Abstract
COPD is a leading cause of morbidity and mortality, characterized by a chronic abnormal inflammatory response to noxious agents. Apoptosis is a physiologic process, critical to cellular homeostasis, in which cell death follows a programmed sequence of events. Apoptosis has been recognized to play an important role in clinical and experimental models of lung diseases. Abnormal apoptotic events in smokers’ and in emphysematous lungs have been shown in epithelial and endothelial lung cells, neutrophils, lymphocytes, and myocytes. Many factors associated with COPD, including cigarette smoke, have the potential to cause apoptosis of alveolar epithelial cells, the main sites of vascular endothelial growth factor (VEGF) production. The decreased expression of VEGF, a known survival factor for endothelial cells, and its receptor, results in lung septal endothelial cell death, leading perhaps to the emphysema observed in COPD. In smokers who develop COPD there is an activation of adaptive immunity, with an infiltration of CD4+ and, especially, CD8 + cells. CD8 + cells are cytotoxic to epithelial cells through the release of granzymes and perforin, which can further induce apoptosis of alveolar cells. Moreover, any reduction in neutrophil apoptosis or dysregulation of macrophage uptake of apoptotic neutrophils could lead to chronic inflammation and tissue injury. Increased rates of T-cell apoptosis may lead to a defective immune response to infective organisms, contributing to the high frequency of infections seen in COPD. Increased apoptosis of skeletal muscle could be responsible for the skeletal muscle atrophy, the main cause of unexplained weight loss in patients with COPD. This paper is a review of the current knowledge on the apoptotic pathways involved in COPD pathogenesis and their interaction with other known contributing factors.
Introduction
COPD is a leading cause of chronic morbidity and mortality worldwide, with a substantial economic and social burden (CitationSiafakas et al 1995; CitationCelli et al 2004; CitationPauwels and Rabe 2004). The 2002 WHO World Health Report listed COPD as the fifth leading cause of death in the world and further increases in its prevalence and mortality are expected in the coming decades (CitationWHO 2002; CitationPauwels and Rabe 2004). Recently, the ATS–ERS Task Force has published a document on the pathogenesis, diagnosis, monitoring, and management of COPD (CitationCelli et al 2004). The latest definition of COPD according to this document notes that COPD is “characterised by airflow limitation that is not fully reversible... is usually progressive and is associated with an abnormal inflammatory response of the lungs to noxious particles or gases, primarily caused by cigarette smoking ... it also produces significant systemic consequences.” (CitationCelli et al 2004). Although significant progress has been made on the pathogenesis of COPD, the overall picture remains unclear.
Apoptosis, programmed cell death, is a highly ordered physiologic process by which useless, irreparably damaged or unwanted cells are eliminated throughout life. This process has attracted great attention in recent years, though the lung has been relatively little investigated. Elucidating the regulating mechanisms of apoptosis in lung cells should contribute to the understanding of the pathophysiology of lung disorders and allow identification of specific therapeutic targets (CitationPlataki et al 2005).
This review focuses on the existing literature on apoptosis in COPD, and the potential role of apoptosis on COPD pathogenesis. A synopsis of the relevant literature is presented in .
Table 1 Studies of apoptosis in COPD
Apoptosis
Apoptosis, “normal” or “programmed” cell death, is an evolutionarily conserved, innate process by which cells systematically inactivate, disassemble, and degrade their own structural and functional components. The term “apoptosis” was first introduced by Kerr et al in 1972 (CitationKerr et al 1972). It is a key mechanism allowing multicellular animals to tightly regulate cell growth (CitationBaetu and Hiscott 2002). Necrosis, “accidental” cell death, is a pathological process which occurs when cells are exposed to a serious physical or chemical insult. During necrosis loss of membrane integrity leads to the influx of water and extracellular ions. Intracellular organelles, most notably the mitochondria, and the entire cell swell and rupture and the cytoplasmic contents are released into the extracellular fluid. During apoptosis coordinated morphological and biochemical changes occur within the nucleus, cytoplasm, organelles, and plasma membrane (CitationCohen 1993). Chromatin condenses and aggregates along the nuclear periphery and fragmentation of the nucleus occurs into chromatin containing fragments. Other cytoplasmic organelles remain structurally intact, although pore formation in the mitochondrial membrane is associated with apoptosis. The cell rounds up, dissociates itself from its neighbors, and shrinks, throwing out protuberances that separate into membrane-bound “apoptotic bodies”, containing cytoplasmic organelles and nuclear material. Within tissues apoptotic bodies are rapidly phagocytosed by neighboring cells (CitationCohen 1993).
