Abstract
A growing body of evidence indicates that matrix metalloproteinases (MMPs) play a role in the pathogenesis of COPD. Therefore, we conducted a candidate gene association study of 4 promoter polymorphisms that are known to modify expression levels of the MMP-1, MMP-2, and MMP-9 genes and a Gln279Arg polymorphism in exon 6 of MMP-9 that modifies the substrate-binding region. We examined the association of each variant and haplotypes in 385 male veterans with greater than 20 pack-years of cigarette smoking whose COPD status was characterized using spirometry. The association of these polymorphisms was also examined with decline of pulmonary function in a subset of participants. Only the 279Arg variant was more common in participants with COPD and the homozygous variant was associated with a 3-fold increased risk for COPD. In the haplotype analysis, the haplotype comprising the 249Arg and the CA promoter polymorphism within the MMP-9 gene was associated with risk, suggesting that either 279Arg or a linked variant on this haplotype underlies the association. No association of this polymorphism was found with decline in pulmonary function. These studies show that variants of the MMP-9 gene are associated with COPD in this cohort of veterans.
Introduction
Genetic factors likely play a critical role in the development of COPD, because only a minority of heavy smokers develops this disease. The first genetic defect associated with COPD was a deficiency in α1-antitrypsin (CitationGanrot et al 1967). However, other genetic variations that affect the proteinase–antiproteinase imbalance (CitationEvans and Pryor 1994) also likely affect the onset and development of COPD, because only <1% of COPD cases are associated with α1-antitrypsin deficiency (CitationSandford et al 2002).
A role for the matrix metalloproteinases (MMPs) has been suggested in the pathogenesis of pulmonary emphysema (CitationBarnes 2000). Immunoreactivity for MMP-1, MMP-2, and MMP-9 is more intense in emphysematous samples compared with normal lung tissue (CitationOhnishi et al 1998; CitationSegura-Valdez et al 2000). Alveolar macrophages obtained from smokers release more MMP-9 than those from non-smokers (CitationLim et al 2000).
There is an insertion–deletion polymorphism in the promoter region of the MMP-1 gene (1G-1607/2G). The addition of G in the MMP-1 promoter creates an Ets binding site, and leads to higher transcription of the protein compared with the common 1G polymorphism (CitationRutter et al 1998). The incidence of the 2G allele rises to 62% in tumor cells in ovarian and endometrial carcinomas (CitationRutter et al 1998). A single nucleotide polymorphism (SNP) in the MMP-2 promoter sequence (−1306 C→T) disrupts a SP1 site and results in lower promoter activity with the T allele (CitationPrice et al 2001), and participants homozygous for C have an overall 2-fold increased risk for developing lung cancer compared with those with the CT or TT genotype (CitationYu et al 2002). We chose to study the association of these SNPs with COPD, because participants with COPD are at higher risk of developing lung cancer (CitationCohen et al 1977; CitationWu et al 1995).
Sequence analysis of the MMP-9 gene revealed a total of 10 variable sites, 4 in the promoter, 5 in the coding region (3 of which alter the amino acid encoded), and 1 in the 3′ untranslated region (CitationZhang et al 1999a). The variable length CA repeat located at −90 bp within the promoter region of MMP-9 may affect promoter activity and creates a sequence-specific DNA-binding protein site (CitationPeters et al 1999). A C→T polymorphism at −1562 bp results in higher transcription rates for the T allele than the C allele promoter (CitationZhang et al 1999b) and dysregulation of MMP-9 function could increase susceptibility for smoking-induced airway obstruction. A polymorphism in exon 6 of MMP-9 is located in the substrate-binding domain of this enzyme and leads to a substitution of an uncharged amino acid (glutamine) by a positively charged amino acid (arginine). This polymorphism likely alters protein conformation, leading to a change in substrate-binding and enzyme activity, while the other two polymorphisms in the coding region that lead to altered amino acids are not likely to have such a profound change in enzyme activity (CitationZhang et al 1999a).
