouse lung buds:interaction of nicotine and superoxide dismutasePharmacol Res503415015225679
- YangDDKuanCYWhitmarshAJ1997Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 geneNature389865709349820
- YaoYLiWWuJ2003Extracellular signal-regulated kinase 2 is necessary for mesoderm differentiationProc Natl Acad Sci U S A100127596414566055
- YokohoriNAoshibaKNagaiA2004Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysemaChest1256263214769747
- ZhangJFujimotoNIwataK1993A one-step sandwich enzyme immunoassay for human matrix metalloproteinase-1 (interstitial collagenase) using monoclonal antibodiesClin Chim Acta2191148306449
- ZhangSDayINYeS2001Microarray analysis of nicotine-induced changes in gene expression in endothelial cellsPhysiol Genomics51879211328964
- ZhangWYanSDZhuA2000Expression of Egr-1 in late stage emphysemaAm J Pathol15713112011021835