Apoptosis can be triggered through two major pathways, one implicating death receptor ligation and the other the release of cytochrome c from mitochondria (CitationBaetu and Hiscott 2002) (). The death receptor pathway is initiated by the ligation of members of the tumor necrosis factor (TNF) family, such as TNF-α and Fasligand, to their respective ligands, TNF receptor 1, and Fas (CitationBaetu and Hiscott 2002). These “death-inducing” receptors recruit the death-inducing signaling complex through interaction with adaptor proteins such as Fas associated death domain protein (FADD). In turn, FADD recruits caspase-8. Caspase-8 activation leads to either direct activation of caspase-3, or to cleaving of Bid, a member of the bcl-2 family, which in turn targets mitochondria for cytochrome c release (CitationBaetu and Hiscott 2002). The mitochondrial pathway can be triggered by both external and internal stimuli. Members of the bcl-2 family of proteins, like bcl-2, bax, and bad, gather at the surface of mitochondria, where they compete to regulate cytochrome c release, which leads to caspase-9 activation. Bcl-2 is an antiapoptotic and bad and bax are proapoptotic members of this family (CitationBaetu and Hiscott 2002). Akt is a serine–threonine kinase that phospholylates and thereby inactivates the proapoptotic proteins bad and caspase 9, and elevates the expression of the antiapoptotic protein bcl-2 (CitationDatta et al 1999). Both pathways converge at the level of caspase-3 activation. Caspases are proteolytic enzymes, the executioners of apoptosis. Caspase activation results in DNA laddering, characteristic of apoptotic cells, and to the cleavage of cytoskeletal proteins. p53 is a critical mediator of two different cellular responses following exposure to DNA damage: growth arrest in the G1 phase of the cell cycle or apoptosis in the case of irreparable damage (CitationCanman and Kastan 1997). 4-Hydroxy-2-nonenal (4-HNE) is an end-product of lipid peroxidation, which is involved in provoking the toxic effects of oxidative stress to cells, promoting apoptosis (CitationRabacchi et al 2004). The frizzled-related protein sFRP-1 plays a part in the disruption of the normal Wnt signaling status and may also promote the apoptotic process (CitationJones and Jomary 2002).
Table 2 Markers upregulating (↑) or downregulating (↓) apoptosis in COPD
A number of methods are used for the detection of apoptosis. These assays try to measure one of the following apoptotic parameters: a) DNA fragmentation (eg, TdT-mediated dUTP nick end labeling, TUNEL); b) membrane alterations, such as phosphatidylserine translocation (eg, annexinV-propidium iodide staining); c) activation of caspases (eg, anti-active caspase 3 antibody); and d) detection of proteolytic cleavage products found in apoptotic cells (eg, anti-poly-ADP-ribose polymerase antibody, anti-fractin antibody). Apoptotic cells are also recognized by electron microscopy due to the characteristic morphological changes. Moreover, the expression of a number of genes and proteins involved in the regulation of apoptosis can also be studied by reverse transcription polymerase chain reaction (RT-PCR) or immunohistochemistry.
Apoptosis of epithelial and endothelial cells
Recently many reports have been published highlighting the presence of apoptosis in human COPD lungs and its role in the pathogenesis of emphysema (CitationShapiro 2000b). Pulmonary emphysema, defined as abnormal, permanent enlargement of airspaces distal to terminal bronchioles, accompanied by destruction of their walls, with or without obvious fibrosis, is a major component of COPD (CitationSnider et al 1985). On the other hand, relatively little attention has been paid to the significance of epithelial or endothelial apoptosis in chronic bronchitis.
Animal and in vitro studies
The importance of apoptosis in the pathogenesis of emphysema stems from animal studies. CitationKasahara, Tuder, Taraseviciene-Stewart, et al (2000) suggested that chronic blockade of vascular endothelial growth factor (VEGF) receptors induces alveolar cell apoptosis and emphysema. Indeed, treatment of rats with a VEGF receptor blocker induced alveolar septal cell apoptosis, a pruning of the pulmonary arterial tree and enlargement of the air spaces, indicative of emphysema. Treatment with a caspase inhibitor prevented the septal cell apoptosis and emphysema caused by the VEGF receptor inhibitor. CitationAoshiba et al (2003) described the induction of emphysematous changes, including airspace enlargement, alveolar wall cell apoptosis (mainly epithelial cells), in mice treated with an intratracheal injection of active caspase-3 and Chariot, a protein transfection reagent, or nodularin, a proapoptotic serine–threonine kinase inhibitor. In addition, epithelial apoptosis and enhanced elastolytic activity were noted in bronchoalveolar lavage (BAL). In vitro findings showed the destruction of the elastin layer beneath the type II cells that were undergoing apoptosis. In both of these studies, fibrosis or increased infiltration of inflammatory cells was absent. Oxidative stress has been shown to have a critical role in alveolar septal cell apoptosis and emphysema caused by VEGF receptor blockade (CitationTuder, Zhen, et al 2003). Rats treated with a VEGF receptor blocker showed increased alveolar septal cell apoptosis, airspace enlargement, and expression of markers of oxidative stress, all of which were prevented by a superoxide dismutase mimetic. Apoptosis predominated in areas of oxidative stress, and treatment with a broad-spectrum caspase inhibitor markedly reduced the expression of markers of oxidative stress. In lungs treated with both the VEGF receptor blocker and the superoxide dismutase mimetic, the preservation of lung structure was associated with enhanced phosphorylation of the prosurvival and antiapoptotic Akt. Moreover it has been reported that i.v. injection of alpha-1 antitrypsin prevents VEGF receptor blockade-induced emphysema in rats (CitationChoe et al 2003). Increased alveolar septal cell apoptosis and pulmonary emphysema, with enlarged air spaces and enhanced pulmonary compliance, were also noted in placenta growth factor (PIGF)–transgenic mice, with constitutive overexpression of PIGF (CitationTsao et al 2004). PIGF is a VEGF homolog, which may function by modulating VEGF activity or by triggering its own intracellular signals, involving proliferation, apoptosis, or angiogenesis (CitationCao et al 1996; CitationAutiero et al 2003). In these mice, apoptotic septal cells were type II pneumocytes. At the same time, the messenger RNA of VEGF and platelet–endothelial cell adhesion molecule-1, an endothelial cell marker, were downregulated indicating a reduced number of endothelial cells and their survival factor VEGF. An inflammatory or fibrotic response was absent in these mice. In addition to an absolute increase in proapoptotic mechanisms, CitationHodge, Hodge, Scicchitano, et al (2003) suggested that impaired capacity for phagocytic clearance of apoptotic material may be a factor further contributing to emphysema pathogenesis. When incubating apoptotic airway epithelial cells with alveolar macrophages from BAL from COPD patients and control subjects, a significantly reduced proportion of alveolar macrophages from COPD subjects ingested apoptotic airway epithelial cells compared with controls.