The major ethnic groups in New Mexico, the area from which the study population was recruited, consist of non-hispanic whites and hispanics. Therefore, the present study analyzed the prevalence of COPD in these ethnic groups and determined whether potentially functional variants that include the promoter regions of MMP-1, MMP -2, and MMP -9 and the Glu279Arg polymorphism within MMP-9 gene are associated with COPD in a cross-sectional study of veteran men and within the major ethnic groups.
Material and methods
Study population
Study participants were drawn from an ongoing cohort study of veterans (n=385). Participants were recruited from the New Mexico Veterans’ Health Care System, which provides medical care for approximately 35 000 veterans in New Mexico, west Texas, and southern Colorado. Inclusion criteria included age 40–75 years and a minimum smoking history of ≥20 pack-years. Informed consent was obtained from each participant prior to completing a standardized clinical examination including spirometry and a standardized questionnaire that provided detailed information on demographics, smoking history, and respiratory health. Phlegm production was assessed by evaluating the St. George’s Respiratory Disease Questionnaire. The same information was obtained at a follow-up visit at 18±3 months for 176 participants. This study was approved by the Institutional Review Boards of the Lovelace Respiratory Research Institute, the New Mexico Veterans Health Care System, and the University of New Mexico Health Sciences Center.
COPD definition
COPD was defined by criteria similar to those suggested as part of the GOLD criteria (CitationPauwels et al 2001) and were used in the Lung Health Study (CitationAnthonisen et al 1994) and recently described by the ATS/ERS Task Force (CitationCelli and MacNee 2004). We did not break down the cohort into various GOLD categories, because of small cell size for GOLD 1–4. However, three definitions for COPD were used: Definition I included all participants with FEV1/FVC<70% as measured by spirometry. Definition II included only participants with FEV1/FVC<70% and who had FEV1<80% as cases and all other participants who were defined as COPD participants in Definition I were excluded from the analyses. Definition III included only participants with FEV1/FVC<70% and FEV1<80%, excluding all who had reversibility of >15% post-bronchodialator exposure. Reversibility was defined as the percent change in percent predicted FEV1 between pre- and post-bronchodialator exposures. For all three definitions, participants were considered to be non-COPD if FEV1/FVC≥70%. Participants who met the criteria for diagnosis of COPD at cohort entry were classified as prevalent cases. Participants with mild COPD were excluded in Definition II to reduce the potential for including participants who have “borderline” airflow obstruction and participants who have reversible airflow obstruction were excluded in Definition III to exclude the majority of participants who have asthma.
Genotyping of polymorphisms in MMP-9
Genomic DNA was isolated from peripheral blood lymphocytes by digestion with proteinase K in sodium dodecyl sulfate (1%), followed by standard phenol–chloroform extraction and ethanol precipitation as described (CitationSambrook et al 1989). The sequence flanking the polymorphic region of each SNP was amplified by PCR using 10 ng of DNA. Negative controls without DNA template were included with each set of reactions. Several methods of genotyping were used to verify the obtained results. Restriction fragment length polymorphism (RFLP) was performed on 50–100 samples to verify the results obtained by the allelic discrimination assay. This assay integrates PCR and detection using allelic discrimination, which applies allele-specific TaqMan probes, a two-dye reporter system, and a reference dye. Wild-type and variant reporter sequences were discriminated using dual labeled reporters and by using mismatched sequences to validate the SNP-genotype. To further ensure quality control, DNA samples from 20 participants whose allelotypes were known were included at random within each of the 96-well plates of the allelic discrimination assay. In addition, approximately 10% of the samples were re-examined by a different person. All analyses were performed blinded with respect to participants’ characteristics. These controls showed 100% concordance with expected results.
The methods of genotyping, including the primers and probes for allelic discrimination of SNPs in MMP-1, MMP-2, and MMP-9 or enzyme used for RFLP, are listed in . The C→T polymorphism was analyzed using a RFLP assay, because several attempts to design primers and probes for this polymorphism failed. The presence of the rare T allele introduces a restriction site for SphI, which is absent when the common C allele is present.
