Abstract
The Alpha One International Registry (AIR), a multinational research program focused on alpha1-antitrypsin (AAT) deficiency, was formed in response to a World Health Organization recommendation. Each of the nearly 20 participating countries maintains a national registry of patients with AAT deficiency and contributes to an international database located in Malmö, Sweden. This database is designed to increase understanding of AAT deficiency. Additionally, AIR members are engaged in active, wide-ranging investigations to improve the diagnosis, monitoring, and treatment of the disease and meet biennially to exchange views and research findings. The fourth biennial meeting was held in Copenhagen, Denmark, on 2–3 June 2005. This review covers the wide range of AAT deficiency-related topics that were addressed encompassing advances in genetic characterization, risk factor identification, clinical epidemiology, inflammatory and signalling processes, therapeutic advances, and lung imaging techniques.
Introduction
Alpha1-antitrypsin (AAT) deficiency was first reported in 1963 by Carl-Bertil Laurell and Sten Eriksson (CitationLaurell and Eriksson 1963) who noted a link between low plasma serum levels of AAT and symptoms of pulmonary emphysema. Since these first cases were described, an understanding of the biochemical mechanisms and genetic abnormalities involved has developed, and AAT deficiency is now thought to be one of the most common hereditary disorders worldwide, comparable in frequency to cystic fibrosis (CitationWHO 1997; Citationde Serres 2002). The most frequent clinical complications of AAT deficiency are pulmonary emphysema of the panacinar type, which may present as early as the third or fourth decade, and liver disease, which typically presents early in infancy as neonatal hepatitis with a diverse degree of liver involvement and outcome (CitationATSERS 2003). The development of AAT augmentation therapy in 1987 provided an option for physicians seeking to improve the course of this disease (CitationWewers et al 1987), and research into other potential treatments such as gene therapy is ongoing.
The Alpha One International Registry (AIR), formed in 1998, is a multinational research project involving nearly 20 countries. AIR was started to fulfill the World Health Organization (WHO) recommendation for the establishment of an international registry for AAT deficiency (CitationWHO 1997). Including subjects identified by a standardized mechanism to create a common database, AIR can be considered central to resolving many of the unanswered questions in AAT deficiency. AIR is committed to organizing scientific meetings every 2 years to update the medical and scientific community on the progress of research in AAT deficiency. The first meeting was held in 1999 in Cernobbio-Como, Italy (CitationStockley 1999), and subsequent meetings have been held in Genoa, Italy, in 2001(CitationLuisetti et al 2002) and Barcelona, Spain, in 2003 (CitationNavickis and Wilkes 2004). The fourth AIR meeting was held in Copenhagen, Denmark, on 2–3 June 2005 and involved 26 speakers in a concentrated 2-day scientific program. The highlights of this meeting are reported in this review.
The Laurell Lecture
In recognition of Carl-Bertil Laurell’s work on AAT deficiency, the AIR Laurell Lecture was established in 2003 to acknowledge those who have made a substantial contribution to the knowledge base in AAT deficiency. Robin W Carrell was the first to present such a lecture (CitationNavickis and Wilkes 2004). The second Laurell Lecture was presented by Professor Noor Kalsheker of the Division of Clinical Chemistry at Queen’s Medical Centre of the University of Nottingham, UK.
Professor Kalsheker discussed the gene regulation (CitationKalsheker et al 2002) and genetic variation of AAT and the relationship with COPD. AAT is a member of the serine proteinase inhibitor (serpin) gene family and is now also referred to as SERPINA1. There are 33 different serpin gene sequences identified in the human genome, and there are 2 discrete clusters (chromosomes 14q 32.1 and 18q 21.3), each consisting of about 11 genes. These clusters contain only serpin genes, and their composition and structure are highly conserved. The way DNA is packaged provides a higher- order structure in terms of gene regulation. It is possible to explore some of the important structural elements associated with chromatin. Some areas of DNA are exposed by the manner of DNA packing, and these chromatin landmarks provide clues to putative regulatory elements and can be assessed by DNase 1 hypersensitivity sites. A region upstream of the AAT gene has been identified that is absolutely critical for expression of the whole gene cluster. Within this 8-kb region, there is 2.3-kb DNA, the locus control region (LCR), which is critical for expression of the entire locus. The LCR controls the whole cluster, but there is also local control within the gene cluster.
Transcription is not a simple process and involves a number of proteins combining to promote gene expression (). Specifically, in AAT expression, 2 main promoters have been found: the major hepatocyte promoter driving expression in the liver and, further upstream, the monocyte promoter that drives expression in other cell types, eg, lung tissue. These promoters are distinct and work via different mechanisms. Promoters regulate basal expression in the steady state, while elements in the DNA known as enhancers modulate expression during inflammation. Several sites of local transcriptional control have been identified upstream of the hepatocyte transcription initiation site, including binding sites for hepatocyte nuclear factor-1α (HNF-1α) and HNF4. Humoral regulation is mediated by cytokines, particularly interleukin-6 (IL-6) and oncostatin-M. These cytokines work via a 3’ enhancer downstream of the hepatocyte promoter, and both oncostatin-M and IL-6 work via different response elements. Therefore, there are discrete binding sites providing distinct pathways for regulating the acute phase response via different cytokines.
