Abstract
Although the prevalence of pulmonary hypertension (PH) in individuals with chronic obstructive pulmonary disease (COPD) is not known precisely, approximately 10%–30% of patients with moderate to severe COPD have elevated pulmonary pressures. The vast majority of PH associated with COPD is mild to moderate and severe PH occurs in <5% of patients. When COPD is associated with PH, both mortality and morbidity are increased. There are no clinical or physical examination findings that accurately identify patients with underlying PH. Radiographic imaging findings are specific but not sensitive indicators of PH. Echocardiography is the principle noninvasive diagnostic test but may be technically limited in a significant proportion of patients with COPD. Right heart catheterization is required for accurate measurement of pulmonary pressures. The combined effects of inflammation, endothelial cell dysfunction, and angiogenesis appear to contribute to the development of PH associated with COPD. Systemic vasodilators have not been found to be effective therapy. Selective pulmonary vasodilators including inhaled nitric oxide and phosphodiesterase inhibitors are promising treatments for patients with COPD associated PH but further evaluation of these medications is needed prior to their routine use.
Keywords:
Introduction
Chronic obstructive pulmonary disease (COPD) is a significant health care burden worldwide and is the only major cause of death in the United States for which both mortality and morbidity are increasing (CitationMurray and Lopez 1997; CitationHurd 2000). This disease process is manifest by progressive airflow limitation, hyperinflation and air trapping, hypoxemia, hypercapnea, and elevations in pulmonary vascular pressures. Clinically, individuals with COPD develop breathlessness, cough, sputum production and disease exacerbations that impair quality of life. Factors that portend a poor prognosis include severity of airflow limitation, ventilatory capacity, hypercapnea, and pulmonary hypertension (CitationBurrows and Earle 1969; CitationWeitzenblum et al 1981; CitationAnthonisen et al 1986). Survival correlates negatively with pulmonary arterial pressure and pulmonary vascular resistance and patients with COPD and PH have increased morbidity and risk for hospitalizations for acute COPD exacerbations (CitationBurrows et al 1972; CitationWeitzenblum et al 1984; CitationKessler et al 1999; CitationBarbera et al 2003).
PH associated with COPD is increasingly recognized as a contributing factor to the clinical manifestations, morbidity, and mortality of the COPD disease process. This recognition has stimulated further research into the cellular and molecular processes contributing to the pathogenesis of PH associated with COPD and the development and testing of new therapeutic interventions. This review will examine the epidemiology of PH associated with COPD, its clinical manifestations, methods of diagnosis, pathophysiology, and treatment strategies.
Prevalence
The prevalence of pulmonary hypertension (PH) in COPD has not been accurately measured in large epidemiologic studies because of the risks and expense of invasive pressure measurement by right heart catheterization. Most studies have utilized noninvasive measures to estimate pulmonary arterial pressures. Estimates of the prevalence of PH in COPD are also confounded by patient selection. Studied patients have varying severity of obstructive lung disease as well as different levels of oxygenation. Finally, over the last several decades, different groups have used various minimal pressures to define PH and severe PH (). Therefore, estimates of the prevalence of PH in patients with COPD vary widely based upon the definition of PH, the methods used to determine pulmonary pressures, and the physiologic characteristics of the studied population.
Table 1 Varying thresholds defining pulmonary hypertension and severe pulmonary hypertension
Earlier autopsy studies demonstrated anatomic evidence of right ventricular hypertrophy in patients with COPD. Two–thirds of patients with chronic bronchitis had evidence of right ventricular hypertrophy demonstrated by increased weight of the right ventricle (CitationMillard and Reid 1974). Similarly, 71% of 20 patients dying of COPD had right ventricular hypertrophy (CitationScott 1976). In contrast, one–third of 104 patients with emphysema had autopsy evidence of right ventricular hypertrophy (CitationLeopold and Gough 1957). Subsequent studies have suggested a correlation between right ventricular hypertrophy and hypoxemia in patients with COPD (CitationCalverley et al 1992). Recent studies utilizing magnetic resonance imaging (MRI) to measure right ventricular wall thickness and volume non–invasively demonstrated a significant increase in right ventricular wall mass that was classified as concentric hypertrophy in patients with severe COPD and either normoxemia or mild hypoxemia (CitationVonk-Noordegraaf et al 2005).
Several studies have determined pulmonary pressures by right heart catheterization in groups of COPD patients with varying levels of physiologic impairment. In a series of 175 patients with moderate to severe COPD (FEV1% = 40.2 ± 11.1%) and mild hypoxemia (40.6% with PaO2 <60 mmHg), 62 (35%) had pulmonary artery pressures >20 mmHg at right heart catheterization (CitationWeitzenblum et al 1981). The mean pulmonary artery pressure (mPAP) in the entire cohort was 19.8 ± 7.6. Similar results were found in a study of 53 patients with severe COPD (FEV1%, 39.8 ± 16.2); the mPAP was 19.0 ± 4.3 and 23 (43%) had PH (mPAP >20 mmHg) (CitationDoi et al 2003). Seventeen of twenty-seven (63%) patients with mild to moderate hypoxemia had PH. The mPAP was slightly greater, 26.9 ± 8.9 mmHg, in 215 patients with severe COPD (FEV1%, 24.3%) who underwent right heart catheterization prior to lung volume reduction surgery or lung transplantation (CitationThabut et al 2005). Approximately half of these patients had PH (mPAP >25 mmHg) (CitationThabut et al 2005).
In a series of 120 patients with severe emphysema (FEV1%, 27% and PaO2, 65.9 ± 10.0 mmHg) who underwent right heart catheterization during evaluation for lung volume reduction surgery in the National Emphysema Treatment trial, Scharf and colleagues (CitationScharf et al 2002) found a mPAP of 26.3 ± 5.2 mmHg. Over 90% of these patients had PH, (mPAP >20 mmHg). Although PaO2 correlated inversely with mPAP, PaO2 was not an independent predictor of mPAP based upon multiple stepwise regression analysis (CitationScharf et al 2002). Severe PH, (mPAP >35 mmHg)occurred in 6 patients, 5%. Thabet and colleagues (CitationThabut et al 2005) found severe PH (mPAP >45 mmHg) in 8 of 215 patients (3.7%) evaluated for lung volume reduction surgery or lung transplant, whereas, (in a retrospective review)only 11 (1.1%) of 998 patients with COPD had this level of PH (CitationChaouat et al 2005). Thus, although the measured prevalence of PH in patients with COPD ranges from approximately 20 to 50%, very few individuals have severe PH.
Measurements of pulmonary hemodynamics in patients with COPD have shown that pulmonary pressures increase significantly with exercise. Right heart catheterizations performed in 151 patients with COPD (FEV1%, 38 ± 12% and PaO2 72 ± 11 mmHg) by Oswald-Mammosser and colleagues (CitationOswald-Mammosser et al 1991) demonstrated PH (mPAP >20 mmHg) in 31 individuals, 21%. The mPAP at rest was significantly greater, 19 ± 4.7 mmHg, in patients with hypoxemia (PaO2 ≤60 mmHg) than in normoxemic patients, 16.8 ± 4.9, p < 0.05. Further, with exercise up to 40 watts, two-thirds, 99 of 151 patients, developed mPAP ≥ 30 mmHg. In the 49 patients in whom pulmonary vascular resistance could be calculated at both rest and with exercise, pulmonary vascular resistance did not change significantly with exercise. Christensen and co-workers (CitationChristensen et al 2004) demonstrated a similar incidence of exercise induced PH, 65% in a series of 17 patients with COPD (FEV1% 35 ± 10%, PaO2 10.6 ± 1.1 kPa) who exercised at a work load of 25 watts. The mPAP was 19.9 ± 4.5 mmHg at rest and increased to 35.0 ± 2.2 mmHg with exercise. Eleven patients, 65%, had mPAP greater than 30 mmHg with exercise. Fujimoto and collaborators (CitationFujimoto et al 2002) studied pulmonary hemodynamics in 75 patients with mild hypoxemia (PaO2 >60 mmHg) at rest and either mild (FEV1% >50%), moderate (FEV1% <50% or >35%), or severe COPD (FEV1% ≤35%) at rest and with exercise. At rest, mPAP’s were 21.5 ± 2.7, 20.0 ± 4.2, and 21.7 ± 1.1 mmHg in the mild, moderate, and severe groups, and increased to 32.7 ± 3.2, 38.1 ± 2.1, and 44.4 ± 2.0 mmHg, respectively, with exercise. Thus, exercise potently elevates PA pressures in individuals with COPD.
Kessler and co-workers (CitationKessler et al 2001) studied the change in pulmonary hemodynamics with time in a group of 131 patients with moderate COPD (FEV1%, 34.6 ± 15.7% and PaO2 67.0 ± 10.4 mmHg). Upon initial right heart catheterization, no patient had a resting mPAP >20 mmHg. After a mean interval of 6.8 ± 2.9 years, 33 (25%) patients had developed PH (mPAP >20) with a range of 20–32.5 mmHg. The average rate of increase in pulmonary arterial pressure was 0.4 mmHg/year. Patients who developed resting PH had higher resting and exercise mPAP and lower resting and exercise PaO2.
Thus, although numerous studies have measured pulmonary hemodynamics in patients with COPD, the prevalence of PH is not accurately known. Analysis of these studies is confounded by different sub-groups of individuals with varying severities of COPD and oxygenation and generally small numbers of patients. Nonetheless, the minimal prevalence of PH in patients with at least 1 hospitalization for COPD has been estimated to be 10%–30% (CitationNaeije 2005) and is as high as 90% in patients undergoing evaluation for lung volume reduction surgery (CitationScharf et al 2002). Exercise induced PH may occur in two-thirds of patients with COPD even when pulmonary pressures are normal at rest (CitationOswald-Mammosser et al 1991; CitationChristensen et al 2004).