Table 1 PCR primers and probes for detecting polymorphisms
The number of CA repeats in the promoter region of MMP-9 was determined by PCR using a fluorescently labeled 6-FAM forward primer and an unlabeled reverse primer. The alleles were separated by a laser-based automated DNA sequencer, ABI PRISM 3100. The product peaks were visualized using GenescanView software (Bio Molecular Research, Padova, Italy). The size of the PCR products was calculated from a standard curve made from internal standards. DNA standards containing 14, 21, 22, and 23 CA repeats were identified by direct sequencing and were included in each experiment as positive controls. For unknown reasons, the polymorphisms of certain samples (1%–5%) could not be assessed even upon repeated attempts, resulting in slight sample size variability for the various polymorphisms.
Statistical analysis
We examined summary information by COPD status for demographic variables (age, gender, ethnicity); clinical risk factors (current smoking status, duration and pack-years of cigarette smoking); and polymorphism status (homozygous-rare, heterozygous, homozygous-common). The chi-square test and Fisher’s exact test were used to compare categorical variables, including gene polymorphisms between participants with and without COPD. Continuous variables such as age and smoking duration were compared using either the two-sample t-test or the Wilcoxon rank sum test. The small number of women (17 in total) enrolled in the larger cohort were excluded from the analysis cohort of 385 veteran men, but this exclusion and inclusion of these women did not change any of the conclusions.
Distributions of genotypes were assessed for Hardy-Weinberg equilibrium using a generalization of Fisher’s exact test (CitationGuo and Thompson 1992). All pairs of MMP-9 loci (T-1562, 279Arg, and CA repeats) were tested for linkage disequilibrium using likelihood tests and permutation procedures to estimate significance levels (CitationGoudet et al 1996; CitationSlatkin and Excoffier 1996) as implemented in PowerMarker v3.12 (Liu and Muse). The degree of disequilibrium was described using D′ (CitationLewontin 1964; CitationZapata et al 2001).
Logistic regression models were fit to estimate odds ratios (OR) for COPD and to allow adjustment for multiple covariates. We estimated effects for dominant, co-dominant, and recessive models for the variant alleles. Covariates in all models included age, ethnicity, and smoking information (former or current, or the continuous forms of smoking duration and pack-years). Logistic regression models for each polymorphism were fitted and the indicators were included as predictors for only those polymorphisms at one region plus the covariates. Since there was linkage disequilibrium between some of the polymorphisms, the polymorphisms were examined separately. Interactions between the genotypes and each covariate, especially cigarette smoking (duration and pack-years), and interactions between the covariates also were investigated. To assess differences by ethnicity, models were fitted separately by ethnic group. Likelihood ratio tests were used to test models and Wald’s chi-square tests were used to evaluate whether individual variables contributed to the model. Linear regression models were used to assess the association between the genetic predictor variables and the predicted FEV1. Covariates and interactions were included in the modeling as done for the logistic regression models above. All standard statistical testing and modeling were conducted using Statistical Analysis System SAS v8.02. (SAS Institute, Inc., Cary, NC, USA). These analyses were done for each of the COPD definitions. Therefore, the analyses ranged from mild to moderate and severe cases as the outcome.
The association between estimated haplotype frequencies and COPD was evaluated with a two-stage process. First, MMP-9 haplotype frequencies based on T-1562, 279Arg, and CA repeats were estimated using Excoffier and Slatkin’s maximum likelihood algorithm (CitationExcoffier and Slatkin 1995). For each possible haplotype, h, an estimate is made of the number of copies of h contained in the true pair of haplotypes carried by that individual conditional on the observed genotype and assuming Hardy-Weinberg equilibrium. The expected haplotype frequencies are analogous to haplotype dosages and were used in the second step as independent variables in logistic regression models that also included adjustment for age, smoking duration, and smoking status. Haplotype dosages for each participant were calculated separately for non-hispanic whites and hispanics. For other and unknown ethnicity, haplotype doses were calculated based on the overall cohort. Second, logistic regression analyses were conducted as outlined previously.
Results
Description of study population
The demographics of the cohort are summarized by COPD status as per definition I, which includes mild to severe cases (). The prevalence of COPD was 32% and, as expected for a disease that increases in frequency with age, overall the age of participants with COPD was older than for non-COPD participants. The median age for participants with and without COPD was 64 and 57 years (p<0.001), respectively. Interestingly, a higher proportion of non-hispanic white participants (36%) had COPD than hispanic participants (25%) (p=0.07). The number of participants included in analyses for COPD definitions II and III decreased to 366 and 339, respectively.