Figure 1 Assembly of general transcription factors required for initiation of transcription. TFIID, a complex of transcription activating factors (TAF) and TATA-binding protein (TBP) binds specifically to a TATA sequence. The presence of a transcriptional inhibitor can block the transcription process at this step. Next, additional transcription factors IIA and IIB assemble into the complex. RNA polymerase II (Pol II), escorted by transcription factor IIF, assemble into the complex along with additional transcription factors. Transcription factor IIH, in the presence of ATP, phosphorylates Pol II, releases the polymerase and initiates transcription. Adapted with permission from: CitationLodish H, Baltimore D, Berk A, et al. 1995. Transcriptional control of gene expression. In Lodish H, Baltimore D, Berk A, et al (eds). Molecular cell biology. 3rd ed. New York: WH Freeman and Company/Worth Publishers. Figure 11–53, Chapter 11, p 454.
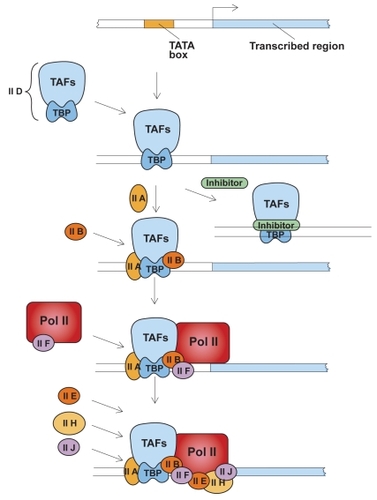
The promoter for other cell types is regulated by a different group of transcription factors known as the Sp1 family. Here it has been suggested that RNA stability also influences expression, as some transcripts may be more stable than others. In the liver, there is only one transcript of AAT; however, in other cell types, there are a number of alternative transcripts. Although most AAT is manufactured in the liver, the tissue in the lung has a huge capacity to make AAT; with appropriate stimuli, expression can be increased approximately 100-fold (CitationBoutten et al 1998). Indeed, it has been estimated that, under inflammatory conditions, the lung could produce as much as one third of the amount of AAT produced by the liver.
Work on identifying molecular variants of AAT and how much genetic variation there is in the AAT gene has recently been undertaken. Using single nucleotide polymorphisms (SNPs) as markers for disease, SNP haplotypes can reveal information on genetic diversity and disease association. A large, case-controlled study of nearly 2000 subjects using both epidemiologic and genetic techniques to correct for confounders such as age and gender assigned individual data to haplotype on a probability basis. The study revealed that haplotypes of AAT, independently of PiZZ, confer an equal or greater risk of COPD than the PiZZ phenotype (CitationChappell et al 2004).
Epidemiology and detection
Screening can have important psychological consequences both on parents and the child. Experience in Sweden (CitationSveger et al 1999) has shown that the mothers of AAT-deficient children suffered increased anxiety compared with controls. Insufficient counselling at the time of identification was reported by the majority of parents, which reinforces the importance of patient–parent education. There were, however, some beneficial effects of screening. Half of the AAT-deficient individuals thought that the knowledge of their high-risk condition had affected their lives, particularly their awareness of the dangers of smoking and environmental pollution. The majority, 88%, knew that they should avoid smoking to protect their lungs (CitationSveger et al 1997). Indeed, the majority of those who were identified through screening and their parents would recommend screening for AAT deficiency.
Screening of blood donors revealed that the prevalence of AAT deficiency in the US was at least 1 in 2750, which equates to more than 80 000 living patients in the US; however, only a few thousand patients are ever diagnosed with AAT deficiency and only 60%–70% of these patients receive augmentation therapy. To improve the efficiency and accuracy of AAT deficiency testing, a central laboratory approach has been adopted, with immunoassay, phenotyping, and genotyping available. A similar approach has also been established outside the US. However, despite clear recommendations from the WHO (CitationWHO 1997) and an ATS–ERS working party (CitationATSERS 2003), physicians’ practices are changing minimally and slowly, and many physicians do not believe that they have undetected patients in their practices. Indeed, many physicians, as well as patients with COPD, remain completely unaware of the disease.
Recently, an approach of testing AAT concentration initially in serum followed by further testing for PiZZ and PiSZ genotypes in patients with AAT levels below 110 mg/dL resulted as the most cost-effective screening method of COPD patients (CitationDe La Roza et al 2005). This approach reduced the cost per sample by about 30% (€13.43 vs (€19.41 [US$16.35 vs US$23.64]) and the cost per PiZZ individual detected also by about 30% ((€3589 vs (€5189 [US$4370 vs US$6319]) compared with screening all samples for genotype (CitationDe La Roza et al 2005).
Italy is one of the European countries least affected by AAT deficiency. Based on calculated allele frequency, approximately 3000 individuals are thought to have the PiZZ phenotype and a further 22 000 the PiSZ phenotype, although hardly any are diagnosed. Registries show a prevalence of rare genotypes between 1.7% and 5.7%; however, in Italy, this number appears to be closer to 15% in severe AAT deficiency, the highest recorded thus far worldwide (CitationFerrarotti et al 2005). AAT serum levels are slightly lower in individuals with rare genotypes compared with PiZZ individuals (23 vs 26 mg/dL). Smoking also appears to be more common in those with rare genotypes, and COPD is the most common manifestation (87%), with 9% exhibiting both COPD and liver disease and 4% exhibiting liver disease alone. In conclusion, rare AAT-deficiency variants may be more frequent in areas characterized by a low prevalence of common AAT-deficiency variants, but these variants are just not detected.