Human studies
CitationSegura-Valdez et al (2000) were the first to show increased apoptosis in the lungs of COPD patients. In their study, endothelial cells from capillaries and arterioles, and occasionally alveolar epithelial, interstitial, and inflammatory cells, exhibited prominent intranuclear staining by in situ end labeling of fragmented DNA. In a report by CitationKasahara et al (2001), there was increased apoptosis of alveolar epithelial and endothelial cells in lung tissue samples obtained from patients with emphysema compared with lung samples from either nonsmokers or smokers without emphysema. Moreover, VEGF, VEGF receptor 2 (VEGF R2) protein, and mRNA expression were significantly reduced in emphysema (CitationKasahara et al 2001). The authors proposed that epithelial and endothelial alveolar septal death due to a decrease of endothelial cell maintenance factors may be part of the pathogenesis of emphysema. It must be noted that in this study the septal structures in the emphysematous areas examined for the presence of apoptotic cells did not show clear signs of acute or chronic inflammation. The same group has reported significantly reduced expression of the VEGF receptor, kinase insert domain-containing receptor (KDR), in emphysematous lungs (CitationKasahara, Tuder, Cool, et al 2000). CitationYokohori et al (2004) also found that the fraction of the total alveolar wall cells undergoing apoptosis and proliferation was significantly higher in patients with emphysema than in asymptomatic smokers and nonsmokers. Patients with emphysema also showed a significant increase in the bax index, suggesting that the overexpression of bax protein may be involved in the apoptosis of the alveolar wall cells in these patients. Most of the apoptotic cells in this study were epithelial type-II cells. A study on tissue from the emphysematous lung areas of human patients by CitationImai et al (2005) showed that the apoptotic index was closely correlated with the severity of disease. They have also identified the expression of the frizzled-related protein sFRP-1 gene, an inducer of apoptosis, in emphysema. In a recent study, CitationImai et al (2005) identified extensive cell death by apoptosis, confirmed with several different techniques, in the lung tissue of patients with emphysema. Apoptosis was inversely correlated with surface area. Bcl-2 expression was not detected in emphysematous lung, whereas bax and bad expression was. Bax staining was localized to alveolar epithelial cells and bad staining to both epithelial and mesenchymal cells (CitationImai et al 2005). CitationRahman et al (2002) investigated the presence of 4-HNE, a highly reactive diffusible product of lipid peroxidation which mediates oxidant-induced cell signaling and apoptosis, in lung tissue from subjects with and without COPD. The 4-HNE-modified protein levels were elevated in airway and alveolar epithelial cells, endothelial cells, and neutrophils in COPD patients, compared with those in subjects without COPD. Moreover, there was a significant inverse correlation between the levels of 4-HNE adducts in alveolar epithelium, airway endothelium, and neutrophils, and FEV1. TNF-α is also considered to play an important role in the induction of apoptosis, and allele 2 of the polymorphism at position −308 in the promoter of the TNF-α gene has been associated with increased TNF-α secretion in vitro (CitationSakao et al 2002). CitationSakao et al (2002) reported that the TNF-α-308*1/2 allele frequency was higher, though not significantly, in COPD patients with more extensive emphysema changes, as assessed by computed tomography (CT). CitationMajo et al (2001) investigated the presence of apoptotic alveolar epithelial cells in the lung, as indirect evidence of T-cell-mediated cytotoxicity, through the cytolytic effect of CD8 + cells by perforin-mediated lysis and apoptosis (CitationChrysofakis et al 2004). It was found that apoptosis of alveolar wall cells in active smokers with emphysema increased together with the amount smoked and the number of CD8 + T-cells after a certain dose of smoking was reached, suggesting that cytotoxicity may play a role in the pathogenesis of emphysema (CitationMajo et al 2001). Recently, in a study by CitationHodge et al (2005) using flow cytometric assays on cell populations obtained from bronchial brushing, both the percentage and absolute number of apoptotic bronchial epithelial cells was significantly higher for patients with COPD than controls. They also noted increased expression of Fas, p53, and activated caspases by brushing-derived bronchial epithelial cells in COPD (CitationHodge et al 2005). Most of the above studies, however, were performed in lung tissue, whereas apoptotic cells lose their attachments with surrounding matrix and might therefore accumulate in airway lumen. Future studies, combining tissue and cytological (induced sputum and BAL) samples could reveal increased number of detached apoptotic cells.