Table 2 Summary of demographic and baseline variables plus information on baseline PFT for all participants and those with two visits
Average smoking duration and pack-years were greater for participants with COPD than for participants without COPD (p<0.001). Furthermore, the unadjusted OR for COPD was 2.1 and 2.0 for every 10 additional years of smoking duration and age, respectively (p<0.001). Finally, COPD was significantly associated with phlegm production when adjusted for age and smoking duration (OR=2.0, 95% CI=1.4 to 3.0; p<0.001). These basic demographic and clinical risk factors are similar to those reported in other studies of COPD (CitationZayas et al 1990) and reinforce the utility of our cohort for evaluating the role of polymorphisms within the MMP-9 gene as risk modifiers for COPD.
In general the percentages for COPD participants, ethnic distribution, smoking duration, and pack-years, and the baseline %FEV1 for the participants with a second visit remained identical to those for the overall cohort who came for the first visit (). Repeat testing was performed on these participants with 90% concordance of those initially classified with chronic airflow obstruction. These tests established the chronicity of airflow limitation through demonstration of FEV1/FVC<70% by spirometry that was not reversed into the normal range after a bronchodilation, but had an overall tendency of decline over time (CitationPauwels et al 2001). No mean linear change in pulmonary function test was found in non-COPD participants within the participants with repeat measurements. However, there was a significant difference in yearly decline of %FEV1 for the COPD participants (2.6 (95% CI: 1.4–3.8)) compared with that of non-COPD participants (0.81 (95% CI: −0.01 to 1.62)) (p<0.02).
Distribution of genotypes
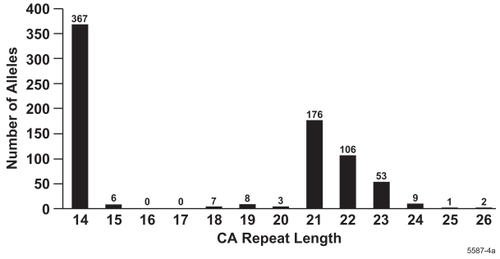
The SNPs were determined in DNA samples from 385 study participants. CA repeats of 14, 21, 22, and 23 were most frequently found while the frequency of other repeat lengths was below 2% (). The CA repeat lengths that were most frequently found as genotypes were 14/14 (27%), 14/21 (20%), 14/22 (16%), 14/23 (8%), 21/21 (11%), and 22/22 (5%). The frequency of all other alleles was below 2% (). Because the frequency distribution of the CA repeat polymorphism showed a bimodal distribution of alleles, the alleles were divided into two subclasses: CA repeats ≤16 and ≥17 were designated as Short and Long, respectively, and treated similarly to genotype data.
Table 3 Distribution of CA repeat alleles
Figure 1 Distribution of CA repeat lengths.
The distributions of homozygotes and heterozygotes for each polymorphism in each ethnic group generally conformed to expectations under the assumption of Hardy-Weinberg equilibrium, with the exception of the CA-repeat among hispanic participants (p=0.02). As a measure for linkage disequilibrium, D′ was determined. The 279Arg was in linkage disequilibrium with both T-1562 (p<0.001) and CA repeat (p<0.001) within the ethnic groups. Of particular note, all participants with the homozygote rare T-1562 (N=20) had rare 279Arg. However, many participants who were homozygous for 279Arg had the C-1562 (). No differences among cases and controls were observed for any of the genotypes in MMP-1 and MMP-2.
Table 4 Distribution of genotypes in COPD and non-COPD participants (%)
A difference in prevalence of the 279Gln (35.0 vs 52.1, p<0.01) and 279Arg (18.7% vs 7.7%, p<0.01) homozygote genotypes was seen in participants with COPD compared with non-COPD participants () and among the COPD and non-COPD participants at the second visit (see ). Significantly different distributions of the homozygote rare 279Arg were also observed within the non-hispanic white participants (p<0.01). This trend in difference was recapitulated within the hispanic participants, but was not significant due to the lower number of hispanic participants with COPD. The rare T-1562 allele, the CA repeat lengths, and the SNPs in MMP-1 and MMP -2 were distributed similarly among the participants with and without COPD in the veterans’ cohort (). The results for COPD definitions II and III remained identical, in that the distribution for the Gln279 was 52% compared with 7.7% for the Arg279. For both COPD definitions II and III, the distribution for Gln279 remained at 47% in non-hispanic white participants compared with 9% for the rare Arg279.