Emphysema in AAT
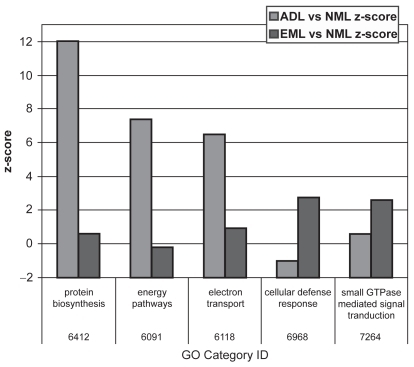
Results of microarray techniques in human emphysematous tissue have been reported in the literature (CitationGolpon et al 2004) (). Severely emphysematous tissue is characterized by an abundance of transcripts encoding proteins involved in inflammation, immune responses, and proteolysis. When compared with “usual” emphysematous tissue, AAT-deficiency-related emphysema has a similar gene expression profile; however, some significant differences exist. Interestingly, the protease caspase 9 is upregulated in AAT deficiency, whereas normal levels are retained in “usual” emphysema. Conversely, cathepsin B is upregulated in “usual” emphysema and remains normal in AAT deficiency. There are further differences between AAT deficiency and “usual” emphysema related to protein biosynthesis, energy pathways, and possibly electron transfer and cellular defense response. These differences suggest that in AAT deficiency, there is impairment in both protein and energy metabolism.
Figure 2 Gene ontology (GO) categories that have a superabundance of differentially expressed genes in either “usual” emphysematous lung tissue (EML) or AAT-deficiency-related emphysematous lung tissue (ADL) when compared with normal lung tissue (NML). Reprinted from CitationGolpon HA, Coldren CD, Zamora MR, et al. 2004. Emphysema lung tissue gene expression profiling. Am J Respir Cell Mol Biol, 31:595–600. Copyright © 2004, with permission from American Thoracic Society.
Several components affect the reaction to cigarette smoke. Microarray analysis in a murine model of cigarette smoke exposure has revealed that early lung response to smoke exposure comprises tissue remodeling, antioxidative response, and fatty acid synthesis and creates a mild inflammatory response. Smoke induces a limited but significant number of hypoxia-responsive genes. All of these genes are associated with the hypoxia-responsive element (HRE) in the gene promoters and are activated by hypoxia-inducible factor (HIF) transcription factors. Therefore, the hypoxia–smoke signature could be targeted by anti-HIF drugs. In addition, different strains of mice have a different gene expression response to smoke exposure with a functional correlation between genotype and phenotype. This work suggests that some genotypes are more susceptible to the effects of hypoxia–smoke.
Clinical observations in siblings with the same PiZZ genotype and similar smoking history but different levels of pulmonary function have suggested that there may be some additional factors influencing the manifestation of emphysema. As almost all smoking AAT-deficient patients will develop emphysema between the third and fifth decades of life, the observation that some do not has aroused interest. Other genetic factors may be involved in the development of pulmonary emphysema in AAT-deficient patients. This is supported in the literature by studies showing an increased prevalence of COPD in relatives of early-onset COPD cases without AAT deficiency compared with relatives of controls. In addition, a heritability of FEV1 approximating 35% suggests that protective or modifier genes may play a role (CitationJoost et al 2002; CitationSilverman et al 2002; CitationPalmer, Celedón, et al 2003). In order to test this hypothesis, a study using the AIR database is currently underway with 96 discordant sibling pairs. It has been calculated that this number of subjects should be able to detect an allele protective for the development of emphysema; however, it will depend on the penetration of the allele and the limits of detection (LOD) scores of the marker(s).
Liver disease in AAT
The characteristic finding in the liver associated with PiZZ AAT deficiency is the accumulation of AAT within hepatocytes as polymers. This manifests as intracytoplasmic inclusion bodies formed at a basal rate in periportal hepatocytes. Fever further induces polymerization rate of type ZZ AAT. In children, the clinical manifestation of this disorder is neonatal hepatitis syndrome with cholestasis. In most cases, spontaneous clinical regression occurs before the age of 6 months, but in 20%–30% of cases with cholestasis, deterioration toward cirrhosis and liver failure occurs. In adults over age 45 years, a wide spectrum of liver disease may develop ranging between mild cirrhosis and hepatocellular carcinoma. Interestingly, significant lung and liver disease in adults rarely coexist in the same individual.
AAT deficiency is the most common metabolic–genetic indication for liver transplantation in children (CitationFrancavilla et al 2000). The majority of cases of PiZZ liver disease present neonatally with prolonged conjugated jaundice, pale stools, and dark urine. From antenatal experience, there do not appear to be obstetric problems or an increased incidence of congenital abnormalities; however, in fetuses aborted at 17–20 weeks’ gestation with the PiZZ phenotype, 4 out of 5 showed a build-up of AAT in the liver (CitationMalone et al 1989). It has been shown that liver involvement occurs in only 17% of PiZZ individuals after 20 years (CitationSveger and Eriksson 1995); however, it is not currently possible to predict which individuals will develop liver disease. Of those who develop neonatal hepatitis syndrome, 22% do not develop chronic liver disease, while 50% develop mild liver disease and the remaining 28% develop severe liver disease necessitating liver transplantation. A recent study investigating the relationship between phenotype and manifestation of liver disease revealed that there was also a significant degree of discordance between sibling pairs regarding the severity of liver disease (CitationHinds et al 2004).
Although PiZZ liver disease is still underdiagnosed in adults, it is being increasingly recognized in those with “cryptogenic cirrhosis”, alcoholic liver disease, and hepatocellular carcinoma and in those receiving liver transplantation. Indeed, the possible relationship with co-morbid conditions such as hepatitis B and C, autoimmune hepatitis, and hemochromatosis has given rise to the “second-hit” theory, where AAT deficiency could make the associated pathology more severe.