Diagnostic testing
Physical examination
In a retrospective analysis of 27 patients with COPD and severe PH, Chaouat and colleagues (CitationChaouat et al 2005) identified 16 individuals with other diseases or processes including appetite suppressant exposure, collagen vascular disease, portal hypertension, left ventricular disease, thromboembolic disease, restrictive lung disease, and sleep apnea syndrome that could cause or contribute to the development of PH. Therefore, a careful clinical history is required to identify or exclude recognized causes of PH in individuals with COPD. A thorough medication history including prior use of anorexigens or chemotherapeutic agents should be elicited. Potential exposures to hepatitis B and C and HIV need to be evaluated with serologies. A high prevalence of pulmonary embolism, 25%, has been detected by spiral computed tomography in patients with unexplained exacerbations of COPD (CitationTillie-Leblond et al 2006). Therefore, a careful evaluation to exclude chronic thromboembolic PH is warranted in all individuals with COPD. Other pulmonary processes such as concurrent interstitial lung disease should be pursued. Cottin and co-workers (CitationCottin et al 2005) described 61 patients with both emphysema of the upper lung zones and diffuse parenchymal lung disease with fibrosis of the lower lung zones, of whom approximately 47% had PH. A careful history of skin, joint, and muscle symptoms should be obtained to exclude connective tissue disease. Smoking is not only a leading cause of COPD, it is also a significant risk factor for the development of ischemic heart disease; therefore, a complete cardiac history and review of symptoms is also important in the evaluation of PH in patients with COPD. Both systolic and diastolic left ventricular function should be assessed.
The signs and symptoms of PH in patients with COPD are subtle and are often obscured by the clinical manifestations of the lung disease. Exertional breathlessness may be due to worsening airflow limitation and air trapping or to the development of PH (CitationSalvaterra and Rubin 1993). Although the predictive value of specific symptoms and physical examination findings in detecting pulmonary vascular disease in a patient with COPD is not known, several signs and symptoms suggests the presence of PH (CitationSalvaterra and Rubin 1993). Decreasing functional capacity with stable pulmonary function testing suggests pulmonary vascular involvement. Signs of right ventricle enlargement or overload such as the presence of a right ventricular lift, prominent P2, right sided S4 gallop, and the murmur of tricuspid regurgitation suggest the presence of PH (CitationSalvaterra and Rubin 1993). Elevated jugular venous pressure, hepatojugular reflux, and a pulsatile liver are often signs of tricuspid insufficiency.
Chest imaging
The characteristic chest radiographic findings of PH are enlargement of the central pulmonary arteries causing hilar prominence and rapid tapering of the arteries in the lung periphery. Co-existing emphysematous changes accentuate oligemia within the lung parenchyma. The hilar thoracic index, the ratio of the distance between the start of the divisions of the right and left pulmonary arteries and the transverse diameter of the chest, has been used to quantify widening of the mediastinum (CitationChetty et al 1982). PH is suggested by a hilar-thoracic index >0.36. Widening of the descending right pulmonary artery to >16 mm or the left descending pulmonary artery to >18 mm suggest PH in patients with COPD (CitationMatthay et al 1981). Chhabra and De (CitationChhabra and De 2004) confirmed the high sensitivity and positive predictive value, 100%, of the findings of increased hilar-thoracic index and width of the descending branch of the right pulmonary artery (>20 mm) in 50 patients with COPD but showed that both measures were insensitive and had a low negative predictive value.
Measurement of pulmonary vessels by chest CT imaging has also been used to detect the presence of pulmonary hypertension. In a study of 36 patients with PH due to variable causes (4 had COPD), enlargement of the main pulmonary artery diameter to ≥29 mm had a sensitivity of 87%, a specificity of 89%, and a positive predictive value of 97% (CitationTan et al 1998). Additionally, the presence of pulmonary hypertension was predicted if the ratio of the diameters of the segmental artery to the corresponding bronchus was >1 in 3 or more lobes (CitationTan et al 1998). Other investigators have suggested that because the diameter of the main pulmonary artery varies normally, an absolute size threshold is not useful for the diagnosis of PH (CitationHaimovici et al 1997). Ng and co-workers (CitationNg et al 1999) determined the ratio of the diameter of the main pulmonary artery to the diameter of the ascending aorta in a group of 50 patients with various pulmonary and cardiovascular diseases who had undergone right heart catheterization. A ratio of the pulmonary artery to aortic diameter >1 was 70% sensitive and 92% specific for PH. The positive predictive value was 96% and the negative predictive value was 52%. Thus, the presence of an enlarged main pulmonary artery with a diameter greater than that of the ascending aorta strongly suggests the presence of PH but a ratio <1 does not exclude the presence of PH.
Electrocardiogram
PH associated with COPD may cause right ventricular hypertrophy or strain. The electrocardiographic manifestations of cor pulmonale are relatively specific, 86%, but not sensitive, 51%, for PH and do not correlate with the severity of PH (CitationOswald-Mammosser et al 1987; CitationHimelman et al 1988). Features of the electrocardiogram in patients with COPD that suggest PH include: A) P pulmonale, P-wave amplitude >2.5 mm in leads II, III, and/or aVF; B) S1, S2, S3 pattern; C) a S1, Q3 pattern; D) incomplete or complete right bundle branch block; E) evidence of RVH, R axis deviation ≥100°, dominant R wave in lead V1 ≥7 mm in amplitude, ST segment depression and T wave inversion in leads V1 to V4, and deep S waves in leads V5, V6, I and aVL with a QRS duration <0.12 s; and F) low voltage QRS (CitationHarrigan and Jones 2002; CitationBarbera et al 2003).
Echocardiography
Noninvasive estimation of systolic pressures by Doppler echocardiography has been studied extensively for the detection of PH in individuals with COPD. Several techniques are used, but most frequently, tricuspid regurgitant flow is detected and the maximum jet velocity is measured by continuous wave Doppler. Using the Bernoulli principle, the peak velocity, v, is used to calculate the trans-tricuspid gradient, 4v2. The systolic PAP is considered equal to the RV systolic pressure as long as there is no evidence of RV outflow obstruction. RV systolic pressure is calculated as the sum of the trans-tricuspid gradient plus the estimated right atrial pressure (RAP). Early studies used a fixed estimation of 15 mmHg for RAP whereas subsequent studies estimate the RAP based upon the size of the inferior vena cava during inspiration: no collapse, 15 mmHg; partial collapse 10 mmHg; and complete collapse 5 mmHg (CitationBredikis and Liebson 1998; CitationArcasoy et al 2003). In addition to these estimations, determination of sPAP by Doppler echocardiography is limited by technical factors. Tricuspid insufficiency must be present to measure a maximal jet velocity. Chest wall configuration, especially the presence of air trapping and cardiac orientation, may affect the utility of this technique in patients with COPD (CitationArcasoy et al 2003).
Doppler echocardiographic success in estimating sPAP in patients with COPD ranges from 26%–66% (CitationLaaban et al 1989; CitationTramarin et al 1991; CitationBach et al 1998; CitationArcasoy et al 2003). In a series of 374 patients undergoing evaluation for lung transplantation, Arcasoy and colleagues (CitationArcasoy et al 2003) were technically able to estimate sPAP in only 38% of patients with obstructive lung disease but were successful in 54% of patients with interstitial lung disease and 67% with pulmonary vascular disease. A residual volume >150% of predicted reduced the success rate of Doppler echocardiography. In an unselected series of 73% patients with COPD seen in an outpatient clinic, sPAP could be estimated in 56, 77% (CitationHigham et al 2001). Although there were no differences in spirometry and diffusing capacity between the measurable and non-measurable groups, lung volumes were not determined.
Laaban and colleagues (CitationLaaban et al 1989) compared the measurement of PAP by Doppler echocardiography with right heart catheterization in 41 patients with stable COPD. The directly measured mean sPAP was 38.5 ± 14.9 mmHg and 21, 51%, had pressures ≥35mmHg. sPAP could be estimated by Doppler echocardiography in 27 patients, 66%, and correlated significantly with the direct hemodynamic measurements, r = 0.65, p < 0.001. In a select subgroup of patients who were undergoing evaluation for lung volume reduction surgery and had both Doppler echocardiography and right heart catheterization, Bach and co-workers (CitationBach et al 1998) did not find a significant correlation between the actual and estimated sPAP but suggested that this difference was due to a single outlying patient. A strong correlation between right heart catheterization and Doppler echocardiographic measurement of sPAP, r = 0.8, was seen a group of 106 patients undergoing evaluation prior to lung transplantation, of whom 45, 42.5%, had obstructive lung disease (CitationBen-Dor et al 2006).
Transcutaneous jugular venous Doppler echo is another sonographic technique to estimate pulmonary pressures (CitationMatsuyama et al 2001). During a breath hold at the end of expiration, jugular venous flow velocities are measured by Doppler echo during diastole and systole. An increasing diastolic to systolic flow ratio correlates significantly with mPAP measured during RHC (CitationMatsuyama et al 2001). Using a diastolic to systolic flow velocity ratio of 1.0, the sensitivity of this technique was 71.4% and the specificity was 95.3%. Limitations of transcutaneous jugular venous Doppler echo include variable ratios in patients with mild PH (25–35 mmHg) and variable flow rates with tachycardias and dysrhythmias.