Pathophysiologic implications
The above studies suggest that apoptosis plays a significant role in emphysema development. The lung is unique because of its exposure to the external environment and high oxygen tension (CitationTuder, Petrache, et al 2003). Many factors associated with COPD are potential causes of apoptosis of alveolar epithelial cells (CitationHodge, Hodge, Scicchitano, et al 2003). These include the 5000-plus chemical compounds and free radical molecules present in cigarette smoke, increased production of cytokines, neutrophil activity, and disruptions of epithelial cell–matrix interactions (CitationHodge, Hodge, Scicchitano, et al 2003; CitationTuder, Petrache, et al 2003). It is possible that lung cell apoptosis, excessive proteolysis, and oxidative stress interact, leading to the destruction seen in emphysema (CitationTuder, Petrache, et al 2003) (). Increased apoptosis of the epithelial cells, which are the main sites of VEGF production, may account for decreased VEGF expression. CitationGerber, Dixit, et al (1998) and CitationGerber, McMurtrey, et al (1998) reported that VEGF induces the expression of anti-apoptotic proteins and CitationAlon et al (1995) that VEGF acts as a survival factor for endothelial cells. If the amount of VEGF is sufficiently reduced and the signal transduction via VEGF R2 is impaired, then alveolar endothelial cells could die as a consequence of a failing VEGF-dependent endothelial cell maintenance program. The compromised microcirculation resulting from endothelial cell damage may further promote pneumocyte cell death, perpetuating a vicious circle (CitationTsao et al 2004). However, it is still unclear whether pneumocytes and endothelial cells use this kind of cell communication. Furthermore, the presence of elevated markers of oxidative stress in smokers’ lung is well documented, especially during exacerbations (CitationTsoumakidou et al 2005). Apoptotic death due to growth factor withdrawal is mediated by oxidative stress (CitationTuder, Zhen, et al 2003). The findings of CitationTuder, Zhen, et al (2003), that apoptosis predominated in areas of oxidative stress, and that apoptosis blockade by a broad-spectrum caspase inhibitor markedly reduced the expression of markers of oxidative stress, suggest that there is a bidirectional relationship and a positive feedback loop between lung alveolar cell apoptosis and oxidative stress (CitationTuder, Zhen, et al 2003). CitationAoshiba et al (2003) provided evidence of detection of increased elastolytic activity in apoptotic lung epithelial cells, which may amplify the destruction of alveolar elastin. The subsequent destruction of the basement membrane and loss of cell–extracellular matrix attachments may lead to further apoptosis of the surrounding cells. This dynamic feedback mechanism could play a pivotal role in the regulation of local tissue proteolysis (CitationTuder, Petrache, et al 2003). Finally, the damaging effects of apoptosis could be amplified by reduced clearance of these cells and persistence of apoptotic cell products, as suggested by CitationHodge, Hodge, Scicchitano et al (2003). Proteases derived from the inflammatory reaction present in COPD can hinder apoptotic cell recognition and proper uptake (CitationVandivier et al 2002; CitationTuder, Petrache, et al 2003). Although cigarette smoking has been shown to induce apoptosis of bronchial epithelial cells in vitro, in a study by CitationHodge et al (2005) cessation of smoking did not reduce the increased percentage of apoptotic cells in COPD. The increased apoptosis that persists after smoking cessation suggests that this might be a reflection of ongoing abnormalities remaining after smoking cessation, once COPD has been established.
Figure 1 Lung epithelial cell apoptosis. Increased apoptosis of epithelial cells may account for the decreased vascular endothelial growth factor (VEGF) expression, resulting in alveolar endothelial cells death. The compromised microcirculation resulting from endothelial cell damage may further promote pneumocyte cell death. Increased elastolytic activity in apoptotic lung epithelial cells amplifies the destruction of alveolar elastin. The subsequent destruction of the basement membrane and the loss of cell–extracellular matrix attachments lead to further apoptosis of the surrounding cells.
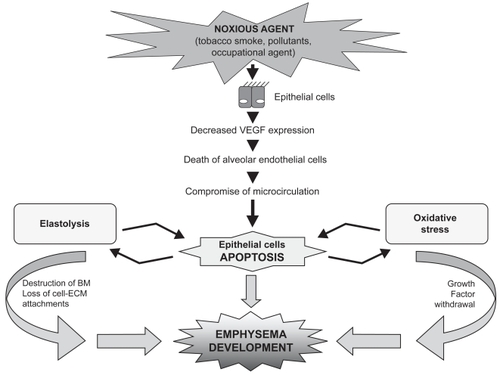
Animal studies have proposed a model of emphysema related to increased apoptosis that excludes inflammation. Animal models, however, do not imitate human diseases and gross lung structure differences in the airways limit extrapolation of findings from animals to humans, eg, smoking mice are not coughing up sputum (CitationShapiro 2000a). Most human studies on epithelial–endothelial cell apoptosis also focus on emphysema. Taking into account that chronic bronchitis and emphysema overlap in COPD and that patients included in human studies had no a1-antitrypsin deficiency, it is difficult to ignore the chronic bronchitis component, although not clearly mentioned in those studies, and the possible implication of inflammation in emphysema. Further studies on COPD including chronic bronchitis are needed to examine the contribution of inflammation and apoptosis in the peripheral airways disease.
Neutrophil apoptosis in COPD
An abnormal inflammatory response, and particularly the persistent presence in tissue of granulocytes and other leukocytes that represent the cellular elements of inflammation, is considered a central process in the pathogenesis of COPD (CitationHaslett 1999). Neutrophil accumulation in the lungs is a characteristic feature of COPD (CitationJeffery 1998; CitationSaetta 1999; CitationCosio Piqueras and Cosio 2001). This could be explained by two mechanisms: 1) increased neutrophil recruitment and chemotaxis, and 2) delayed tissue clearance, due either to delayed apoptosis or defective clearance of these cells. Apoptosis provides an injury-limiting neutrophil clearance mechanism and promotes resolution, rather than persistence, of inflammation (CitationHaslett 1999). The dysregulation of neutrophil apoptosis in the lungs of COPD patients has not been extensively studied.