Polymerization is identified as the main pathological event occurring in the liver. Disappointingly, very few papers have been published in the last few years addressing either liver disease or cell biology in AAT deficiency, especially considering that such research could shed light on a number of conditions. In the area of liver cell biology, using differential antibody binding has shown that the AAT polymer conformations formed by the Z, Siiyama, and Mmalton phenotypes are different (CitationJanciauskiene et al 2004). Another study showed that by using point mutation within a surface hydrophobic cavity at a particular site, polymer formation could be reduced in the Z-type AAT molecule (CitationParfrey et al 2003). Investigation of the polymerization process itself has revealed a kinetic lag phase during which short oligomers are formed prior to the formation of heterogeneous mixtures of longer polymers (CitationPurkayastha et al 2005). These and other studies have increased overall understanding of protein conformation; however, this needs to be linked to pathogenesis and clinical outcomes.
One exciting development is in the area of protein processing. If small compounds that are able to act in the liver cell could prevent the process of polymerization, biochemical functional ZZ protein would be generated for secretion into the peripheral circulation. It has been shown in a tissue culture system that endoplasmic reticulum mannosidase I (ERManI) can contribute to the preferential selection of misfolded AAT monomers for proteasomal degradation (CitationHosokawa et al 2003; CitationWu et al 2003), which could provide the basis for a treatment. Six of the key genes involved in Z-type AAT degradation have also been identified by a group at the University of Nevada, providing further potential therapeutic targets (CitationPalmer, Kruse, et al 2003). Lawless et al demonstrated the “second-hit” phenomenon in vitro; activation of the unfolded protein response pathway requires an additional insult such as an increase in temperature, adding to understanding of the pathogenesis of AAT-deficiency-related liver disease (CitationLawless et al 2004).
Lung disease
AAT is the major serpin in the human circulation with a wide spectrum of antiproteinase activity, including neutrophil elastase, proteinase 3, cathepsin G, and plasminogen activator. AAT is an acute phase protein, and plasma concentrations increase by more than 2 mg/mL during inflammation, infections, cancer, liver disease, or pregnancy (). AAT has an important role in the resolution of inflammation, the inhibition of the immune response in vivo and in vitro, and the stimulation of tissue repair and matrix production. AAT has been shown to block neutrophil activation by inhibiting adhesion to fibronectin. In addition, AAT inhibits nasal interleukin-8 (IL-8) release in response to challenge with lipopolysaccharide (LPS), while in vitro AAT has been shown to inhibit the release of IL-8 by neutrophils (CitationNita et al 2005). It has also been suggested that AAT has antibacterial activities as well as anti-inflammatory effects. AAT has also been shown to inhibit the LPS-induced release of TNFα, IL-1β, and monocyte chemotractant protein (MCP-1), while conversely the release of IL-10, an anti-inflammatory cytokine, is increased after 18 hours. However, after only 2 hours, the release of both TNFa and IL-8 is significantly increased in the presence of AAT. Therefore, it appears that AAT enhances the early response to LPS but acts as a suppressor in the long term. AAT might be extremely important, not only for tissue protection against protease damage, but also for the regulation of cellular responses to inflammatory stimuli.
Figure 3 Structure of AAT: 3 β-sheets, 8 α-helixes. Reprinted from CitationJanciauskiene S. 2001. Conformational properties of serine proteinase inhibitors (serpins) confer multiple pathophysiological roles. Biochim Biophys Acta, 1535:221–35. Copyright © 2001, with permission from Elsevier.
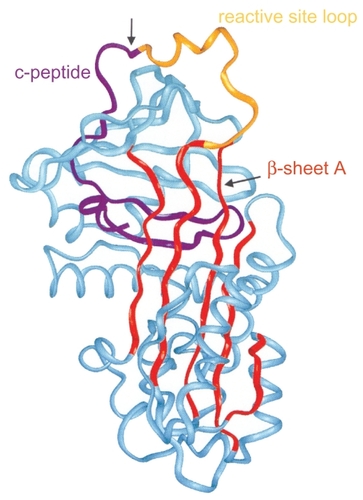
The major biological role of AAT is to inhibit neutrophil elastase, an enzyme that degrades elastin as well as basement membrane and other matrix components (CitationATSERS 2003). It remains to be determined how lung development and function is affected by AAT deficiency. Elastin drives septation and the formation of alveoli and at birth in normal-term infants; there are 150 million alveoli, which increases to an average of 400 million in adults. In many ways, the development of the lung in the antitrypsin deficiency state is normal, demonstrating that at 6 years of age, FEV1 is close to 100% predicted in both PiZZ and PiSZ phenotypes. Further investigation at age 18 years also revealed that phenotype made no contribution to differences in FEV1, residual volume (RV), or total lung capacity (TLC); however, a significant difference in carbon monoxide diffusion was revealed (p = 0.01). In order to explain the apparently normal lung function in PiZZ individuals, factors that may compensate for the protease–protease inhibitor imbalance are sought. Increases in alpha-2 macroglobulin (A2M) levels at ages 8 and 18 years and secretory leukocyte protease inhibitor at age 26 years accompanied by decreases in elastase–AAT complex and neutrophil activity at ages 18 and 26 years were found compared with normal controls. It is hypothesized that a corresponding increase in elastase–A2M complex is likely and that this would downregulate neutrophil activity, providing a compensatory mechanism (CitationSveger et al 1998).
