Abstract
Chronic obstructive pulmonary disease (COPD) is a global health problem. Being a progressive disease characterized by inflammation and predominantly caused by tobacco smoking, it deteriorates pulmonary and skeletal muscle functioning, and reduces physical behavior, societal participation and quality of life. During the last two decades studies were focused on the airway and systemic inflammation, oxidative stress, and airway and/or parenchymal remodeling. Macrophages, neutrophils and T cells are thought to be important key players, as well as structural cells like fibroblasts, epithelial, endothelial and smooth muscle cells. Mediators and proteins including cytokines, chemokines, growth factors, proteinases, and oxidants seem to be involved differentially in its pathogenesis. Current pharmacological treatments are directed to reducing airway inflammation, boosting the endogenous levels of anti-oxidants and relieving airway contraction and sputum production. Most agents were primarily used for treating asthma. But in contrast to asthma, these treatments are not very effective in COPD. As a result, novel more specifically acting interventional drugs with less side effects are being developed to treat chronic inflammatory diseases, including COPD. This review highlights studies on novel or potential drug antioxidants such as dietary antioxidants supplementation, N-acetyl-L-cysteine, N-acystelyn, endosteine, antioxidant enzyme mimetics, and anti-inflammatory agents like antagonists of cytokines, such as tumor necrosis factor (TNF)-α, CXCL8, and CCL2, and inhibitors of signal transduction proteins including phosphodiesterase 4, MAPK p38, Pl-3k, and NFκB.
Introduction
Chronic obstructive pulmonary disease (COPD) is a global health problem. According to estimates of the World Health Organization (WHO) about 80 million patients over the world suffered from COPD in 2005, whereas it accounts for 5% of all deaths. COPD is currently the 4th major cause of death worldwide. The prevalence is estimated to be about 1% worldwide, but is about 2 times higher in Western societies, and often underestimated due to un- or misdiagnosis (CitationPauwels and Rabe 2004; CitationMenezes et al 2005; CitationLindberg et al 2006). The burden of COPD for the patient is high as patients experience a poorer quality of life, suffer from comorbidites (3.7 comorbidities per patient), and direct healthcare costs range from 0.28 billion euros in the Netherlands (in 2000) to 20.9 billion dollars in the USA (in 2004) (CitationHynninen et al 2005; CitationHoogendoorn et al 2006; CitationJones 2006; CitationSin et al 2006).
COPD is a progressive disease which is yet not curable. In Western countries the major cause is tobacco smoking, whereas in developing countries also indoor pollution eg, from cooking fuel biomass burning is a cause. Other risk factors for COPD include genetic predisposition, occupational and environmental exposure, and asthma. More than 90% of patients with COPD are smokers (CitationSnider 1989), but at least 10%–20% of the smokers develop COPD pointing at an additional risk factor, eg, gene susceptibility. Among the genetic susceptibility factors are polymorphisms in genes coding for (anti-) proteases like alpha-1 antitrypsin (A1AT) (accounting for at least 5% of COPD cases) and a disintegrin and metalloproteinase 33 (ADAM33), genes coding for antioxidant enzymes like glutamate cysteine ligase, epoxide hydrolase, glutathione-S-transferase, and superoxide dismutase (SOD) 3, or genes coding for cytokines like tumor necrosis factor alpha (TNFα) and transforming growth factor beta 1 (TGFβ1) (CitationHarrison et al 1997; CitationKeatings et al 2000; CitationSandford et al 2001; CitationKucukaycan et al 2002; CitationCeledon et al 2004; CitationGosman, Boezen et al 2006; CitationYoung et al 2006).
Since chronic pulmonary inflammation and oxidative stress are important characteristics in the pathogenesis of COPD, this paper discusses the role of inflammatory mediators and oxidants and rational of anti-inflammatory and anti-oxidant therapeutic intervention in the management of COPD.
Pathophysiology
COPD is an inflammatory chronic lung disease that shows a largely irreversible lung function decline, and can be classified into 4 classes of severity based on lung function [GOLD Guidelines]. Emphysema, chronic bronchitis with airway obstruction, and small airways disease are the distinct phenotypes of COPD, but most patients show a combination. Emphysema is characterized by a Th1-type inflammation, destruction of the alveolar septa, loss of elastic recoil, airspace enlargement and hence loss of gas diffusion capacity (CitationWright and Churg 2006). Chronic bronchitis affects the airways by airway inflammation, goblet cell hyperplasia and mucus hypersecretion. In addition to decreased lung function, these patients experience chronic sputum production, coughing and often dyspnoea. Ciliated airway epithelium lining the airway lumen is impaired in its function due to the hypersecreted mucus, and in activation and damage by tobacco compounds. As a result, the ciliated epithelium may be partly replaced by other epithelial cell types like squamous and goblet cells, further contributing to the airway dysfunction. The inflammatory phenotype in chronic bronchitis is neutrophilic or eosinophilic. In eosinophilic chronic bronchitis the sputum or bronchoalveolar lavage (BAL) fluid presents more eosinophils whereas in neutrophilic airway inflammation predominates the neutrophilic phenotype. The eosinophilic phenotype exhibits a higher reversible forced expiratory volume in 1 second (FEV1) as compared to the neutrophilic phenotype, and is more responsive to corticosteroid treatment (CitationChanez et al 1997; CitationPizzichini et al 1998; CitationBrightling et al 2005). Small airways disease mainly affects the bronchioles featuring airway inflammation and metaplasia of Clara cells.