Human and animal studies
CitationPletz et al (2004) assessed the apoptosis of peripheral blood neutrophils in COPD patients during acute exacerbation and found reduced apoptosis of neutrophils in hospitalized patients with an acute exacerbation of COPD. The apoptosis rate increased progressively during the recovery. The apoptosis rates in healthy volunteers were significantly higher than in COPD subjects at admission, but there was no significant difference in apoptosis rate between controls and COPD patients at discharge (CitationPletz et al 2004). CitationNoguera et al (2004) found that peripheral blood neutrophil apoptosis in stable COPD patients occurred at a rate similar to that found in healthy individuals and smokers with normal lung function. Qualitatively, however, there was increased surface expression of Mac-1 and decreased surface expression of L-selectin in the apoptotic neutrophils of COPD patients (CitationNoguera et al 2004). Using flow cytometric techniques, there was a nonsignificant trend for reduced apoptosis of neutrophils in COPD (CitationHodge et al 2005). In a study by CitationTurlej et al (2001), broncoalveolar lavage fluid (BALF) granulocytes from heaves-affected horses demonstrated a significant delay in apoptosis compared with blood granulocytes from the same horses and blood and BALF granulocytes from healthy horses (CitationTurlej et al 2001). Heaves is considered to be an animal model for COPD (CitationRaulo et al 2001). The rate of apoptosis in blood granulocytes from healthy and heaves-affected horses was similar. The enhanced survival of BALF granulocytes from sick horses was suppressed in the presence of anti-GM-CSF receptor antibodies.
Pathophysiologic implications
Apoptosis is an important mechanism controlling functional longevity of neutrophils at inflamed sites (CitationHaslett 1999). During apoptosis the neutrophil surface membrane remains intact and neutrophils lose the ability to degranulate on deliberate external stimulation. Macrophages can clear large number of apoptotic neutrophils without leakage of potential injurious neutrophil contents into the surrounding milieu (CitationHaslett 1999). The uptake of apoptotic neutrophils may not only suppress the release of proinflammatory agents, such as interleukin (IL)-1β, GM-CSF, IL-8, and TNF, but increase macrophage release of agents, such as transforming growth factor β (TGF-β), and prostaglandin E2, that have suppressive influences on the inflammatory response (CitationFadok et al 1998; CitationHaslett 1999). It has been demonstrated in vitro that nicotine prolongs neutrophil survival in a dose-dependent manner by suppressing apoptosis (CitationAoshiba et al 1996). Several inflammatory agents known to be elevated in COPD, such as lipopolysaccharide (LPS) and GM-CSF, or hypoxic conditions inhibit neutrophil apoptosis. Thus, any reduction in neutrophil apoptosis or dysregulation of macrophage uptake of apoptotic neutrophils could lead to chronic inflammation and tissue injury, such as that observed in COPD. The observation by CitationNoguera et al (2004) of higher surface expression of Mac-1 and lower expression of L-selectin in apoptotic neutrophils of COPD patients compared with controls is compatible with a higher activation status during the apoptosis process. This suggests that neutrophils may still contribute to tissue damage in COPD patients even during apoptosis (CitationNoguera et al 2004). Moreover, increased surface expression of Mac-1 may alter the ability of macrophage recognition and removal of apoptotic neutrophils (CitationNoguera et al 2004).
A significant limitation of the studies by CitationPletz et al (2004) and CitationNoguera et al (2004) is that they studied neutrophil apoptosis in vitro and in circulating, not lung-derived, cells. Peripheral blood neutrophils are expected to exhibit different apoptotic behavior than the cells exposed to the complex inflammatory processes of the COPD lung, and in vivo neutrophils are under the dynamic influence of several pro-and anti-apoptotic factors simultaneously. The above-mentioned studies do not provide enough evidence to rule out delayed neutrophil apoptosis as a cause of neutrophil accumulation and chronic inflammation in COPD lungs.
Lymphocyte apoptosis in COPD
Studies quantifying the inflammation of the airways in smokers and defining their phenotype found that the predominant inflammatory cell present was the CD8 + T-cell and that their number correlated inversely with the degree of airflow limitation (CitationSaetta etal 1993; CitationJeffery 1998; CitationHodge, Hodge, Reynolds, et al 2003; CitationHogg et al 2004; CitationChrysofakis et al 2004; CitationTsoumakidou et al 2004). These findings suggest an important role for activated T-cells in the pathogenesis of airway abnormalities in COPD. A possible mechanism in order to prevent permanent damage and remodeling in COPD could be removal of excess inflammatory cells, including T-cells, by apoptosis, as has been suggested in other disease models (CitationPonomarev and Dittel 2005).
Human studies
CitationHodge, Hodge, Reynolds, et al (2003) assessed apoptosis of T lymphocytes isolated from peripheral blood in COPD patients, and found significantly increased apoptosis of stimulated T-cells, CD4, and CD8, compared with controls. There was no significant difference in levels of apoptosis between CD4 and CD8 T-cell subsets in COPD patients. An upregulation of Fas, TNF-α, TNF receptor 1, and TGF receptor (TGFR) was also observed. Monocyte production of TGF-β and release of active TGF-β in the peripheral blood were increased. There was a decrease in anti-apoptotic bcl2 and IL-7R expression in the peripheral blood T-cells in COPD (CitationHodge et al 2005). The authors concluded that the increased T-cell death may be associated with both an increase in pro-apoptotic and a decrease in anti-apoptotic mediators in the peripheral blood in COPD. The same group investigated apoptosis of lung-derived T-cells (CitationHodge et al 2005). They found an increased percentage and absolute number of apoptotic BAL-derived T-cells in COPD. In addition, there was no significant change in the percentage of T-cells expressing bcl2 or bax, although p53 expression and activated caspases by BAL-derived CD3 + T-cells were significantly increased in COPD (CitationHodge et al 2005).