Conformational changes in the Z-type AAT molecule can lead to spontaneous polymerization, with long chains of AAT molecules formed. These chains can be seen under electron microscopy in the endoplasmic reticulum (ER) of hepatocytes in AAT-deficient individuals with liver disease. The Z mutation destabilizes a key beta-sheet in the AAT molecule, allowing the reactive loop of a second AAT molecule to insert itself and form a dimer. This highly precise, specific linkage results in reactive-loop beta-sheet polymers being formed of AAT (). The mechanisms of emphysema in PiZZ individuals consist of 2 main factors: the activity of Z-type AAT is reduced in comparison with M-type AAT, and reduced levels of AAT reaching the lungs allow unopposed neutrophil elastase activity, thereby increasing proteolytic destruction. It is also important to note that the inflammatory cell profile is different in PiZZ emphysema compared with emphysema in PiMM individuals, that the AAT–elastase complex is chemotactic to neutrophils, and that both the AAT–elastase complex and AAT polymers share epitopes.
Figure 4 Loop-sheet polymer of AAT molecules. Reproduced from CitationLomas DA, Mahadeva R. 2002. α1-Antitrypsin polymerization and the serpinopathies: pathobiology and prospects for therapy. J Clin Invest, 110:1585–90. Copyright © 2002, with permission from American Society for Clinical Investigation.
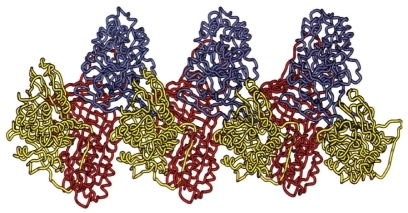
Polymerization occurs in the lungs of PiZZ individuals and contributes to inflammation. It was observed that polymers are co-localized with neutrophils in alveolar walls; further investigation revealed that the presence of AAT polymers acts as a chemoattractant to neutrophils. Conformational transition of AAT from monomer to polymer converts an anti-inflammatory molecule into a pro-inflammatory molecule, providing an additional mechanism for the development of lung disease.
Neutrophil elastase causes direct damage to epithelial cells as well as hyperplasia of mucus-producing cells, enhancing mucus secretion. It also has effects on the host defense by impairing ciliary clearance, cleaving complement and immunoglobulins, and impairing the killing of microorganisms. The problem in AAT deficiency is not too little antitrypsin but too much in the wrong place. The accumulation of misfolded Z-type AAT in the endoplasmic reticulum (ER) causes ER stress. Cells respond to ER stress by inducing the expression of novel genes whose products are designed to restore proper ER function but can also be pro-inflammatory. The ER stress pathways, including the unfolded protein response, ER overload, and apoptosis, appear to increase IL-8 production and neutrophil chemoattraction. AAT-deficiency-related lung disease clearly is not due purely to a lack of antiprotease on the respiratory epithelial surface.
It can be seen that in sputum, the activity of elastase is greater in PiZZ patients, as would be expected. However, similar experiments also reveal higher levels of tumor necrosis factor-alpha (TNFα) and leukotriene B4 (LTB4) in AAT-deficient patients, while there is no significant increase in IL-1β or IL-8 levels compared with physiologically matched controls. Sputum samples from PiZZ individuals also show greater chemotactic activity compared with PiMM samples. Investigations have shown that LTB4 provides a significant proportion of the chemotactic activity associated with AAT deficiency. This suggests that elastase may be driving macrophages to produce LTB4 as a mechanism for neutrophil recruitment.
Antimicrobial peptides (AMPs) are produced by epithelial cells, neutrophils, and macrophages and, in addition to exerting an in vitro antimicrobial effect, they have also been shown to stimulate the epithelium, angiogenesis, and both the innate and adaptive immune systems. It has been shown that levels of the AMPs, also known as α-defensins, are higher in those with AAT-deficient lung disease (CitationSpencer et al 2004). Exact functions of AMPs are not yet clear, but they could be acting as inflammatory mediators and further research was needed.
There is considerable interest in discovering an easily measurable biomarker for pulmonary emphysema in humans, and work on biomarkers of elastin degradation in body fluids is undergoing a revival. The problem with elastin peptides is that they are hard to evaluate and characterize and thus special attention has been focused on desmosines. This is an unusual, tetrafunctional, pyridinium ring-containing amino acid involved in elastin cross-linking that is unique to mature, cross-linked elastin. A number of studies show a relationship between excretion of desmosines in the urine and COPD (CitationStone et al 1995; CitationGottlieb et al 1996). However, plasma is thought to be a better fluid for the determination of desmosine concentration, and good correlation between urine and plasma concentrations has been demonstrated (CitationAnnovazzi et al 2004). It is reported that in 12 patients with emphysema associated with PiZZ AAT deficiency assessed over 7 weeks, there was a good correlation between lung diffusion capacity (% predicted KCO) and plasma desmosine (p < 0.01) and urine desmosine (p < 0.05) levels (CitationStolk et al 2005). Further investigation of the usefulness of desmosine as a biomarker for emphysema is ongoing.
Imaging
COPD has been defined as airflow obstruction that is not fully reversible (CitationPauwels et al 2001). FEV1 is a nonspecific measure that fails to identify the relative contribution of the component parts of the COPD. This has led to the development of computed tomography (CT) lung densitometry because it is an extremely good tool for identifying changes in the lung, which can provide objective quantification of parenchmyal changes. Emphysema involves a reduction in lung tissue density, and this is often shown by an increase in the voxel index, representing a shift in the voxel distribution histogram to more low-density voxels. CT densitometry has been validated in a number of studies and, importantly, the relation of progression in CT to deterioration in health status has been assessed in a longitudinal study (CitationStolk et al 2003). It has, however, been noted that although FEV1 and KCO correlate well in a large patient cohort, some patients are discordant for these measures. It has therefore been hypothesized that this discordance represents differences in the distribution of emphysema within the lung. A recent study showed that the emphysema distribution in PiZZ patients is more heterogeneous than conventional basal predominance descriptions indicate (CitationParr et al 2004a). It was also found that on average, those with basal emphysema have lower FEV1 scores, while those with apical predominant disease have lower KCO scores. Using a single physiologic index to assess emphysema may introduce systematic bias depending on where the emphysema is located.