Pathogenesis
The pathogenesis of COPD is not known yet. However, pathological features of COPD include lung tissue and vascular remodeling, and pulmonary and systemic inflammation (CitationBarnes et al 2003; CitationLangen et al 2003; CitationHogg 2004; CitationDe Boer 2005; CitationWright and Churg 2006; CitationDe Boer et al 2007). Clinical research in patients with established COPD showed inflammation with cells involved in innate immunity including macrophages, neutrophils, and T cells (predominantly CD8+, but less prominent in severe COPD) (CitationGrashoff et al 1997; CitationDi Stefano et al 2001; CitationHogg 2004; CitationBarnes and Stockley 2005; CitationDe Boer 2005). Some studies also showed higher lung tissue numbers of mast cells, and also eosinophils during exacerbations or in patients with COPD showing reversible lung function (CitationGrashoff et al 1997; CitationPapi et al 2000; CitationBarnes and Stockley 2005; CitationDe Boer 2005). Differences between the outcomes of studies may be due to inclusion criteria, numbers of participating patients, the COPD phenotype studied or the pulmonary location of sampled tissue. Recent studies also point to a link between the innate and acquired immune system. Studies with COPD patients demonstrated the presence of B-cell follicles in lung tissue (CitationGosman, Willemse et al 2006; Citationvan der Strate et al 2006) while studies on smoke-exposed mice show a role for dendritic cells in the pathogenesis of emphysema (CitationBracke et al 2006). A limited number of studies suggested an autoimmune component in emphysema (CitationAgusti et al 2003). This suggestion was based on observations showing the presence of T cells (CD8+and CD4+), B cell follicles in the airway walls, and clonal expansion of CD4+T cells in lung tissue, the presence of anti-idiotypic antibodies with tobacco-like activity, and induction of emphysema in rats via a CD4+ and in mice via a CD8+ cell-specific mechanism (CitationSaetta et al 1999; CitationKoethe et al 2000; CitationSullivan et al 2005; CitationTaraseviciene-Stewart et al 2005; CitationGosman, Willemse et al 2006; Citationvan der Strate et al 2006; CitationMaeno et al 2007).
Oxidants and proteinases
Mechanisms that may contribute to the pathogenesis involve disturbed oxidant-antioxidant and proteinase-antiproteinase balances. Lungs are continuously exposed to oxidants, either generated endogenously by metabolic reactions (eg, from mitochondrial electron transport during respiration or during activation of phagocytes) or exogenously, such as air pollutants or cigarette smoke. Tobacco smoke constitutes about 5000 compounds and 1017 free radicals per puff, many of which are able to induce reactive oxygen or nitrogen species that can be inactivated by endogenous molecules like SOD, catalase, cytochrome P450, flavanoids and glutathione. In patients with COPD this balance may be disturbed due to mutations in genes encoding these enzymes or enzymes related to these pathways (CitationLangen et al 2003; CitationRahman and Adcock 2006). Production of reactive oxygen species (ROS) has been directly linked to oxidation of proteins, DNA, and lipids, which may cause direct lung injury or induce a variety of cellular responses, through the generation of secondary metabolic reactive species (CitationRepine et al 1997). ROS may alter remodeling of extracellular matrix (ECM) and blood vessels, stimulate mucus secretion, inactivate antiproteases, cause apoptosis, and regulate cell proliferation (CitationRahman and MacNee 1996a, Citation1996b, Citation1999). Furthermore, increased levels of ROS have been implicated in initiating inflammatory responses in the lungs through the activation of transcription factors such as nuclear factor-kappaB (NF-κB) and activator protein-1 (AP-1), signal transduction, chromatin remodeling and gene expression of pro-inflammatory mediators (CitationRahman and MacNee 1998).
4-Hydroxy-2-nonenal (4-HNE) is a highly reactive and specific diffusible end-product of lipid peroxidation, and is found to induce the COX-2 genes in RAW264.7 cells (CitationKumagai et al 2004), thus reflecting the potential role of 4-HNE as perpetrator of inflammation. Furthermore exogenous micromolar levels of 4-HNE increases the expression of several genes eg, heme oxygenase-1, collagen α1(I), and aldose reductase (CitationParola et al 1993; CitationBasu-Modak et al 1996; CitationSpycher et al 1997). Also 4-HNE has been reported to have chemotactic, cytotoxic and immunogenic properties both in vitro and in vivo (CitationSchaur et al 1994; CitationSteinerova et al 2001), and these effects were achieved in vitro with 4-HNE concentrations as low as 2.5 μM (CitationMuller et al 1996). Data from the authors’ laboratories indicate increased 4-HNE-modified protein levels in airway and alveolar epithelial cells, endothelial cells and neutrophils in subjects with airway obstruction compared to subjects without airway obstruction (CitationRahman and MacNee 2000b; CitationRahman et al 2002). An important outcome of 4-HNE generation is its interaction with the important thiol antioxidant glutathione (GSH) (CitationTjalkens et al 1999). The conjugation of 4-HNE with GSH might be one of the important mechanism whereby a cell may lose its antioxidant pool leading to oxidative stress. Interestingly, increased formation of 4-HNE has also been reported to induce expression of glutamyl cysteine ligase (GCL) gene which increases synthesis of GSH. This might be an important cellular antioxidant adaptation during oxidative stress. Inhibition of lipid peroxidation, specifically the pathways leading to the production of 4-HNE and F2-isoprostane, may therefore be important and novel targets for antioxidant therapy in inflammation and injury in patients with COPD.