Pathophysiologic implications
Several observations can be made based on the findings by CitationHodge, Hodge, Reynolds, et al (2003). The increased numbers of T-cells in COPD lungs may result from local proliferation of T-cells in the lung or enhanced trafficking from the bloodstream. This trafficking also occurs from the airways to the bloodstream. Therefore, the authors think that it is possible that increased apoptosis of T-cells in the peripheral blood in COPD may result either from local apoptotic stimuli in the airways or from apoptotic stimuli in the peripheral blood (CitationHodge, Hodge, Reynolds, et al 2003; CitationHodge et al 2005). Increased numbers of apoptotic T-cells in the lung, if not effectively cleared, may lead to secondary necrosis, which has a pro-inflammatory effect. Furthermore, the increased secretion of TGF-β by monocytes may be attributed to ingestion of increased numbers of apoptotic cells (CitationFadok et al 1998). Increased rates of T-cell apoptosis may lead to a defective immune response to infective organisms, contributing to the high frequency of infections seen in COPD (CitationHodge et al 2005).
The decrease in the CD4/CD8 ratio seen in COPD cannot be explained by a preferential increase in the apoptosis of CD4 cells. Recent studies in mice showed that cigarette smoke exposure specifically prevented the expansion and maximal activation of CD4 T-cells and reduced the number of both activated CD4 and CD8 T-cells. Consequently, smoke exposure decreased the activated CD4:CD8 T-cell ratio from 3 to 1.5 (CitationRobbins et al 2004). The mutagenic effect of cigarette smoke on “susceptible” smokers could predispose dendritic cells to production of cytotoxic CD8 + cells, which in turn induce epithelial cell death and apoptosis by perforin (CitationChrysofakis et al 2004; CitationTzanakis et al 2004) ().
Figure 2 Oxidative stress of cigarette smoke in “susceptible” smokers activates adaptive immunity, predisposing dendritic cells to clonal expansion of CD4 + and especially of cytotoxic CD8 + cells, inducing epithelial cell death by perforin.
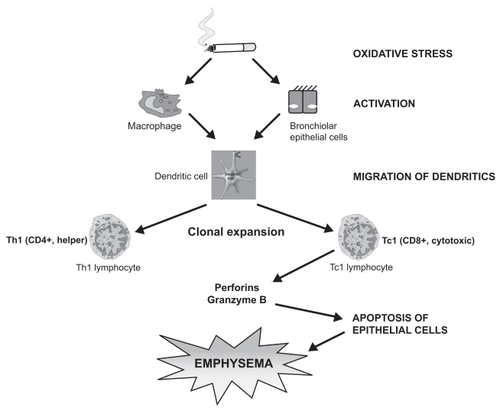
In conclusion, the observation of increased peripheral blood and BAL T lymphocyte apoptosis is not sufficient to establish a causal relationship with the pathogenesis of COPD. Further studies investigating the apoptotic pathways in lymphocyte subsets in both airways (induced sputum) and peripheral blood in COPD are needed in order to draw safe conclusions.
Skeletal muscle wastage and apoptosis in COPD
COPD is considered to be a complex lung disease with systemic consequences, involving several organs (CitationOudijk et al 2003). Unexplained weight loss and peripheral muscle weakness is a major problem for patients with COPD, seriously affecting their quality of life. Whereas apoptosis in mononucleated cells leads to cell death, in multinucleated cells, like the myocyte, it causes cell atrophy (CitationAllen et al 1997). Increased apoptosis of skeletal muscle could be responsible for the skeletal muscle atrophy, the main cause of unexplained weight loss in patients with COPD. CitationGosker et al (2003) reported absence of apoptosis of muscle fibers, at least not at a higher rate in stable COPD patients compared with controls (CitationGosker et al 2003). On the other hand, CitationAgusti et al (2002) found that skeletal muscle apoptosis is increased in patients with COPD and low body mass index (BMI) compared with patients with COPD and normal BMI, healthy volunteers, and sedentary subjects undergoing orthopedic surgery. They also found that in patients with COPD, BMI is inversely related to skeletal muscle apoptosis and that this skeletal muscle apoptosis is associated with a lower exercise tolerance despite a similar degree of lung function impairment. A simple explanation for the different findings of these two studies is that COPD patients included in the study by Gosker et al were in a weight stable condition with a similar BMI to the control group. Increased skeletal muscle apoptosis could not be attributed to aging, inactivity, lung function impairment, smoking history, and oral steroids (CitationAgusti et al 2002). Underweight patients with COPD were younger than those with normal BMI, they had similar lung function impairment (including arterial hypoxemia) and smoking history, and none had received oral steroids. Oxidative stress and systemic inflammation are both prominent features in COPD and have the potential to induce apoptosis (CitationBoots et al 2003; CitationOudijk et al 2003). Moreover, mitochondria play a key role in apoptosis, and mitochondrial abnormalities have been reported in skeletal muscle in COPD. An increase in the activity of cytochrome oxidase (CitationSauleda et al 1998) and the presence of significant bioenergetic abnormalities (CitationATS-ERS 1999) have been demonstrated in skeletal muscle in COPD. A central cytokine in the loss of muscle mass could also be TNF-α by stimulating apoptosis via interaction with the TNF-α receptor present on the muscle cells (CitationOudijk et al 2003). The role of apoptosis in muscle wasting in COPD is an attractive concept that needs further investigation to confirm the presence of apoptosis in these cells and elucidate possible pathogenetic pathways.