Investigation of the validation of CT data over time revealed a number of potential factors influencing these types of measurements, including variability between scanners and the importance of calibration (CitationParr et al 2004). Standardized protocols are needed to improve agreement between different centers. Further validation of CT as a measure of disease progression in 34 PiZZ individuals characterized annually over 3 years confirmed a correlation between annual change in CT densitometry and FEV1. Recent advances in magnetic resonance imaging (MRI) of the lung using hyperpolarized helium-3 (3He) have been reported. Conventional proton MRI in the lung could be problematic due to the large number of air–tissue interfaces giving rise to susceptibility gradients and the lower proton density of the lungs leading to a weak MRI signal. However, hyperpolarized 3He can also produce a magnetic resonance signal, and this nontoxic gas can be inhaled for lung imaging. Images taken from lungs of AAT-deficient, “usual” emphysema, and healthy control subjects showed that distribution of the gas was not as homogeneous in the lungs of the AAT-deficient patient as in the lungs of a healthy control. The lower lobe predominance of emphysema in the AAT-deficient patient was also noticeable compared with a smoke-induced COPD patient where defects appeared more evenly spread throughout the lung. Good correlation between percent ventilation defect as measured from 3He MRI and lung function tests has been shown. In such studies, the apparent diffusion coefficient (ADC) varies according to the size of the alveolus and expresses the mean area–velocity of the nuclei in a voxel. Calculating the ADC can provide a map of diffusion gradients throughout the lung. In a longitudinal study comparing ADC maps of the lung obtained via 3He MRI in 8 PiZZ and 8 PiMM patients with emphysema and 8 healthy volunteers, it was found that there was a significant difference in mean ADC between PiZZ patients compared with PiMM patients. However, there was only a minimal, nonsignificant difference in carbon monoxide diffusing capacity (DLCO) between the 2 groups. This suggests that measurement of ADC with hyperpolarized 3He diffusion MRI is a sensitive method for the assessment of emphysema and may make the detection and monitoring of early emphysema and small changes possible.
Treatment
A meta-analysis of clinical trials investigating the efficacy of augmentation therapy in AAT deficiency pulmonary disease was presented during the meeting (). Five studies were included in the meta-analysis (CitationSeersholm et al 1997; CitationAATDRSG 1998; CitationDirksen et al 1999; CitationWencker et al 2001; CitationChapman et al 2005) with UK registry data used for a comparison with untreated patients. The results suggested a reduced rate of FEV1 decline in AAT-deficient individuals, with baseline FEV1 being an important predictor of benefit. There were limitations in this analysis, including the lack of randomized controlled trials, variable dosing schedules of the augmentation therapy, and an increasing belief that FEV1 is not necessarily the best endpoint to measure.
Figure 5 Rate of FEV1 decline: augmentation vs control (CitationSeersholm et al 1997; CitationAATDRSG 1998; CitationDirksen et al 1999; CitationWencker et al 2001; CitationChapman et al 2005).
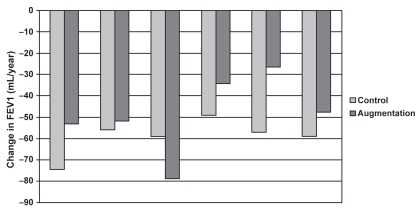
It is known that an AAT infusion of 60 mg/kg every 7 days can maintain serum total AAT concentrations higher than 0.5 g/L during the interval between doses (CitationATSERS 2003) The inconvenience of life-long weekly infusions of AAT has led to the suggestion that extended dosage regimens (every 14, 21, or 28 days) may be preferable. Population pharmacokinetic studies could assist in determining the biochemical efficacy of prolonged regimens. Using a Monte-Carlo simulation, a number of doses and frequencies were tested to see if total serum concentrations of AAT higher than 0.5 g/L could be maintained throughout the dosing interval. Preliminary results show that AAT dosage regimens every 2 or 3 weeks may be adequate, but to extend the interval between doses to 21 days requires a large increase in the dose of AAT administered in order to maintain total serum AAT concentrations above the recommended target.
AAT augmentation therapy appears to be associated with a marked reduction in the frequency and severity of lung infections (CitationLieberman 2000) as well as a reduction in the loss of lung tissue as measured by CT scanning (CitationDirksen et al 1999) compared with untreated patients. As a result of these observations, the EXACTLIE (Exacerbations and CT Scanning in Laurell’s Syndrome as Investigational Endpoints) trial has been designed. This is a multicenter, randomized, placebo-controlled, double-blind, exploratory trial with parallel groups that will investigate the progression of disease in 80 patients with AAT deficiency treated with AAT or placebo for 2 years. The efficacy of weekly intravenous AAT (Prolastin®; Talecris, Clayton, NC, USA) infusions in AAT-deficient patients with regard to lung density and exacerbations compared with placebo will also be assessed. Since October 2003, 76 patients have been enrolled in the study. To date, few serious adverse events have occurred and none were thought to be related to the study drug.