Histopathology
In 1965, CitationHicken et al (1965) described the histopathologic changes in the pulmonary vasculature of patients with chronic bronchitis or emphysema and evidence of right ventricular hypertrophy. They noted a circular layer of smooth muscle in the media of the pulmonary arterioles that was not present in the smaller pulmonary arteries. Both the pulmonary arterioles and small pulmonary arteries had a layer of longitudinal smooth muscle beneath the internal elastic lamina but neither one of the vessels had significant intimal fibrosis. Because these vascular changes were similar to the alterations reported in PH associated with high altitude, CitationHicken et al (1965) hypothesized that PH associated with hypoxic lung diseases such as COPD was caused solely by hypoxic vasoconstriction and arteriolar muscular hyperplasia. Subsequent observations that high altitude PH resolves after descent to lower altitudes (Grover et al 1996) suggested that PH associated with COPD might reverse with correction of hypoxemia. Contrary to the expected result, pulmonary pressures decreased but did not normalize in individuals with PH associated with COPD who were treated with supplemental oxygen therapy (CitationAbraham et al 1968). Subsequent studies of acute and chronic oxygen therapy in patients with COPD also failed to show resolution of PH (CitationLejeune et al 1984; CitationTimms et al 1985). These physiologic findings suggested that the PH associated with COPD that persists after correction of hypoxemia is due to fixed vascular remodeling and spurred further, more detailed histopathological studies (CitationWilkinson et al 1988; CitationPeinado et al 1999; CitationHale et al 1980). In 1988, Wilkinson (CitationWilkinson et al 1988) observed similar histopathologic findings in lung tissue from 10 patients with hypoxic cor pulmonale and COPD regardless of treatment with supplemental oxygen. The media of the muscular pulmonary arteries was normal or atrophic but the intima revealed an active deposition of longitudinal muscle, fibrosis and elastosis. The media of the normally poorly muscularized arterioles revealed a circular muscular coat bounded by a new internal elastic lamina. Luminal narrowing was present with frequent recanalization of the arteriolar lumen (CitationWilkinson et al 1988). Subsequent studies of lung tissue from individuals with moderate to severe COPD have consistently reported similar findings (CitationWright et al 1983; CitationHale et al 1984; CitationWright et al 1992). Interestingly, when lung sections from smokers with COPD were compared with non-smokers, the density of the fully muscularized (0–300 μm diameter) pulmonary arteries and the thickness of the medial muscle layer was doubled and the depth of intimal fibrosis was tripled (CitationHale et al 1984). This intimal thickening is due to both smooth muscle cell proliferation and increased elastin and collagen deposition (CitationSantos et al 2002).
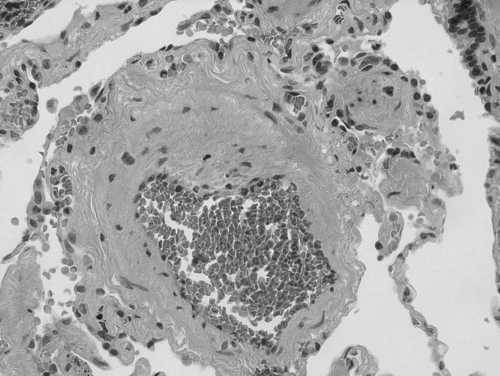
Importantly, similar findings of vascular remodeling are present in the pulmonary vasculature of smokers (CitationSantos et al 2002; CitationHale et al 1980) and patients with mild COPD (CitationSantos et al 2002). The earliest vascular changes are intimal thickening, luminal narrowing, and arteriolar muscularization (CitationHale et al 1980; CitationMagee et al 1988; CitationBarbera et al 1994; CitationPeinado et al 1998). The degree of intimal thickening correlates with the severity of small airways disease and emphysema (CitationHale et al 1980). These very early histopathologic findings suggest that the morphologic changes in the pulmonary arteries are initiated by the toxic effects of tobacco smoke and progress in parallel with the parenchymal changes of COPD (CitationHale et al 1980) ().
Figure 1 Histologic evidence of vascular changes in a small pulmonary artery in a patient with COPD and associated pulmonary hypertension. Hematoxylin and eosin staining demonstrates marked medial and intimal thickening with resulting luminal narrowing (40X, original magnification). Photomicrograph was provided courtesy of Hai Bui, MD, Department of Pathology, Cincinnati VAMC, Cincinnati Ohio.
Pathophysiology
The pulmonary vasculature of patients with COPD associated PH is markedly abnormal and shows increased intimal and medial thickening that cause luminal narrowing and vascular obstruction of the small pulmonary arteries (CitationWright et al 1992). These vascular changes lead to an increase in pulmonary vascular resistance (PVR) and elevation of pulmonary artery pressures (PAP). The severity of vascular abnormalities does not correlate directly with the pulmonary pressure at rest (CitationWilkinson et al 1988; CitationWright et al 1992). However, the degree of intimal thickening is proportional to the increase in pulmonary pressure during exercise and this relationship is thought to be due in part to decreased distensability and recruitment within the abnormal pulmonary vasculature (CitationKubo et al 2000).
In most cases, PH associated with COPD develops slowly over time and the pressure increases approximately 0.4 mmHg yearly (CitationKessler et al 2001). Moderately severe COPD is associated with PH in 10%–35% of patients (CitationScharf et al 2002; CitationEddahibi et al 2003). PH leads to pressure overload of the right ventricle (RV). RV muscular hypertrophy occurs in response to the increased pulmonary pressures (CitationDias et al 2002) and occurs in up to 40%–70% of patients with COPD at autopsy (11,62). Hypertrophy is followed by contractile dysfunction of the RV (CitationVoelkel et al 2006). The contractile dysfunction leads to RV dilation, a decrease in cardiac output, and an increase in right sided filling pressures (CitationVoelkel et al 2006). The dilation and pressure overload of the RV causes left ventricular (LV) diastolic dysfunction (CitationLouie et al 1995). Eventually, the ability of the RV to compensate is overwhelmed and RV failure ensues.
Pathogenesis
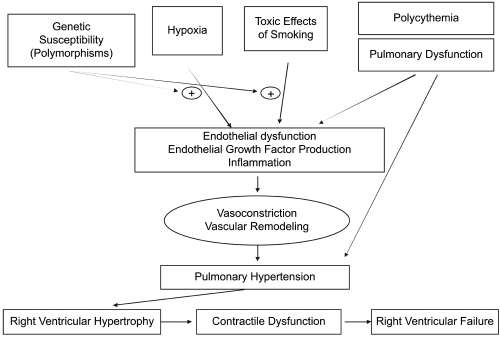
The pathogenesis of the vascular abnormalities associated with COPD have not been fully elucidated but appear to be caused by the combined effects of hypoxia (CitationBurrows 1974), pulmonary dysfunction with air trapping (CitationWright 1993) and the toxic effects of smoking (CitationSantos et al 2002; CitationHale et al 1980) leading to inflammation (CitationPeinado et al 1999), endothelial dysfunction (CitationDinh-Xuan et al 1991; CitationPeinado et al 1998), and angiogenesis (CitationSantos et al 2003) ().
Figure 2 Pathophysiology of PH and right ventricular dysfunction associated with COPD. The combined action of hypoxemia, toxic tobacco smoke, pulmonary dysfunction, and polycythemia lead to endothelial dysfunction, growth factor production, and inflammation in the pulmonary vasculature. These processes cause elevation of pulmonary arterial pressures. PH may cause RV hypertrophy and subsequent RV contractile dysfunction and RV failure.
Hypoxia
Unlike the systemic circulation, acute hypoxia causes vasoconstriction in the pulmonary circulation (CitationVon Euler and Liljerstarand 1946) and a transient increase in pulmonary vascular resistance (CitationPease et al 1982). Although the effect of recurrent, transient episodes of hypoxia in the development of pulmonary vascular dysfunction is unclear, chronic hypoxia has been studied extensively and is a known cause of PH (CitationHeath et al 1973; CitationThompson et al 1989; CitationVender 1994; CitationReeves and Grover 2005). The pathophysiologic mechanisms of hypoxic pulmonary vasoconstriction are not fully understood but are believed due to direct and indirect effects of lowered oxygen levels on the pulmonary vascular smooth muscle cells (CitationHida et al 2002). The direct action occurs through potassium and calcium channels (CitationWeir and Archer 1995). In the presence of hypoxia, potassium channels within pulmonary smooth muscle cells close causing membrane depolarization and an influx of calcium, stimulating smooth muscle cell contraction (CitationWeir and Archer 1995). Hypoxia may also act indirectly on the pulmonary vasculature by stimulating the production of transcription factors (CitationYan et al 1998; CitationMechtcheriakova et al 1999) such as hypoxia-inducible factor-1 (HIF-1) (CitationSemenza 2000; CitationYu et al 1998)and the release of multiple endogenous mediators including angiotensin II (CitationMorrell et al 1995), endothelin-1 (CitationChen et al 1995; CitationDiCarlo et al 1995; CitationElton etal 1992; CitationHu et al 1998), and growth factors (CitationMechtcheriakova et al 1999; CitationSantos et al 2003). These mediators trigger vasoconstriction, vascular remodeling, and angiogenesis.
Disturbances in pulmonary function/air trapping
Animal models of smoke-induced emphysema and PH suggest that PH associated with COPD is due in some part to hyperinflation and gas trapping that compress the pulmonary vessels (CitationWright 1993). However, PH has not been shown to improve after lung volume reduction surgery (LVRS) despite improvement in pulmonary function and reduction in gas trapping (CitationHaniuda et al 2003).