Conclusion
Despite the now widely recognized harmful effects of tobacco, COPD remains a leading cause of morbidity and mortality worldwide. Apoptosis is a highly regulated process playing a protective role in physiologic conditions. The mechanisms controlling apoptosis act differently on different cell populations and different tissue settings. Dysregulation of apoptosis has been implicated in many lung diseases.
The reviewed studies indicate that apoptosis might play a significant role in COPD pathogenesis, together with oxidative stress, protease–antiprotease imbalance, and genetic susceptibility. COPD pathology deteriorates even after smoking cessation. This observation may be related to increased apoptosis in the airways of COPD patients even after smoking cessation. These studies provide evidence of activation of different apoptotic mechanisms in COPD compared with controls, which could also represent a protective reaction of the lung to deleterious stimuli. The link between apoptosis and COPD pathogenesis remains to be proven and extensive research, including also the chronic bronchitis component of COPD, is still needed in order to verify the above issues. Elucidating the pathogenetic mechanisms leading to this condition will help to develop new therapeutic strategies against this highly prevalent and often fatal disease.
References
- AgustiAGNSauledaJMirallesC2002Skeletal muscle apoptosis and weigth loss in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med166485912186825
- AllenDLLindermanJKRoyRR1997Apoptosis: a mechanism contributing to remodeling of skeletal muscle in response to hindlimb unweightingAm J Physiol273C579879277355
- AlonTHemoIItinA1995Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurityNature Med1102487489357
- [ATS-ERS] American Thoracic Society and European Respiratory Society1999Skeletal muscle dysfunction in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med159S14010194189
- AoshibaKNagaiAYasuiS1996Nicotine prolongs neutrophil survival by suppressing apoptosisJ Lab Clin Med127186948636647
- AoshibaKYokohoriNNagaiA2003Alveolar wall apoptosis causes lung destruction and emphysematous changesAm J Respir Cell Mol Biol285556212707011
- AutieroMWaltenbergerJCommuniD2003Role of PIGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1Nat Med99364312796773
- BaetuTMHiscottJ2002On the TRAIL to apoptosisCyt Growth Factor Rev13199207
- BootsAWHaenenGRMMBastA2003Oxidant metabolism in chronic obstructive pulmonary diseaseEur Respir J22S1427
- CanmanCEKastanMB1997Role of p53 in apoptosisAdv Pharmacol41429609204155
- CaoYLindenPShimaD1996In vivo angiogenic activity and hypoxia induction of heterodimers of placenta growth factor/vascular endothelial factorJ Clin Invest982507118958213
- CelliBRMacNeeW committee members2004Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paperEur Respir J239324615219010
- ChoeKHTaraseviciene-StewartLScerbaviciusR2003Alpha-1 antitryspin prevents VEGF receptor blockade-induced emphysema in adult rats [abstract]Amer J Respir Crit Care Med167A71
- ChrysofakisGTzanakisNKyriakoyD2004Perforin expression and cytotoxic activity of sputum CD8+ lymphocytes in patients with COPDChest12571614718423
- CohenJJ1993ApoptosisImmunol Today14126308466628
- Cosio PiquerasMGCosioMG2001Diseases of the airways in chronic obstructive pulmonary diseaseEur Respir J34SupplS419
- DattaSRBrunetAGreenbergME1999Cellular survival: A play in three AktsGenes Dev1329052710579998
- FadokVABrattonDLKonowaiA1998Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGFβ, PGE2 and PAFJ Clin Invest90151322
- GerberHPDixitVFerraraN1998Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cellsJ Biol Chem2731331369582377
- GerberHPMcMurtreyAKowalskiJ1998Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3-kinase/Akt signal transduction pathway: requirement for Flk-1/KDR activationJ Biol Chem27330336439804796
- GoskerHRKubatBSchaartG2003Myopathological features in skeletal muscle of patients with chronic obstructive pulmonary diseaseEur Respir J22280512952261
- HaslettC1999Granulocyte apoptosis and its role in the resolution and control of lung inflammationAm J Respir Crit Cre Med160S511
- HodgeSHodgeGHolmesM2005Apoptosis in COPDCurrent Respiratory Medicine Reviews13341
- HodgeSHodgeGScicchitanoR2003Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cellsImmunol Cell Biol812899612848850
- HodgeSJHodgeGLReynoldsPN2003Increased production of TGF-β and apoptosis of T lymphocytes isolated from peripheral blood in COPDAm J Physiol Lung Cell Mol Physiol285L492912851215
- HoggJCChuFUtokaparchS2004The nature of small-airway obstruction in chronic obstructive pulmonary diseaseN Engl J Med35026455315215480
- ImaiKD’ArmientoJ2002Differential gene expression of sFRP-1 and apoptosis in pulmonary emphysemaChest1217S11893652
- ImaiKMercerBASchulmanLL2005Correlation of lung surface area to apoptosis and proliferation in human emphysemaEur Respir J25250815684288
- JefferyPK1998Structural and inflammatory changes in COPD: a comparison with asthmaThorax53129369624299
- JonesSEJomaryC2002Secreted frizzled-related proteins: searching for relationships and patternsBioessays248112012210517