Gene therapy is still considered to be of potential use. Currently the approach of an intramuscular injection of the AAT gene using a recombinant adeno-associated virus (rAAV) vector, AAV-2, is being studied. The long-term, high-level expression of AAT from muscle in mice has been previously demonstrated (CitationSong et al 1998). A phase I, single- site, open-label, single-dose safety study is currently underway involving 4 cohorts of 3 subjects with dose escalation between cohorts. Preliminary findings indicate that the treatment is safe with no serious vector-related adverse effects and no evidence of cell-mediated immunity; however, significant changes in serum levels of muscle-derived AAT have yet to be detected. Future directions for research may include the use of different vectors, such as AAV-1, which may increase the level of AAT expression. Another AAV vector, AAV-8, shows promise in treating liver disease because it shows great potential in transducing hepatocytes, not only for secreting AAT, but also for transducing a greater number of hepatocytes compared with other AAV vectors.
Conclusion
Currently available intravenous replacement therapy is aiming only at the prevention of lung disease progression. It has no effect on liver disease and does not induce repair of lung injury. While few new treatments for AAT deficiency are on the near horizon (such as inhaled AAT or synthetic oral elastase inhibitors), understanding the biochemistry of PiZZ AAT and its downstream effects may lead to new concepts of treatment. For example, trying to prevent the polymerization of PiZZ AAT by inserting small compounds into a hydrophobic cavity bounded by the D and E helices in AAT would allow release of functional active PiZZ AAT to the circulation and prevent the development or progression of cirrhosis. Likewise, at the level of cellular systems, small compounds that release type ZZ AAT from hepatocytes are currently being explored as chaperone-based therapy. Modifier genes that clarify the difference in disease phenotype of sib pairs with equal smoking history and equal genotype could provide insight into new therapeutic concepts and the clinical epidemiology of individuals who will or will not develop lung and/or liver disease. Finally, the aim to switch on repair mechanisms in the lung is currently being studied in a proof-of-concept study in the PiZZ population with emphysema.
References
- [AATDRSG] Alpha-1-Antitrypsin Deficiency Registry Study Group1998Survival and FEV1 decline in individuals with severe deficiency of α1-antitrypsinAm J Respir Crit Care Med15849599655706
- [ATSERS] American Thoracic Society, European Respiratory Society2003American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiencyAm J Respir Crit Care Med16881890014522813
- AnnovazziLViglioSGheduzziD2004High levels of desmosines in urine and plasma of patients with pseudoxanthoma elasticumEur J Clin Invest341566414764080
- BouttenAVenembrePSetaN1998Oncostatin M is a potent stimulator of alpha1-antitrypsin secretion in lung epithelial cells: modulation by transforming growth factor-beta and interferon-gammaAm J Respir Cell Mol Biol18511209533938
- ChapmanKRBradiACPatersonD2005Slower lung function decline during augmentation therapy in patients with alpha1-antitrypsin deficiency (A1ATD): results from the Canadian Air RegistryAm J Respir Crit Care Med1703824
- ChappellSGuetta-BaranesTBatowskiK2004Haplotypes of the alpha-1 antitrypsin gene in healthy controls and Z deficiency patientsHum Mutat24535615532029
- De La RozaCRodriguez-FriasFLaraB2005Results of a case-detection program for α1-antitrypsin deficiency in COPD patientsEur Respir J266162216204591
- de SerresFJ2002–Worldwide racial and ethnic distribution of α1-antitrypsin deficiency: summary of an analysis of published genetic epidemiologic surveysChest12218182912426287
- DirksenADijkmanJHMadsenF1999A randomized clinical trial of α1-antitrypsin augmentation therapyAm J Respir Crit Care Med16014687210556107
- FerrarottiIBaccheschiJZorzettoM2005Prevalence and phenotype of subjects carrying rare variants in the Italian registry for alpha1-antitrypsin deficiencyJ Med Genet42282715744045
- FrancavillaRCastellanetaSPHadẑićN2000Prognosis of alpha-1-antitrypsin deficiency-related liver disease in the era of paediatric liver transplantationJ Hepatol329869210898319
- GolponHAColdrenCDZamoraMR2004Emphysema lung tissue gene expression profilingAm J Respir Cell Mol Biol3159560015284076
- GottliebDJStonePJSparrowD1996Urinary desmosine excretion in smokers with and without rapid decline of lung function: the Normative Aging StudyAm J Respir Crit Care Med154129058912738
- HindsRHadchouelACheesemanP2004Phenotypic expression of liver disease in families with PiZZ Alpha-1-antitrypsin deficiencyJ Pediatr Gastroenterol Nutr39Suppl 1S139
- HosokawaNTremblayLOYouZ2003Enhancement of endoplasmic reticulum (ER) degradation of misfolded null Hong Kong α1-antitrypsin by human ER mannosidase IJ Biol Chem278262879412736254
- JanciauskieneS2001Conformational properties of serine proteinase inhibitors (serpins) confer multiple pathophysiological rolesBiochim Biophys Acta15352213511278163
- JanciauskieneSErikkssonSCalleaF2004Differential detection of PAS-positive inclusions formed by the Z, Siiyama, and Mmalton variants of alpha1-antitrypsinHepatology4012031015486938
- JoostOWilkJBCupplesA2002Genetic loci influencing lung function: a genomewide scan in the Framingham studyAm J Respir Crit Care Med165795911897646