Polycythemia
Secondary polycythemia occurs in COPD with hypoxia and causes an increase in blood viscosity. However, to date, there are few studies evaluating the relationship between blood viscosity, polycythemia, and PH. A retrospective analysis of 41 patients with emphysema revealed that increasing levels of hemoglobin independently correlated with increasing PVR and PAP (CitationNakamura et al 2000). Polycythemia also inhibits the vasodilatory effect of acetylcholine in patients with COPD. This effect is believed to be due to the inactivation of endothelial derived NO by hemoglobin (CitationDefouilloy et al 1998).
Toxic effects of cigarette smoke
Tobacco smoke is not only toxic to the airways and lung parenchyma but also has an effect on the pulmonary vasculature and may play a significant role in the development of PH. The small pulmonary arteries of smokers develop intimal thickening due to elastin deposition, increased collagen production, and smooth muscle proliferation regardless of the development of obstructive lung disease (CitationSantos et al 2002; CitationHale et al 1980). The etiology of these changes is not fully understood; however, tobacco smoke does induce vascular inflammation (CitationPeinado et al 1999) and impairs endothelial function (CitationBarbera et al 2001). The number of CD8+ T-lymphocytes infiltrating the adventitia of the small muscular pulmonary arteries is increased in both smokers and patients with COPD. The degree of inflammatory infiltrate correlates positively with pulmonary arterial intimal thickness in patients with COPD (CitationPeinado et al 1999). It is possible that this inflammatory infiltrate serves as a source of cytokines and growth factors leading to the medial hypertrophy, thickening, and luminal obstruction observed in smokers (CitationPeinado et al 1999).
Vasoactive mediators also appear to play a role in smoking related pulmonary vascular changes. Brief exposure to tobacco smoke up-regulates gene expression of the vasoconstrictor/vasoproliferative agents endothelin-1 (ET-1) and vascular endothelial growth factor (VEGF) in Sprague-Dawley rats (CitationWright et al 2002). The expression of VEGF is also increased in the pulmonary arteries of smokers and correlates with the thickness of the pulmonary arteries and the degree of obstructive lung disease (CitationSantos et al 2003). Smoke exposure also alters NO production in endothelial cells. When the lungs of smokers are compared with those of non-smokers, smokers have lower endothelial nitric oxide synthase (eNOS) levels (CitationBarbera et al 2001). Further, in vitro exposure of pulmonary artery endothelial cells to tobacco smoke causes irreversible inhibition of eNOS activity (CitationSu et al 1998).
Genetics
Multiple genetic polymorphisms within several different genes correlate with the development of pulmonary vascular disease in patients with COPD associated PH. The angiotensin converting enzyme (ACE) gene contains insertion (I) or deletion (D) polymorphisms that create three genotypes (DD, II, and DI) (CitationRigatet al 1992). The deletion polymorphism is associated with increases in circulating angiotensin converting enzyme (ACE) levels (CitationRigat et al 1990). The DI polymorphism is associated with the development of PH in Caucasians with COPD (CitationTkacova et al 2005). Additionally, the DI genotype may confer susceptibility to exercise induced PH in patients with COPD (CitationKanazawa et al 2000). Polymorphisms of the endothelial nitric oxide synthase (eNOS) gene are also present in patients with COPD. The BB polymorphism of the eNOS gene is associated with PH in patients with COPD. (CitationYildiz et al 2003). The serotonin transporter (5-HTT) plays a role in smooth muscle hyperplasia and vascular remodeling (CitationEddahibi et al 2001). L allelic polymorphisms of the 5-HTT gene promoter cause 5-HTT over-expression. LL and LS 5-HTT are more common in patients with idiopathic pulmonary arterial hypertension (IPAH) than in controls (CitationEddahibi et al 2001). In patients with COPD, the LL 5-HTT gene polymorphism is associated with the presence of PH in hypoxemic COPD patients and correlates with the severity of PH (CitationEddahibi et al 2003). The GG polymorphism of the interleukin-6 (IL-6) gene leads to increased levels of IL-6 and occurs more commonly in COPD patients with PH than in those with COPD alone (CitationYildiz et al 2003).
Pathobiology
Endothelial dysfunction
The production of vasoactive chemokines by pulmonary endothelial cells is dysregulated during hypoxic stress (CitationChen et al 1995; CitationDiCarlo et al 1995; CitationElton et al 1992) and toxin exposure (CitationWright et al 2002; CitationWright et al 2004; CitationSanto et al 2003). Endothelial dysfunction also occurs in patients with COPD (CitationDinh-Xuan et al 1991; CitationPeinado et al 1999) and with COPD associated PH (CitationGiaid et al 1993; CitationClini et al 2000).
Nitric oxide (NO) is a potent vasodilating mediator with antiproliferative effects (CitationMurad 1997). NO is synthesized in the pulmonary vasculature by endothelial nitric oxide synthase (eNOS). eNOS expression is reduced in smokers (CitationBarbera et al 2001) and patients with COPD (CitationGiaid and Saleh 1995). Fractional exhaled nitric oxide (FENO) is also reduced in patients with COPD and PH. FENO levels correlate inversely with the degree of PH and the development of cor pulmonale (CitationClini et al 2000). It is postulated that the reduction in FENO seen in patients with COPD, PH, and cor pulmonale is due to a decrease in endothelial production of NO.
Prostacyclin is another potent endothelial cell-derived vasodilating mediator that inhibits platelet aggregation (CitationGerber et al 1980) and has antiproliferative effects (CitationHara et al 1995). It is synthesized by prostacyclin synthase via the arachidonic acid pathway (CitationAlhenc-Gelas et al 1982). Prostacyclin synthase is reduced in patients with IPAH (CitationTuder et al 1999) and in patients with emphysema (CitationLee et al 2005; CitationNana-Sinkam et al 2006). However, little is known about the role that prostacyclin plays in COPD associated PH.
Endothelin (ET-1) is a potent vasoconstricting (CitationLeDouceur et al 1993) and mitogenic mediator produced by the endothelium (CitationDubin et al 1989; CitationBoscoe et al 2000; CitationWedgewood et al 2001). Endothelin levels are increased in patients with COPD (CitationSpiropoulos et al 2003), and rise further during COPD exacerbations (CitationRoland et al 2001). Endothelin levels are increased in patients with pulmonary arterial hypertension (PAH) and PH associated with COPD, interstitial lung disease (ILD) and congestive heart failure (CHF) (CitationGiaid et al 1993).
Growth factors
It is currently unclear what role growth factors play in the development and maintenance of the pulmonary vascular abnormalities observed in COPD and PH (CitationPapaioannou et al 2006). VEGF is an endothelial growth factor that stimulates angiogenesis and permeability (CitationPapaioannou et al 2006). VEGF expression is increased in the pulmonary arteries of patients with COPD (CitationSantos et al 2003). Hypoxia also stimulates VEGF production via the transcription factor Hypoxia-Inducible Factor-1 (HIF-1) (CitationCarmeliet et al 1998; CitationIyer et al 1998; CitationSemenza 2000). Although VEGF is a potent endothelial cell mitogen and is increased in smokers and patients with COPD and hypoxia, VEGF may also have a protective role in the pulmonary vasculature. When animals treated with a VEGF inhibitor are exposed to hypoxic conditions, severe PH develops (CitationTaraseviciene-Stewart et al 2001). Additional studies are needed to understand VEGF’s potential protective role in the development of pulmonary vascular changes in patients with COPD.
Platelet derived growth factor (PDGF) is another mediator of angiogenesis that may be involved in vascular remodeling in hypoxia and COPD. PDGF is a potent mitogen for smooth muscle cells (CitationHannink and Donoghue 1989). Indirect evidence that PDGF is important in the development of PH comes from rat models of hypoxic and monocrotaline induced PH that demonstrate reversal of PH after inhibition of PDGF (CitationSchermuly et al 2005).
Inflammation
The role of inflammation in the pathogenesis of COPD associated PH is uncertain. The degree of inflammatory infiltrate within the pulmonary vascular adventitia of patients with COPD correlates with the severity of the intimal thickening in the pulmonary arteries (CitationPeinado et al 1999). Additionally, systemic inflammatory markers, C-reactive protein (CRP), and tumor necrosis factor alpha (TNF-alpha), are increased in COPD patients with PH (CitationJoppa et al 2006) when compared with COPD patients without PH. However, the significance of these findings is unclear and additional investigations are needed to determine the etiologic significance of inflammation in the vascular remodeling that occurs with smoking, COPD, and COPD associated PH.
Management
The treatment of patients with COPD associated PH requires a multifaceted approach that includes smoking cessation, optimization of COPD management, and correction of hypoxemia. Additionally, recent advances in the understanding of the pathogenesis of COPD associated PH provide several potential biologic targets for future therapies.
Smoking cessation
Toxic effects of tobacco smoke (CitationSantos et al 2002; CitationHale et al 1980) play a role in the development of the pulmonary vascular disease that occurs in COPD associated PH. Smoking cessation is the single most important intervention to slow progression of COPD (CitationAnthonisen et al 1994; CitationAnthonisen et al 2002) and thus likely decreases the risk of developing COPD associated PH. Once an individual quits smoking, resumption of cigarettes should be continually discouraged as there is a steeper rate of decline in FEV1 in patients who restart smoking than in those who smoke without interruption (CitationSherrill et al 1996).