- KasaharaYTuderRMCoolCD2000Expression of 15-lipoxygenase and evidence for apoptosis in the lungs from patients with COPDChest117S260
- KasaharaYTuderRMCoolCD2001Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysemaAm J Respir Crit Care Med1637374411254533
- KasaharaYTuderRMTaraseviciene-StewartL2000Inhibition of VEGF receptors causes lung cell apoptosis and emphysemaJ Clin Invest1061311911104784
- KerrJFWyllieAHCurrieAR1972Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kineticsBr J Cancer26239574561027
- MajoJGhezzoHCosioMG2001Lymphocyte population and apoptosis in the lungs of smokers and their relation to emphysemaEur Respir J179465311488331
- NogueraASalaEPonsAR2004Expression of adhesion molecules during apoptosis of circulating neutrophils in COPDChest12518374215136398
- OudijkEJDLammersJWJKoendermanL2003Systemic inflammation in chronic obstructive pulmonary diseaseEur Respir J22S513
- PauwelsRARabeKF2004Burden and clinical features of chronic obstructive pulmonary disease (COPD)Lancet3646132015313363
- PlatakiMKoutsopoulosAVDarivianakiK2005Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosisChest1272667415653994
- PletzMWRIoanasMDe RouxA2004Reduced spontaneous apoptosis in peripheral blood neutrophils during exacerbation of COPDEur Respir J23532715083750
- PonomarevEDDittelBN2005Gamma delta T-cells regulated the extent and duration of inflammation in the central nervous system by a Fas ligand-dependent mechanismJ Immunol17446788715814692
- RabacchiSAFriedmanWJShelanskiML2004Divergence of the apoptotic pathways induced by 4-hydroxynonenal and amyloid β-proteinNeurobiol Aging2510576615212831
- RahmanIvan SchadewijkAAMCrowtherAJL20024-Hydroxy-2-Nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med166490512186826
- RauloSMSorsaTTervahartialaT2001MMP-9 as a marker of inflammation in tracheal epithelial lining fluid (TELF) and in bronchoalveolar fluid (BALF) of COPD horsesEquine Vet J33113511266058
- RobbinsCSDaweDEGoncharovaSI2004Cigarette smoke decreases pulmonary dendritic cells and impacts antiviral immune responsivenessAm J Respir Cell Mol Biol302021112920055
- SaettaMDi StefanoAMaestrelliP1993Activated T-lymphocytes and macrophages in bronchial mucosa of subjects with chronic bronchitisAm Rev Respir Dis14730168430952
- SaettaM1999Airway inflammation in chronic obstructive pulmonary diseaseAm J Respir Crit Cre Med160SupplS1720
- SakaoSTatsumiKIgariH2002Association of tumor necrosis factor- α gene promoter polymorphism with low attenuation areas on high-resolution CT in patients with COPDChest1224162012171811
- SauledaJGarcia-PalmerFJWiesnerR1998Cytochrome oxidase activity and mitochondrial gene expression in skeletal muscle of patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med157141379603116
- Segura-ValdezLPardoAGaxiolaM2000Upregulation of gelatinases A and B, collagenases 1, and 2, and increased parenchymal cell death in COPDChest1176849410712992
- ShapiroS2000Animal models for chronic obstructive pulmonary diseaseAm J Respir Cell Mol Biol224710615058
- ShapiroSD2000Vascular atrophy and VEGFR-2 signaling: old theories of pulmonary emphysema meet new dataJ Clin Invest10613091011104783
- SiafakasNMVermeirePPrideNB1995Optimal assessment and management of chronic obstructive pulmonary disease (COPD). The European Respiratory Society Task ForceEur Respir J813984207489808
- SniderGLKleinermanLJThurlbeckWM1985The definition of emphysema: report of a National, Heart, Lung and Blood Institute. Division of Lung Diseases WorkshopAm Rev Respir Dis13218254014865
- TakabatakeNNakamuraHInoueS2000Circulating levels of soluble Fas ligand and soluble Fas in patients with chronic obstructive pulmonary diseaseRespir Med9412152011192958
- TsaoPNSuYNLiH2004Overexpression of placenta growth factor contributes to the pathogenesis of pulmonary emphysemaAm J Respir Crit Care Med1695051114644931
- TsoumakidouMTzanakisNChrysofakisG2005Nitrosative stress, heme oxygenase-1 expression and airway inflammation during severe exacerbations of COPDChest1271911815947302
- TsoumakidouMTzanakisNKyriakouD2004Inflammatory cell profiles and T-lymphocyte subsets in chronic obstructive pulmonary disease and severe persistent asthmaClin Exp Allergy342344014987303
- TuderRMPetracheIEliasJA2003Apoptosis and emphysema. The missing linkAm J Respir Cell Mol Biol28551412707010
- TuderRMZhenLChoCY2003Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockadeAm J Respir Cell Mol Biol29889712600822
- TurlejRKFievezLSandersenCF2001Enhanced survival of lung granulocytes in an animal model of asthma: evidence for a role of GM-CSF activated STAT5 signalling pathwayThorax5669670211514690
- TzanakisNChrysofakisGTsoumakidouM2004Induced sputum CD8+ T-lymphocytes subpopulations in chronic obstructive pulmonary diseaseRespir Med98576514959815
- VandivierRWFadokVAHoffmannPR2002Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasisJ Clin Invest1096617011877474
- WHO2002World Health Report [online]Accessed 21 May 2005 URL: http://www.who.int/whr/2002
- YasudaNGotohKMinatoguchiS1998An increase of soluble Fas, an inhibitor of apoptosis, associated with progression of COPDRespir Med9299399893764
- YokohoriNAoshibaKNagaiAthe Respiratory Failure Research Group in Japan2004Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysemaChest1256263214769747