Table 5 Association of genotypes with COPDTable Footnotea
Table 6 MMP-9 C/T-1562, Gln279Arg, and CA repeat haplotype frequencies
Appendix Table 4 Distribution of polymorphisms by COPD status (definition I)
Association of allelotype with COPD
The rare 279Arg variant was significantly associated with COPD (). This table gives the odds ratios which is the odds for COPD relative to the odds for those without COPD. After adjustment for age, smoking duration, and current smoking status, the OR for COPD participants who were homozygous for 279Arg variant was 3.8 relative to those who were homozygous for the common allele (p<0.01). In addition, participants who were heterozygous also showed a significant increased risk for the disease (OR=1.8; p<0.05), indicating a co-dominant association between 279Arg and COPD. This association was significant within the non-hispanic white but not within the hispanic participants, likely because of limited sample size for this ethnic group. Among the non-hispanic white participants, significant associations between COPD and the homozygous rare T-1562 (OR=4.5; p<0.05) and the Long/Long form of CA repeat (OR=2.3, p<0.05) were observed. Similar results were obtained for COPD definitions II and III (see ). For example, for COPD definition II for the Gln279Arg the OR for the homozygote rare genotype remained significant at 3.13 (95% CI: 1.4–6.9) and in non-hispanic white participants at 2.8 (95% CI: 1.1–6.8). For COPD definition III, the OR was 3.1 (95% CI: 1.4–7.3) and 3.3 (95% CI: 1.3–8.4) overall and for non-hispanic whites, respectively. No association with COPD was found for the variants in MMP-1 and MMP -2. The similarity of results between the ethnic groups and among the definitions of COPD indicates that multiple comparison adjustments are not needed for these factors. All models showed that smoking variables, including duration, pack-years, and current status, were significantly associated with COPD, but none of the variables was a co-founder with the genetic variables.
Appendix Table 5 Association of genotypes with COPD
Gene–gene and gene–environment interaction effects were evaluated by adding interaction terms in logistic regression models that included adjustments for age and smoking duration. To assess differences by ethnicity, analyses were done separately for non-hispanic white and hispanic participants. The gene–environment interactions used cigarette smoking measures as the environmental factors. We found no significant gene–gene or gene–environment interaction effects.
Association of SNPs with the decline in pulmonary function–longitudinal analyses
While we did not see a decline in pulmonary function over time when analyzing the data of the entire population, non-hispanic white participants and those who smoked more than 40 pack-years showed a statistically significant decrease per year when the raw or percentage difference of the baseline value in FEV1 was analyzed. Non-hispanic whites showed an average decrease of 1.2 (95% CI: 0.5–1.9) or 1.7% (95% CI: 0.7–2.6), respectively, per year. Those who smoked ≥40 pack-years showed an average decrease of 1.6 (95% CI: 0.6–2.7) or 2.1% (95% CI: 0.8– 3.4%), respectively per year. However, we did not see any association of the polymorphisms tested with decline in pulmonary function assessed over 18 months, while the distribution of polymorphisms by COPD status was identical to that observed for all participants.