- KalshekerNMorleySMorganK2002Gene regulation of the serine proteinase inhibitors α1-antitrypsin and α1-antichymotrypsinBiochem Soc Trans3093812023832
- LawlessMWGreeneCMMulgrewA2004Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z α1-antitrypsin deficiencyJ Immunol1725722615100318
- LaurellCBErikssonS1963The electrophoretic pattern α1-globulin pattern of serum in α1-antitrypsin deficiencyScan J Clin Lab Invest1513240
- LiebermanJ2000Augmentation therapy reduces frequency of lung infections in antitrypsin deficiency: a new hypothesis with supporting dataChest1181480511083705
- LodishHBaltimoreDBerkALodishHBaltimoreDBerkA1995Transcriptional control of gene expressionMolecular cell biology3rd edNew YorkWH Freeman and Company/Worth Publishers
- LomasDAMahadevaR2002α1-Antitrypsin polymerization and the serpinopathies: pathobiology and prospects for therapyJ Clin Invest11015859012464660
- LuisettiMMiravitllesMStockleyRA2002α1-antitrypsin deficiency: a report from the 2nd meeting of the Alpha One International Registry, Rapallo (Genoa, Italy), 2001Eur Respir J201050612412702
- MaloneMMieli-VerganiGMowatAP1989The fetal liver in PiZZ alpha-1-antitrypsin deficiency: a report of five casesPediatr Pathol9623312481300
- NavickisRJWilkesMM2004Update on research, diagnosis and management of alpha1-antitrypsin deficiency. Highlights from the 3rd international Alpha One International Registry Meeting, June 2003, Barcelona, SpainInt J COPD127992
- NitaIHollanderCWestinU2005Prolastin, a pharmaceutical preparation of purified human α1-antitrypsin, blocks endotoxin-mediated cytokine releaseRespir Res61215683545
- PalmerLJCeledónJCChapmanHA2003Genomewide linkage analysis of bronchodilator responsiveness and post-bronchodilator spirometric phenotypes in chronic obstructive pulmonary diseaseHum Mol Genet12119921012719384
- PalmerEAKruseKBFewellSW2003Differential requirements of novel A1PiZ degradation deficient (ADD) genes in ER-associated protein degradationJ Cell Sci11623617312711700
- ParfreyHMahadevaRRavenhillNA2003Targeting a surface cavity of α1-antitrypsin to prevent conformational diseaseJ Biol Chem27833060612807889
- ParrDGStoelBCStolkJ2004aPattern of emphysema distribution in α1-antitrypsin deficiency influences lung function impairmentAm J Respir Crit Care Med1701172815306534
- ParrDGStoelBCStolkJ2004bInfluence of calibration on densitometric studies of emphysema using computed tomographyAm J Respir Crit Care Med1708839015271692
- PauwelsRABuistASCalverleyPM2001Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summaryAm J Respir Crit Care Med16312567611316667
- PurkayasthaPKlemkeJWLavenderS2005Antitrypsin polymerization: a fluorescence correlation spectroscopic studyBiochemistry442642915709777
- SeersholmNWenckerMBanikN1997Does α1-antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary α1-antitrypsin deficiency?Eur Respir J10226039387950
- SilvermanEKPalmerLJMosleyJD2002Genomewide linkage analysis of quantitative spirometric phenotypes in severe early-onset chronic obstructive pulmonary diseaseAm J Hum Genet7012293911914989
- SongSMorganMEllisT1998Sustained secretion of human alpha-1-antitrypsin from murine muscle transduced with adeno-associated virus vectorsProc Natl Acad Sci U S A951438489826709
- SpencerLTPaoneGKreinPM2004Role of human neutrophil peptides in lung inflammation associated with α1-antitrypsin deficiencyAm J Physiol Lung Cell Mol Physiol286L5142014594730
- StockleyR1999Alpha-1-antitrypsin deficiency: First scientific meeting of the alpha-1 international registry [online]Accessed 04 October 2005 URL: http://www.alfa1.org/deteccion_registro_air_meeting_1999.htm
- StolkJNgWHBakkerME2003Correlation between annual change in health status and computer tomography derived lung density in subjects with α1-antitrypsin deficiencyThorax5810273014645966
- StolkJVeldhuisenBAnnovazziL2005Short-term variability of biomarkers of proteinase activity in patients with emphysema associated with type Z alpha-1-antitrypsin deficiencyRespir Res64715927063
- StonePJGottliebDJO’ConnorGT1995Elastin and collagen degradation products in urine of smokers with and without chronic obstructive pulmonary diseaseAm J Respir Crit Care Med15195297697272
- SvegerTThelinTMcNeilTF1999Neonatal alpha1-antitrypsin screening: parents’ views and reactions 20 years after the identification of the deficiency stateActa Paediatr88315810229044
- SvegerTThelinTMcNeilTF1997Young adults with alpha 1-antitrypsin deficiency identified neonatally: their health, knowledge about and adaptation to the high-risk conditionActa Paediatr8637409116423
- SvegerTErikssonS1995The liver in adolescents with alpha 1-antitrypsin deficiencyHepatology2251477635419
- SvegerTOhlssonKPiitulainenE1998Adolescents with alpha1-antitrypsin deficiency have high alpha2-macroglobulin and low neutrophil lipocalin and elastase levels in plasmaPediatr Res44939419853931
- WHO1997α1-antitrypsin deficiency: memorandum from a WHO meetingBull World Health Organ753974159447774
- WenckerMFuhrmannBBanikN2001Longitudinal follow-up of patients with α1-protease inhibitor deficiency before and during therapy with IV α1-protease inhibitorChest1197374411243951
- WewersMDCasolaroMASellersSE1987Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysemaN Engl J Med3161055623494198
- WuYSwuliusMTMoremenKW2003Elucidation of the molecular logic by which misfolded α1-antitrypsin is preferentially selected for degradationProc Natl Acad Sci U S A10082293412815101