Supplemental oxygen
Correction of hypoxemia with supplemental oxygen is also recommended therapy for patients with COPD and PH. Long term oxygen therapy is recommended for patients with COPD whose resting PaO2 is <55 mmHg or is between 56 and 59 mmHg during rest, sleep, or exertion and who have clinical evidence of cor pulmonale or polycythemia (CitationCelli et al 2004). Long term oxygen therapy (LTOT) improves survival in hypoxic COPD patients and is associated with a mild improvement in pulmonary hemodynamics (CitationMedical Research Council Working Party 1981; CitationNocturnal Oxygen Therapy Group 1980). Additionally, supplemental oxygen during exercise decreases PAP (CitationBurrows et al 1972; CitationFujimoto et al 2002), increases exercise tolerance (CitationFujimoto et al 2002) and improves RV function (CitationOlvey et al 1980). Pulmonary pressures are known to increase with sleep in patients with COPD associated PH (CitationRaeside et al 2002). Acutely, oxygen therapy abolishes this nocturnal rise in pulmonary pressures (CitationRaeside et al 2002). Long term nocturnal oxygen decreases pulmonary pressures in patients with moderate to severe COPD, PH, and daytime PaO2 >60 mmHg but who experience nocturnal desaturation (CitationFletcher et al 1992). A 3 year randomized trial involving 51 patients with moderate to severe COPD and PH demonstrated that patients with nocturnal desaturation treated with supplemental oxygen experienced a 3.7 mm Hg decrease in PAP whereas those patients with nocturnal desaturation who were treated with room air experienced a 3.9 mm Hg increase in PAP (CitationFletcher et al 1992). LTOT also ameliorates the progression of COPD associated PH. In 1985, Weitzenblum and colleagues (CitationWeitzenblum et al 1985) evaluated the progression of COPD associated PH before and after 2–4 years of LTOT. Prior to LTOT, the PAP increased by approximately 1.47 ± 2.3 mmHg per year whereas the PAP decreased by 2.15 ± 4.4 mmHg per year with oxygen therapy. Despite the beneficial effects of oxygen therapy in patients with hypoxemia and COPD, LTOT does not lead to the normalization of pulmonary pressures (CitationAbraham et al 1968) or the reversal of morphologic derangements in the pulmonary vasculature (CitationWilkinson et al 1988). Therefore, additional interventions are needed to treat the fixed vascular remodeling in COPD associated PH.
Systemic vasodilators
Calcium channel blockers (CCB) have been studied extensively in COPD associated PH. Acutely, the administration of a short acting CCB (nifedipine) decreases the PAP, increases cardiac index (CI) (3.7 ± 0.2 to 4.6 ± 0.3 L/min/m2)and decreases PVR (426 ± 52 to 294 ± 28 dyne•s−1•cm−5) (CitationSturani et al 1983). The acute improvement in hemodynamics with CCB has been reproduced by several other investigators (CitationGarzaniti et al 1985; CitationMuramoto et al 1985). Despite the acute beneficial effects of CCB’s, they are ineffective long term pulmonary vasodilators (CitationAgostoni et al 1989) and do not effect progression of COPD associated PH (CitationSturani et al 1983). Additionally, CCB’s can alter ventilation perfusion (VQ) matching deleteriously and worsen hypoxemia (CitationMelot et al 1984). Due to these negative side effects, CCB’s are not recommended in the routine management of COPD associated PH.
Hydralazine has been tested in stable patients with PH secondary to COPD. Acutely, hydralazine improved pulmonary hemodynamics and oxygenation in a small group of hypoxemic COPD patients (CitationKeller et al 1984). However, nearly one quarter of the patients (3/14) could not tolerate hydralazine due to tachycardia. Hydralazine neither improves pulmonary hemodynamics in patients during COPD exacerbations nor increases exercise capacity in patients with severe COPD and associated PH (CitationCerda et al 1985; CitationDal Nogare and Rubin 1986).
Modulation of the renin-angiotensin systems has also been studied in COPD associated PH. Animal studies suggested that angiotensin converting enzyme (ACE) inhibition may be beneficial in the treatment of COPD associated PH (CitationNong et al 1996); however the intravenous administration of captopril did not result in an acute change on pulmonary hemodynamics in patients with COPD (CitationPatakas et al 1988). Furthermore, chronic treatment of patients with COPD associated PH with losartan did not result in a statistically significant change in echocardiography, exercise capacity, or respiratory symptoms (CitationMorrell et al 2005).
Theophylline
Theophylline is a phosphodiesterase inhibitor used in the treatment of COPD. In a small study, it has been shown to reduce pulmonary artery pressures acutely with intravenous bolus administration (CitationLiu et al 1992). Further studies are needed to validate this finding and to assess the effect of chronic theophylline therapy on COPD associated PH.
Future therapeutic targets
The most likely targets of future therapies in COPD associated PH are the endothelium and pulmonary vascular remodeling. Interventions aimed at these targets include nitric oxide (NO), phosphodiesterase inhibitors (PDEI) and statins.
NO is a selective pulmonary vasodilator that does not cause systemic hypotension (CitationPepke-Zaba et al 1991) and is effective treatment for idiopathic PH (CitationPepke-Zaba et al 1991). In patients with COPD associated PH, 3 months of therapy with inhaled pulsed NO via nasal cannula improved pulmonary hemodynamics. The mean pulmonary artery pressure (mPAP) decreased from 27.6 to 20.6 mmHg, PVR decreased from 276.9 to 173 dyne•s−1•cm−5 and no worsening of hypoxemia occurred (CitationVonbank et al 2003). However, when Barbera and colleagues (CitationBarbera et al 1996) administered inhaled NO (40 ppm) to patients with COPD, VQ matching worsened.
Phosphodiesterase inhibitors (PDEI) have been tested in small numbers of patients with COPD associated PH. PDEI prolong the effect of NO by inhibiting the degradation of NO’s second messenger (cGMP) and promoting smooth muscle relaxation (CitationRabe et al 1994). In 6 patients with COPD associated PH, pulmonary hemodynamics improved after the initial dose of intravenous sildenafil and, after 3 months of oral therapy with sildenafil 50mg twice daily, the mPAP decreased from 30.2 ± 5.5 to 24.6 ± 4.2 mmHg, PVR declined from 401 ± 108 to 264 ± 52 dyne•s−1•cm−5 and the six minute walk distance increased from 351 ± 49 to 433 ± 52 m (CitationAlp et al 2006).
Despite these promising, preliminary results demonstrating beneficial effects of NO and PDEI in COPD associated PH, larger studies are needed to determine their safety and efficacy in COPD associated PH. Prostacyclin and endothelin antagonists are also potential targets for therapy; however, very limited information exists on the use of these agents in COPD associated PH.
3-hydroxy-3-methyl-glutaryl – coenzyme-A (HMG-CoA) reductase inhibitors (statins) have anti-inflammatory, antioxidant, and antithrombogenic effects, restore endothelial function (CitationBonetti et al 2003), and may ameliorate the deleterious effects of tobacco smoking on the lung parenchyma and vasculature (CitationLee et al 2005). In rats exposed to 16 weeks of tobacco smoke with or without concomitant statin treatment, parenchymal destruction, vascular changes, and inflammation were attenuated by statin administration (CitationLee et al 2005). Further studies of statins are needed to determine if they promote vascular remodeling in COPD and PH.
Summary
Pulmonary hypertension is a common feature of advanced COPD and is estimated to affect ≥20% of patients with advanced COPD (CitationWeitzenblum et al 1981; CitationScharf et al 2002; CitationOswald-Mammosser et al 1991). The vast majority of PH associated with COPD is mild to moderate (mPAP 20–35 mmHg). Severe PH (mPAP ≥40 mmHg) occurs in <5% of patients with COPD (CitationChaouat et al 2005). Severe PH in patients with COPD reduces median survival by approximately 40 months (CitationChaouat et al 2005). The pathogenesis of PH associated with COPD has not been clearly elucidated but is likely due to the combined effects of inflammation (CitationPeinado et al 1999), endothelial cell dysfunction (CitationDinh-Xuan et al 1991; CitationPeinado et al 1998), and angiogenesis (CitationSantos et al 2003) that lead to intimal thickening, luminal narrowing, and arteriolar muscularization (CitationBarbera et al 1994; CitationPeinado et al 1998; CitationMagee et al 1988; CitationHale et al 1980). Long-term oxygen therapy is a proven therapy for COPD associated PH (CitationMedical Research Council Working Party 1981; CitationNocturnal Oxygen Therapy Group 1980). Systemic vasodilators have not been found to be effective therapy in COPD related pulmonary hypertension (CitationSturani et al 1983). Selective pulmonary vasodilators are currently being evaluated. Inhaled NO (CitationVonbank et al 2003) and PDEI (CitationAlp et al 2006) are promising treatments for patients with COPD associated PH. However, further evaluation of the safety, efficacy, and optimal dosing of these medications is needed prior to routine use of these therapies in the management of PH associated with COPD.
Case study
This 54 year old man smoked up to 3 to 4 packs per day for the past 36 years and continues to smoke occasional cigarettes. He is breathless walking down the hallway and has dyspnea with most daily activities. He has no cough or sputum production. His medications include: a long acting beta agonist, short acting beta agonist, short acting anticholinergic, and an inhaled corticosteroid. The alpha-1 antitrypsin level was 218. Physical examination revealed a resting SaO2 of 85%. The chest was hyperresonant with markedly reduced breath sounds with a prolonged expiratory phase but no wheezes. The left ventricular impulse was subxiphoid and the second heart sound was increased. There was no clubbing or peripheral edema.