Haplotype analyses
Haplotype (common [C], rare [R], long [L], short [S]) frequencies formed from MMP-9 C/T-1562, MMP-9 Gln279Arg, and CA repeats were estimated for each cohort and ethnic group and evaluated for differences with respect to COPD status (). ORs adjusted for age, smoking duration, and current smoking status were calculated relative to the common C-1562, Gln279, and short CA repeats (C-C-S) for haplotypes with frequencies >0.005 (). The haplotypes with significant association with COPD were C-R-L (1.8, 95% CI, 1.1–2.9) and R-R-L (1.7, 95% CI, 1.1–2.7) compared with the C-C-S haplotype. Participants with the haplotype including the T-1562 and the rare 279Arg showed the same OR as participants with the haplotype, including the C-1562 and the rare 279Arg, indicating that either the 279Arg polymorphism is the main determinant or another variant on this RL haplotype may underlie the increased association by MMP-9 and risk for COPD. Models with and without covariates were compared using likelihood ratio tests. Models with haplotype dosages were no better than covariate-only models for predicting COPD status of hispanic veterans. Haplotype information was useful for predicting COPD in non-hispanic white veterans; however, the only adjusted OR different from the reference haplotype, C-C-S, was R-R-L (p=0.02, OR=2.0, 95% CI=1.1–3.4).
Discussion
This cross-sectional study provides evidence that the tested polymorphisms in the MMP-9 gene are associated with susceptibility for onset of COPD in men. The increased risk for COPD may stem from the rare allele in exon 6, which replaces the non-charged glutamine (MMP-9Q) with the charged amino acid, arginine (MMP-9R).
MMP-9R could increase degradation of the ECM leading to emphysematous lungs. MMP-9 is expressed and secreted from cells as inactive zymogen through multiple mechanisms. This inactive form of MMP-9 is maintained by a cysteine and additional domains within the highly conserved region of the pro-peptide and the essential zinc atom in the catalytic domain (CitationPark et al 1991). This tight regulation may be diminished in MMP-9R, especially in the presence of inflammatory responses that are induced by the presence of cigarette smoke. Alternatively, the affinity of MMP-9R to tissue inhibitors of metalloproteinases (TIMP)-1, the major inhibitor of MMP-9, may be lower compared with that of MMP-9Q, because TIMP-1 forms a complex at the carboxy-terminus of the pro-enzyme and the catalytic domain of the active form (CitationGoldberg et al 1992).
A previous study comparing smokers with COPD showed an association with a 5-year rate of decline with MMP-1 G-1607GG and haplotypes consisting of alleles from the MMP-1 G-1607GG and MMP-12 Asn357Ser polymorphisms (CitationJoos et al 2002). In contrast, the CA repeat in MMP-9 was not associated with the rate of decline in lung function. The differences in findings from our study may be because the Joos et al study was designed to focus on the rate of decline in lung function rather than overall risk for developing COPD. That study also did not include the Gln279Arg SNP of MMP-9. As expected, the frequencies of the A (65%) and G (35%) alleles in our study were very similar to those reported previously (CitationZhang et al 1999a). In general the frequencies of the alleles for each of the SNPs in this study were very similar to those reported in the NCBI website or in manuscripts (CitationZhang et al 1999a; CitationHinoda et al 2002).
The CA repeat length that was observed with high frequency in this cohort was 14 repeats. The second allele in these individuals had a CA repeat of 21, 22, or 23 repeats. The next most frequent alleles were between 21 and 22 CA repeats. While a similar distribution in CA repeats was observed in a Caucasian population (CitationJoos et al 2002), this distribution of CA repeats is vastly different from that reported for 223 Japanese who had primarily MMP-9 alleles that contained 20 or more CA repeats. Only 2.8% of the Japanese population studied had 1 or 2 alleles with fewer than 20 CA repeats (CitationShimajiri et al 1999). This difference in the distribution of CA repeat length may be significant, because promoters with 21 CA repeats appeared to have a higher promoter activity compared with those with shorter repeats (CitationShimajiri et al 1999). In contrast, MMP-9 promoter containing 14 repeats had 53% promoter activity seen with 24 CA repeats (CitationHuang et al 2003). Similarly, CitationPeters et al (1999) found that the promoter with 23 CA repeats showed a 1.5-fold higher promoter activity compared with those containing 21 or 22 repeats. Consistent with these findings, non-hispanic white participants in particular with the Long/Long genotype had an increased risk for COPD compared with those with a Short/Short genotype in our study.
The -1562T genotype was significantly associated with increased risk for COPD in non-hispanic whites only. This finding supports the finding by CitationMinematsu et al (2001) that the allelic frequency of the MMP-9 C-1562T promoter polymorphism was significantly increased in 45 smokers with emphysema compared with 65 matched smokers without emphysema, when emphysematous changes were scored from chest computer tomography scans. However, they did not find significant differences when COPD was characterized by FEV1. Differences may be due to the association being specific for a specific ethnic group.