Pulmonary function studies:
| TLC | 8.35 | 110% | |
| FRC | 6.48 | 177% | |
| RV | 4.41 | 190% | |
| FEV1 | 1.34 | 32% (Prebronchodilator) | 1.35 32% (Postbronchodilator) |
| FVC | 3.84 | 73% (Prebronchodilator) | 4.62 88% (Postbronchodilator) |
| DLCO | 3.30 | 8.6% |
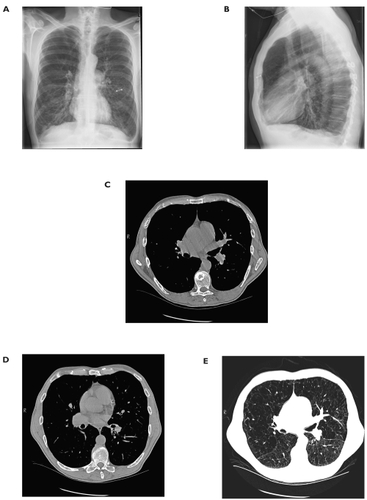
Echocardiogram showed normal left ventricular size and function with a left ventricular ejection fraction of 50%. The left ventricular wall thickness was normal and there were no regional wall motion abnormalities. Systolic and diastolic ventricular septal flattening was present suggesting right ventricular (RV) pressure overload. The RV was mildly dilated and systolic function was mildly to moderately reduced. The systolic pulmonary artery pressure was calculated to be 51–56 mmHg assuming a right atrial pressure of 5–10 mmHg. Agitated saline contrast study did not reveal an intracardiac shunt ().
Figure 3 A. Posterior-anterior chest X-ray and B. Lateral chest X-ray demonstrating enlarged pulmonary arteries and severe hyperinflation. The metallic opacification is a left nipple ring. C. Chest CT scan at the level of the pulmonary outflow tract demonstrating a main pulmonary artery diameter greater than the adjacent aorta. D. Chest CT scan at the level of the lower lobes demonstrating segmental pulmonary arteries that are larger than the accompanying bronchi. E. Chest CT at the same level as Figure 3C using parenchymal windows to demonstrate severe emphysematous changes throughout the lung parenchyma.
Assessment for coexisting etiologies of pulmonary hypertension was performed. Connective tissue disease serologies and HIV testing were negative. Ventilation perfusion scanning was interpreted as low probability for pulmonary embolism. Arterial blood gas sampling confirmed resting hypoxemia but did not show hypercarbia. Liver function testing was normal and there was no clinical evidence of hepatic dysfunction.
Treatment with long acting beta agonist, short acting beta agonist, short acting anticholinergic, and an inhaled corticosteroid was continued. Supplemental oxygen was instituted for resting hypoxemia. Smoking cessation was strongly encouraged and interventions were successful.
The patient’s dyspnea improved with smoking cessation, supplemental oxygen and inhaled therapies for COPD. He completed pulmonary rehabilitation and is now enrolled in a pulmonary rehabilitation maintenance program.
Acknowledgements
The authors thank Faye Warner-Jones for her expert secretarial assistance.
References
- AbrahamASColeRBBishopJMReversal of pulmonary hypertension by prolonged oxygen administration to patients with chronic bronchitisCirc Res196823147575661934
- AgostoniPDoriaEGalliCNifedipine reduces pulmonary pressure and vascular tone during short- but not long-term treatment of pulmonary hypertension in patients with chronic obstructive pulmonary diseaseAm Rev Respir Dis198913912052912331
- Alhenc-GelasFTsai CallahanKSStimulation of prostaglandin formation by vasoactive mediators in cultured human endothelial cellsProstaglandins198224723426819604
- AlpSSkryganMSchmidtWESildenafil improves hemodynamic parameters in COPD – an investigation of six patientsPulm Pharmacol Ther2006193869016291503
- AnthonisenNRConnettJEKileyJPThe effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline in FEV1: the Lung Health StudyJAMA199427214975057966841
- AnthonisenNRConnettJEMurrayRPSmoking and lung function of Lung Health Study participants after 11 yearsAm J Respir Crit Care Med2002166675912204864
- AnthonisenNRWrightECHodgkinJEIPPD Trial Group. Prognosis in chronic obstructive pulmonary diseaseAm Rev Respir Dis198613314203510578
- ArcasoySMChristieJDFerrariVAEchocardiographic assessment of pulmonary hypertension in patients with advanced lung diseaseAm J Respir Crit Care Med20031677354012480614
- BachDSCurtisJLChristensenPJPreoperative echocardiographic evaluation of patients referred for lung volume reduction surgeryChest1998114972809792564
- BarberaJAPeinadoVISantosSReduced expression of endothelial nitric oxide synthase in pulmonary arteries of smokersAm J Respir Crit Care Med20011647091311520741
- BarberaJAPeinadoVISantosSPulmonary hypertension in chronic obstructive pulmonary diseaseEur Respir J20032189290512765440
- BarberaJARiverolaARocaJPulmonary vascular abnormalities and ventilation-perfusion relationships in mild chronic obstructive pulmonary diseaseAm J Respir Crit Care Med199414942398306040
- BarberaJARogerNRocaJWorsening of pulmonary gas exchange with nitric oxide inhalation in chronic obstructive pulmonary diseaseLancet1996347436408618485
- Ben-DorIMordechaiMRaccahACheocardiography versus right-sided heart catheterization among lung transplantation candidatesAnn Thorac Surg20068110566016488722
- BonettiPOLermanLONapoliCStatin effects beyond lipid lowering: are they clinically relevant?Eur Heart J2003242254812590901
- BoscoeMJGoodwinATAmraniMEndothelins and the lungInt J Biochem Cell Biol200032416210661893
- BredikisAJLiebsonPRThe echocardiograpm in COPD: estimating right heart pressuresJ Respir Dis1998191918
- BurrowsBEarleRHCourse and prognosis of chronic obstructive lung disease: a prospective study of 200 patientsN Engl J Med19692803974045763088
- BurrowsBKettelLJNidenAHPatterns of cardiovascular dysfunction in chronic obstructive lung diseaseN Engl J Med1972286912185013974
- BurrowsBArterial oxygenation and pulmonary hemodynamics in patients with chronic airways obstructionAm Rev Respir Dis197411064704613234
- CalverleyPMAHowatsonRFlenleyDCClinicopathological correlations in cor pulmonaleThorax19924749481412090
- CarmelietPDorYHerbertJMRole of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesisNature1998394485909697772
- CelliBRMacNeeWcommittee membersStandards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paperEur Respir J2004239324615219010
- CerdaEEstebanAde la CalMAHemodynamic effects of vasodilators on pulmonary hypertension in decompensated chronic obstructive pulmonary diseaseCrit Care Med19851322133979067
- ChaouatABugnetASKadaouiNSevere pulmonary hypertension and chronic obstructive pulmonary diseaseAm J Respir Crit Care Med20051721899415831842
- ChenSJChenYFMengQCEndothelin-receptor antagonist bosentan prevents and reverses hypoxic pulmonary hypertension in ratsJ Appl Physiol1995792122318847282
- ChettyKGBrownSELightRWIdentification of pulmonary hypertension in chronic obstructive pulmonary disease form routine chest radiographsAm Rev Respir Dis1982126338417103259
- ChhabraSKDeSClinical significance of hilar thoracic index and width of right descending branch of pulmonary artery in chronic obstructive pulmonary diseaseChest200446917
- ChristensenCCRygMSEdvardsenARelationship between exercise desaturation and pulmonary haemodynamics in COPD patientsEur Respir J200424580615459136
- CliniECremonaGCampanaMProduction of endogenous nitric oxide in chronic obstructive pulmonary disease and patients with cor pulmonale. Correlates with echo-Doppler assessmentAm J Respir Crit Care Med20001624465010934068
- CottinVNunesHBrilletPYCombined pulmonary fibrosis and emphysema: a distinct underrecognised entityEur Respir J20052658659316204587
- Dal NogareARRubinLJThe effects of hydralazine on exercise capacity in pulmonary hypertension secondary to chronic obstructive pulmonary diseaseAm Rev Respir Dis198613338593954247
- DefouilloyCTeigerESediameSPolycythemia impairs vasodilator response to acetylcholine in patients with chronic hypoxemic lung diseaseAm J Respir Crit Care Med19981571452609603123
- DiasCAAssadRSCaneoLFReversible pulmonary trunk banding. II. An experimental model for rapid pulmonary ventricular hypertrophyJ Thorac Cardiovasc Surg2002124999100612407385
- DiCarloVSChenSJMengQCETA-receptor antagonist prevents and reverses chronic hypoxia-induced pulmonary hypertension in ratAm J Physiol1995269L69077491990
- Dinh-XuanATHigenbottamTWClellandCAImpairment of endothelium-dependent pulmonary-artery relaxation in chronic obstructive lung diseaseN Engl J Med19913241539472027358
- DoiMNakanoKHiramotoTSignificance of pulmonary artery pressure in emphysema patients with mild-to-moderate hypoxemiaRespiratory Medicine2003979152012924518
- DubinDPrattREKookeJPEndothelin, a potent vasoconstrictor, is a vascular smooth muscle mitogenJ Vasc Biol Med198911504
- EddahibiSChaouatAMorrellNPolymorphism of the serotonin transporter gene and pulmonary hypertension in chronic obstructive pulmonary diseaseCirculation200310818394414530202
- EddahibiSChaouatATuLInterleukin-6 gene polymorphism confers susceptibility to pulmonary hypertension in chronic obstructive pulmonary diseaseProc Am Thorac Soc20063475616921112
- EddahibiSHumbertMFadelESerotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertensionJ Clin Invest200110811415011602621
- EltonTSOparilSTaylorGRNormobaric hypoxia stimulates endothelin-1 gene expression in the ratAm J Physiol1992263R126041481936
- FishmanAPState of the art: chronic cor pulmonaleAm Rev Respir Dis197611477594788575