Non-hispanic whites were more likely to have COPD compared with hispanics and the decline in FEV1 was more pronounced in these participants compared with hispanics. In addition, all polymorphisms within MMP-9 that were tested were significantly associated with COPD in the non-hispanic whites. The -1562T genotype was associated with COPD among the non-hispanic whites, but the genotype is rare and was not observed in the hispanics. The association for 279Arg and Long/Long form of CA repeat was similar for both ethnic groups, but the result was not statistically significant for the hispanics, which may be a result of the smaller sample size. Collectively, these findings suggest that, while the distribution of the genotypes may vary by ethnic group, the association between genotype and COPD is not affected by ethnicity. Thus, confounding by population admixture is unlikely to account for the observed associations. Whether this association is also found for women with COPD will be addressed in the future.
Many studies have suggested the involvement of the MMPs in COPD (CitationOhnishi et al 1998; CitationVignola et al 1998; CitationSegura-Valdez et al 2000; CitationImai et al 2001). Overexpression of MMP-1 results in emphysema in transgenic mice (CitationD’Armiento et al 1992) and MMP-12 deficient mice are less susceptible to cigarette smoke-induced emphysema compared with wild-type mice (CitationHautamaki et al 1997). However, the relevance of these animal models to human disease has not been proven. Our study supports the hypothesis that MMP-9 may be a protease that is related to the pathogenesis of COPD and polymorphisms in MMP-9 may act as intrinsic factors to affect sensitivity to cigarette smoke-induced onset of COPD.
Acknowledgments
This research was funded by the State of New Mexico as a direct appropriation from the Tobacco Settlement Fund, the NIEHS Center (P30 ES012072) and by the VA grant V501P-2638. This material is based upon work supported by the Office of Research and Development (R&D), Medical Research Service, GrantV501P-2638.
References
- AnthonisenNRConnettJEKileyJP1994Effects of smoking on intervention and use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1JAMA27214975057966841
- BarnesPJ2000Chronic obstructive pulmonary diseaseN Engl J Med3432698010911010
- CelliBRMacNeeW2004Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paperEur Respir J239324615219010
- CohenBHDiamondELGravesCG1977A common familial component in lung cancer and chronic obstructive pulmonary diseaseLancet2523695731
- D’ArmientoJDalalSSOkadaY1992Collagenase expression in the lungs of transgenic mice causes pulmonary emphysemaCell71955611458541
- EvansMDPryorWA1994Cigarette smoking, emphysema, and damage to alpha 1-proteinase inhibitorAm J Physiol266L5936118023948
- ExcoffierLSlatkinM1995Maximum-likelihood estimation of molecular haplotype frequencies in a diploid populationMol Biol and Evolution129217
- GanrotPOLaurellCBErikssonS1967Obstructive lung disease and trypsin inhibitors in alpha-1-antitrypsin deficiencyScand J Clin Lab Invest1920586048625
- GoldbergGIStronginACollierIE1992Interaction of 92-kDa type IV collagenase with the tissue inhibitor of metalloproteinases prevents dimerization, complex formation with interstitial collagenase, and activation of the proenzyme with stromelysinJ Biol Chem2674583911311314
- GoudetJRaymondMMeeüsD1996Testing differentiation in diploid populationsGenetics1441933408978076
- GuoSThompsonE1992Performing exact tests of Hardy-Weinberg proportion for multiple allelesBiometrics48361721637966
- HautamakiRDKobayashiDKSeniorRM1997Requirement for macrophage elastase for cigarette smoke-induced emphysema in miceScience277200249302297
- HinodaYOkayamaNTakanoN2002Association of functional polymorphisms of matrix metalloproteinase (MMP)-1 and MMP-3 genes with colorectal cancerInt J Cancer102526912432557