- FletcherECLuckettRAGoodnight-WhiteSA double-blind trial of nocturnal supplemental oxygen for sleep desaturation in patients with chronic obstructive pulmonary disease and a daytime PaO2 above 60 mm HgAm Rev Respir Dis1992145107061586049
- FujimotoKMatsuzawaYYamaguchiSBenefits of oxygen on exercise performance and pulmonary hemodynamics in patients with COPD with mild hypoxemiaChest20021224576312171817
- GarzanitiNel AllafDD’OrioVHemodynamic effects of nifedipine on secondary pulmonary hypertension in manActa Cardiol198540207153873156
- GerberJGVoelkelNFNiesNModeration of hypoxic vasoconstriction by infused arachidonic acid: role of PGI2J Appl Physiol198049107126995413
- GiaidASalehDReduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertensionN Engl J Med1995333214217540722
- GiaidAYanagisawaMLanglebenDExpression of endothelin-1 in the lungs of patients with pulmonary hypertensionN Engl J Med1993328173298497283
- GroverRFVogelJHVoigtGCReversal of high altitude pulmonary hypertensionAm J Cardiol196618928325924007
- HaimoviciJBATrotman-DickensonBHalpernEFRelationship between pulmonary artery diameter at computed tomography and pulmonary artery pressures at right-sided heart catheterizationAcad Radiol199743279156228
- HaleKAEwingSLGosnellBALung disease in long-term cigarette smokers with and without chronic air-flow obstructionAm Rev Respir Dis1984130716216497154
- HaleKANiewoehnerDECosioMGMorphologic changes in the muscular pulmonary arteries: relationship to cigarette smoking, airway disease, and emphysemaAm Rev Respir Dis198012227387416604
- HaniudaMKuboKFujimotoKEffects of pulmonary artery remodeling on pulmonary circulation after lung volume reduction surgeryThorac Cardiovasc Surg200351154812833205
- HanninkMDonoghueDJStructure and function of platelet-derived growth factor (PDGF) and related proteinsBiochim Biophys Acta19891981102546599
- HaraSMorishitaRToneYOverexpression of prostacyclin synthase inhibits growth of vascular smooth muscle cellsBiochem Biophys Res Commun199521686277488205
- HarriganHAJonesKABC of clinical electrocardiography: conditions affecting the right side of the heartBMJ20023241201412016190
- HeathDEdwardsCWinsonMEffects on the right ventricle, pulmonary vasculature, and carotid bodies of the rat of exposure to, and recovery from, simulated high altitudeThorax1973282484685208
- HickenPHeathDBrewerDBThe small pulmonary arteries in emphysemaJ Pathol Bacteriol196590107145843931
- HidaWTunYKikuchiYPulmonary hypertension in patients with chronic obstructive pulmonary disease: recent advances in pathophysiology and managementRespirology2002731311896895
- HighamMADawsonDJoshiJUtility of echocardiography in assessment of pulmonary hypertension secondary to COPDEur Respir J200117350511405510
- HimelmanRBStruveSNBrownJKImproved recognition of cor pulmonale in patients with severe chronic obstructive pulmonary diseaseAmer J Med19888489183364448
- HuJDischerDJBishopricNHHypoxia regulates expression of the endothelin-1 gene through a proximal hypoxia-inducible factor-1 binding site on the antisense strandBiochem Biophys Res Commun199824589499588211
- HurdSThe impact of COPD on lung health worldwide: epidemiology and incidenceChest2000117S14
- IyerNVKotchLEAganiFCellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alphaGenes Dev199812149629436976
- JoppaPPetrasovaDStancakBSystemic inflammation in patients with COPD and pulmonary hypertensionChest20061303263316899829
- KanazawaHOkamotoTHirataKDeletion polymorphisms in the angiotensin converting enzyme gene are associated with pulmonary hypertension evoked by exercise challenge in patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med20001621235811029323
- KellerCAShepardJWJrChunDSEffects of hydralazine on hemodynamics, ventilation, and gas exchange in patients with chronic obstructive pulmonary disease and pulmonary hypertensionAm Rev Respir Dis1984130606116486560
- KesslerRFallerMFourgautGPredictive factors of hospitalization for acute exacerbation in a series of 64 patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1999159158649872834
- KesslerRFallerMWeitzenblumE“Natural history” of pulmonary hypertension in a series of 131 patients with chronic obstructive lung diseaseAm J Respir Crit Care Med20011642192411463591
- KuboKGeRLKoizumiTFujimotoKPulmonary artery remodeling modifies pulmonary hypertension during exercise in severe emphysemaRespir Physiol200012071910786646
- LaabanJPDieboldBZelinskiRNoninvasive estimation of systolic pulmonary artery pressure using Doppler echocardiography in patients with chronic obstructive pulmonary diseaseChest1989961258622582830
- LaDouceurDMFlynnMAKeiserJAETA and ETB receptors coexist on rabbit pulmonary artery vascular smooth muscle mediating contractionBiochem Biophys Res Commun1993196209158216294
- LeeJDTaraseviciene-StewartLKeithRThe expression of prostacyclin synthase is decreased in the small pulmonary arteries from patients with emphysemaChest2005128575S16373836
- LeeJHLeeDSKimEKSimvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungsAm J Respir Crit Care Med20051729879316002570
- LejeunePMolsPNaeijeRAcute hemodynamic effects of controlled oxygen therapy in decompensated chronic obstructive pulmonary diseaseCrit Care Med198412103256510000
- LeopoldJGGoughJThe centrilobular form of hypertrophic emphysema and its relation to chronic bronchitisThorax1957122193513467881
- LiuSChenWLuoYRelationship among dosages of aminophylline, plasma levels of theophylline and variation of pulmonary arterial pressureHua Xi Yi Ke Da Xue Xue Bao19922387901398633
- LouieEKLinSSReynertsonSIPressure and volume loading of the right ventricle have opposite effects on the left ventricle ejection fractionCirculation199592819247641362
- MageeFWrightJLWiggsBRPulmonary vascular structure and function in chronic obstructive pulmonary diseaseThorax19884318393406902
- MatsuyamaWOhkuboRMichizonoKUsefulness of transcutaneous Doppler jugular venous echo to predict pulmonary hypertension in COPD patientsEur Respir J20011711283111491154
- MatthayRASchwarzMIEllisJHJrPulmonary artery hypertension in chronic obstructive pulmonary disease: determination by chest radiographyInvest Radiol198116951007216709
- MechtcheriakovaDWlachosAHolzmullerHVascular endothelial cell growth factor-induced tissue factor expression in endothelial cells is mediated by EGR-1Blood19999338112310339488
- Medical Research Council Working PartyLong term domiciliary oxygen therapy in chronic hypoxic cor pulmonale complicating chronic bronchitis and emphysema. Report of the Medical Research Council Working PartyLancet1981168166110912
- MelotCHallemansRNaeijeRDeleterious effect of nifedipine on pulmonary gas exchange in chronic obstructive pulmonary diseaseAm Rev Respir Dis1984130612166091508
- MillardJReidJRight ventricular hypertrophy and its relationship to chronic bronchitis and emphysemaBrit J Dis Chest197468103104277638
- MorrellNWHighamMAPhillipsPGPilot study of losartan for pulmonary hypertension in chronic obstructive pulmonary diseaseRespir Res2005611015631627
- MorrellNWMorrisKGStenmarkKRRole of angiotensin-converting enzyme and angiotensin II in development of hypoxic pulmonary hypertensionAm J Physiol1995269H1186947485548
- MuradFWhat are the molecular mechanisms for the antiproliferative effects of nitric oxide and cGMP in vascular smooth muscle?Circulation199795110139054833
- MuramotoACaldwellJAlbertRKNifedipine dilates the pulmonary vasculature without producing symptomatic systemic hypotension in upright resting and exercising patients with pulmonary hypertension secondary to chronic obstructive pulmonary diseaseAm Rev Respir Dis198513296364062050
- MurrayCJLopezADAlternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease StudyLancet199734914985049167458
- NaeijeRPulmonary hypertension and right heart failure in chronic obstructive pulmonary diseaseProc Am Thorac Soc2005220216113464
- NakamuraAKasamatsuNHashizumeIEffects of hemoglobin on pulmonary arterial pressure and pulmonary vascular resistance in patients with chronic emphysemaRespiration200067502611070452
- Nana-SinkamSPLeeJDStearmanRProstacyclin synthase in smoking-related lung diseaseProc Am Thorac Soc2006351716921134
- NgCSWellsAUPadleySPA CT sign of chronic pulmonary arterial hypertension: the ratio of main pulmonary artery to aortic diameterJ Thoracic Imaging1999142708
- Nocturnal Oxygen Therapy Trial GroupContinuous or nocturnal oxygen therapy in hypoxemic chronic obstructive lung disease: a clinical trialAnn Intern Med19809339186776858
- NongZStassenJMMoonsLInhibition of tissue angiotensin-converting enzyme with quinapril reduces hypoxic pulmonary hypertension and pulmonary vascular remodelingCirculation199694194178873672
- OlveySKRedutoLAStevensPMFirst pass radionuclide assessment of right and left ventricular ejection fraction in chronic pulmonary disease. Effect of oxygen upon exercise responseChest198078497471843