- HuangTSLeeCCChangAC2003Shortening of microsatellite deoxy(CA) repeats involved in GL331-induced down-regulation of matrix metalloproteinase-9 gene expressionBiochem Biophys Res Commun300901712559958
- ImaiKDalalSSChenES2001Human collagenase (matrix metalloproteinase-1) expression in the lungs of patients with emphysemaAm J Respir Crit Care Med1637869111254539
- JoosLHeJQShepherdsonMB2002The role of matrix metalloproteinase polymorphisms in the rate of decline in lung functionHum Mol Genet115697611875051
- LewontinRC1964The interaction of selection and linkage. I. General considerations; heterotic modelsGenetics49496717248194
- LimSRocheNOliverBG2000Balance of matrix metalloprotease-9 and tissue inhibitor of metalloprotease-1 from alveolar macrophages in cigarette smokers. Regulation by interleukin-10Am J Respir Crit Care Med16213556011029344
- LiuKMuseSFree program distributed by the author over the internet URL: http://www.powermarker.net
- MinematsuNNakamuraHTatenoH2001Genetic polymorphism in matrix metalloproteinase-9 and pulmonary emphysemaBiochem Biophys Res Commun2891161911708786
- OhnishiKTakagiMKurokawaY1998Matrix metalloproteinase-mediated extracellular matrix protein degradation in human pulmonary emphysemaLab Invest781077879759652
- ParkAJMatrisianLMKellsAF1991Mutational analysis of the transin (rat stromelysin) autoinhibitor region demonstrates a role for residues surrounding the “cysteine switch”J Biol Chem2661584901988438
- PauwelsRABuistASCalverleyPM2001Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summaryAm J Respir Crit Care Med16312567611316667
- PetersDGKassamASt JeanPL1999Functional polymorphism in the matrix metalloproteinase-9 promoter as a potential risk factor for intracranial aneurysmStroke3026121610582986
- PriceSJGreavesDRWatkinsH2001Identification of novel, functional genetic variants in the human matrix metalloproteinase-2 gene: role of Sp1 in allele-specific transcriptional regulationJ Biol Chem27675495811114309
- RutterJLMitchellTIButticeG1998A single nucleotide polymorphism in the matrix metalloproteinase-1 promoter creates an Ets binding site and augments transcriptionCancer Res58532159850057
- SambrookJFritschEFManiatisT1989Molecular Cloning A Laboratory Manual2nd edNew YorkCold Spring Harbor Press
- SandfordAJJoosLParePD2002Genetic risk factors for chronic obstructive pulmonary diseaseCurr Opin Pulm Med8879411845002
- Segura-ValdezLPardoAGaxiolaM2000Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPDChest1176849410712992
- ShimajiriSArimaNTanimotoA1999Shortened microsatellite d(CA)21 sequence down-regulates promoter activity of matrix metalloproteinase 9 geneFEBS Letts45570410428474
- SlatkinMExcoffierL1996Testing for linkage disequilibrium using the EM algorithmHeredity76377838626222
- VignolaAMRiccobonoLMirabellaA1998Sputum metalloproteinase-9/tissue inhibitor of metalloproteinase-1 ratio correlates with airflow obstruction in asthma and chronic bronchitisAm J Respir Crit Care Med1581945509847290
- WuAHFonthamETReynoldsP1995Previous lung disease and risk of lung cancer among lifetime nonsmoking women in the United StatesAm J Epidemiol1411023327771438
- YuCPanKXingD2002Correlation between a single nucleotide polymorphism in the matrix metalloproteinase-2 promoter and risk of lung cancerCancer Res626430312438229
- ZapataCCarolloCRodriguezS2001Sampling variance and distribution of the D’ measure of overall gametic disequilibrium between multiallelic lociAnn Human Gen65395406
- ZayasJGManGCKingM1990Tracheal mucus rheology in patients undergoing diagnostic bronchoscopy. Interrelations with smoking and cancerAm Rev Respir Dis1411107132339832
- ZhangBHenneyAErikssonP1999aGenetic variation at the matrix metalloproteinase-9 locus on chromosome 20q12.2–13.1Hum Genet1054182310598806
- ZhangBYeSHerrmannSM1999bFunctional polymorphism in the regulatory region of gelatinase B gene in relation to severity of coronary atherosclerosisCirculation9917889410199873