- Oswald-MammosserMApprillMBachezPPulmonary hemodynamics in chronic obstructive pulmonary disease of the emphysematous typeRespiration199158304101792422
- Oswald-MammosserMOswaldTNyankiyeENon-invasive diagnosis of pulmonary hypertension in chronic obstructive pulmonary disease. Comparison of ECG, radiological measurements, echocardiography and myocardial scintigraphyEur J Respir Dis198771419293443164
- PapaioannouAIKostikasKKolliaPClinical implications for vascular endothelial growth factor in the lung: friend or foe?Respir Res2006712817044926
- PatakasDGeorgopoulosDRodiniHEffects of captopril in patients with chronic obstructive pulmonary disease and secondary pulmonary hypertensionPostgrad Med J19886419353050941
- PeaseRDBenumofJLTrousdaleFRPAO2 and PVO2 interaction on hypoxic pulmonary vasoconstrictionJ Appl Physiol19825313497118626
- PeinadoVIBarberaJAAbatePInflammatory reaction in pulmonary muscular arteries of patients with mild chronic obstructive pulmonary diseaseAm J Respir Crit Care Med199915916051110228134
- PeinadoVIBarberaJARamirezJEndothelial dysfunction in pulmonary arteries of patients with mild COPDAm J Physiol1998274L908139609729
- Pepke-ZabaJHigenbottamTWDinh-XuanATInhaled nitric oxide as a cause of selective pulmonary vasodilatation in pulmonary hypertensionLancet1991338117341682593
- PilatesNDJacobsLERerkpattanapipatPClinical predictors of pulmonary hypertension in patients undergoing liver transplant evaluationLiver Transpl20006859110648583
- RabeKFTenorHDentGIdentification of PDE isozymes in human pulmonary artery and effect of selective PDE inhibitorsAm J Physiol199426653643
- RaesideDABrownAPatelKRAmbulatory pulmonary artery pressure monitoring during sleep and exercise in normal individuals and patients with COPDThorax2002571050312454300
- ReevesJTGroverRFInsights by Peruvian scientists into the pathogenesis of human chronic hypoxic pulmonary hypertensionJ Appl Physiol200598384915591308
- RigatBHubertCAlhenc-GelasFAn insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levelsJ Clin Invest199086134361976655
- RigatBHubertCCorvolPPCR detection of the insertion/deletion polymorphism of the human angiotensin converting enzyme gene (DCP1) (dipeptidyl carboxy peptidase 1)Nucl Acids Res19922014331313972
- RolandMBhowmikASapsfordRJSputum and plasma endothelin-1 levels in exacerbations of chronic obstructive pulmonary diseaseThorax20015630511120901
- SalvaterraCGRubinLJInvestigation and management of pulmonary hypertension in chronic obstructive pulmonary diseaseAm Rev Respir Dis19931481414178239185
- SantosSPeinadoVIRamirezJEnhanced expression of vascular endothelial growth factor in pulmonary arteries of smokers and patients with moderate chronic obstructive pulmonary diseaseAm J Respir Crit Care Med20031671250612615615
- SantosSPeinadoVIRamirezCharacterization of pulmonary vascular remodeling in smokers and patients with mild COPDEur Respir J200219632811998991
- ScharfSMIqbalMKellerCNational Emphysema Treatment Trial (NETT) Group. Hemodynamic characterization of patients with severe emphysemaAm J Respir Crit Care Med20021663142212153963
- SchermulyRTDonyEGhofraniHAReversal of experimental pulmonary hypertension by PDGF inhibitionJ Clin Invest200511528112116200212
- ScottKWMA clinicopathological study of fatal chronic airways obstructionThorax197631693701138210
- SemenzaGLHIF-1: mediator of physiological and pathophysiological responses to hypoxiaJ Appl Physiol20008814748010749844
- SemenzaGLOxygen-regulated transcription factors and their role in pulmonary diseaseRespiratory Research200011596211667980
- SherrillDLEnrightPClineMRates of decline in lung function among subjects who restart cigarette smokingChest1996109100158635322
- SpiropoulosKTrakadaGNikolaouEEndothelin-1 levels in the pathophysiology of chronic obstructive pulmonary disease and bronchial asthmaRespir Med200397983912924528
- StevensSMSharmaKSzidonPSevere pulmonary hypertension associated with COPDAnn Transplant2000581211147031
- SturaniCBasseinLSchiavinaMOral nifedipine in chronic cor pulmonale secondary to severe chronic obstructive pulmonary disease (COPD)Chest198384135426872591
- SuYHanWGiraldoCEffect of cigarette smoke extract on nitric oxide synthase in pulmonary artery endothelial cellsAm J Respir Cell Mol Biol199819819259806747
- TanRTKuzoRGoodmanLRLUtility of CT scan evaluation for predicting pulmonary hypertension in patients with parenchymal lung diseaseChest199811312509596302
- Taraseviciene-StewartLKasaharaYAlgerLInhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertensionFASEB J2001154273811156958
- ThabutGDauriatGSternJBPulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantationChest20051271531615888824
- ThompsonBTHassounPMKradinRLAcute and chronic hypoxic pulmonary hypertension in guinea pigsJ Appl Physiol19896692082708221
- Tillie-LeblondIMarquetteCHPerezTPulmonary embolism in patients with unexplained exacerbation of chronic obstructive pulmonary disease: prevalence and risk factorsAnn Intern Med2006144390616549851
- TimmsRMKhajaFUWilliamsGWHemodynamic response to oxygen therapy in chronic obstructive pulmonary diseaseAnn Intern Med198510229363966742
- TkacovaRJoppaPStancakBThe link between angiotensin-converting enzyme genotype and pulmonary artery pressure in patients with COPDWien Klin Wochenschr2005117210415875760
- TramarinRTorbickiAMarchandiseBDoppler echocardiographic evaluation of pulmonary artery pressure in chronic obstructive pulmonary disease. A European multicentre study. Working Group on Noninvasive Evaluation of Pulmonary Artery Pressure. European Office of the World Health Organization, CopenhagenEur Heart J199112103112044542
- TuderRMCoolCDGeraciMWProstacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertensionAm J Respir Crit Care Med199915919253210351941
- van DijkAPTorbickiAMore on the noninvasive diagnosis of pulmonary hypertension: Doppler echocardiography revisitedEur Respir J19958144598575567
- VenderRLChronic hypoxic pulmonary hypertension. Cell biology to pathophysiologyChest1994106236438020277
- VoelkelNFQuaifeRALeinwandLARight ventricular function and failure: report of a national heart, lung, and blood institute working group on cellular and molecular mechanisms of right heart failureCirculation200611418839117060398
- Von EulerULiljerstrandGObservations on the pulmonary arterial blood pressure in the catAct Physiol Scand19461230120
- VonbankKZiescheRHigenbottamTWControlled prospective randomized trial on the effects on pulmonary haemodynamics of the ambulatory long term use of nitric oxide and oxygen in patients with severe COPDThorax2003582899312668787
- Vonk-NoordegraafAMarcusJTHolverdaSEarly changes of cardiac structure and function in COPD patients with mild hypoxemiaChest2005127189890315947300
- WedgwoodSDettmanRWBlackSMET-1 stimulates pulmonary arterial smooth muscle cell proliferation via induction of reactive oxygen speciesAm J Physiol Lung Cell Mol Physiol2001281L10586711597896
- WeirEKArcherSLThe mechanism of acute hypoxic pulmonary vasoconstriction: the tale of two channelsFASEB J1995918397781921
- WeitzenblumEHirthCDucoloneAPrognostic value of pulmonary artery pressure in chronic obstructive pulmonary diseaseThorax19813675287330793
- WeitzenblumESautegeauAEhrhartMLong-term oxygen therapy can reverse the progression of pulmonary hypertension in patients with chronic obstructive pulmonary diseaseAm Rev Respir Dis198513149383922267
- WeitzenblumESautegeauAErhartMLong-term course of pulmonary arterial pressure in chronic obstructive pulmonary diseaseAm Rev Respir Dis198413099386439091
- WilkinsonMLanghorneCAHeathDA pathophysiological study of 10 cases of hypoxic cor pulmonaleQ J Med19886665853174923
- WrightJLLawsonLParePDThe structure and function of the pulmonary vasculature in mild chronic obstructive pulmonary disease. The effect of oxygen and exerciseAm Rev Respir Dis198312870276625346
- WrightJLPettyTThurlbeckWMAnalysis of the structure of the muscular pulmonary arteries in patients with pulmonary hypertension and COPD: National Institutes of Health nocturnal oxygen therapy trialLung1992170109241501507
- WrightJLTaiHChurgACigarette smoke induces persisting increases of vasoactive mediators in pulmonary arteriesAm J Respir Cell Mol Biol200431501915242846
- WrightJLTaiHDaiJCigarette smoke induces rapid changes in gene expression in pulmonary arteriesLab Invest2002821391812379773
- WrightJLRelationship of pulmonary arterial pressure and airflow obstruction to emphysemaJ Appl Physiol199374132048482673
- YanSFZouYSGaoYTissue factor transcription driven by EGR-1 is a critical mechanism of murine pulmonary fibrin deposition in hypoxiaProc Natl Acad Sci USA19989582983039653181
- YildizPOflazHCineNGene polymorphisms of endothelial nitric oxide synthase enzyme associated with pulmonary hypertension in patients with COPDRespir Med2003971282814682408
- YuAYFridMGShimodaLATemporal, spatial, and oxygen-regulated expression of hypoxia-inducible factor 1 in the lungAm J Physiol1998275L818269755115