One of deleterious outcomes of oxidative stress is the remodeling of ECM leading to lung injury. ROS activate latent proforms of matrix metalloproteinase (MMP) (CitationLindholt et al 2003), and antioxidant species decrease MMP expression and activation (CitationRajagopalan et al 1996). Cigarette smoke treatment of alveolar macrophages from subjects with COPD induced increased release of MMP-9 compared to that of non-smokers. MMP-9 has an ECM degrading activity, thus suggesting the role of oxidative components of cigarette smoke in increased elastolytic enzyme activity (CitationRussell et al 2002). Increased proteolytic load due to MMP-9 has been attributed to increased neutrophil recruitment in the lungs that triggers degradation of ECM and basement membrane in the airways and lungs. Antioxidants N-acetylcysteine and pyrrolidine dithiocarbamate, and the NADPH oxidase inhibitors diphenylene iodonium chloride and apocynin decreased the production of MMP-2 and -9 in alveolar macrophages from surfactant protein D-deficient mice (CitationYoshida et al 2001). However, a recent study showed that mice lacking gp91phox, a phagocyte-specific component of the NADPH oxidase, developed extensive, spontaneous emphysematous destruction of their peripheral air spaces (CitationKassim et al 2005). In addition, peritoneal macrophages from gp91phox-null mice had greater MMP-12 activity than macrophages from wild type mice (CitationKassim et al 2005). These findings indicate that reactive intermediates provide a physiological mechanism to protect tissues from excessive macrophage-mediated damage during inflammation. Factors other than oxidative stress, such as ozone and lipid peroxides also induce collagen I and MMP-1 gene expression (CitationChoi et al 1994). Other forms of oxidative stress derived from tert-butyl hydroperoxide and iron can also modify collagen synthesis, by a mechanism presumably involving redox sensor/receptor.
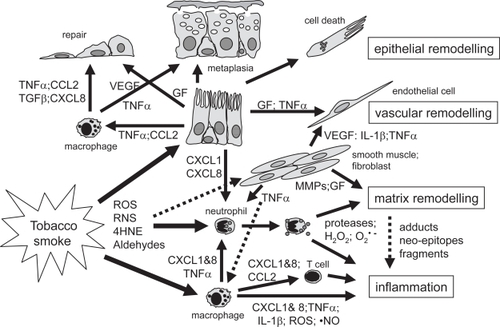
The proteinase-antiproteinase dysbalance is thought to be related to the increased proteolytic activity or protease expression observed in sputum, BAL fluid or tissue of patients with COPD, and tissue remodeling or destruction as seen in emphysema (CitationBarnes et al 2003; CitationHogg 2004). Several studies reported enhanced levels or gene mutations of MMPs like MMP-1, MMP-9 or MMP-12 associated with COPD and lung function decline (CitationJoos et al 2002; CitationCulpitt et al 2005; CitationDemedts et al 2006), the presence of fragments of ECM proteins like elastin or collagen (CitationDillon et al 1992; CitationStone et al 1995; CitationWeathington et al 2006), and/or altered levels of ECM molecules in sputum, BAL fluid or lung tissue of patients with COPD (CitationLang et al 1994; CitationDentener et al 2005; CitationKranenburg et al 2006; CitationMartin-Mosquero et al 2006). Extracellular matrix hyaluronan (HA) has a pro-inflammatory role and HA levels were found to be increased in sputum of COPD patients (CitationDentener et al 2005). Two categories of COPD subjects have been identified: one group having high HA levels and the other having moderate levels. COPD subjects exhibiting higher HA levels had low FEV1 as compared to moderated and control categories. Increased breakdown and therefore increased HA levels were further correlated with an increased expression of hyaluronidase 2 gene. Furthermore, increased HA breakdown has been associated with local inflammation and severity of COPD. Yet, a recent study demonstrated that aerosolized HA limits airspace enlargement in a mouse model of cigarette smoke-induced pulmonary emphysema (CitationCantor et al 2005). In addition, treatment with HA partially blocked LPS (1 ng/ml) induced TNFα release by blood cells from COPD patients (CitationDentener et al 2006). Thus the high levels of HA in COPD subjects would be a consequence of degradation of ECM, which in turn can bind to lung elastic fibers, thereby adaptively preventing their further degradation by protease (CitationCantor et al 1997, Citation2000). Targeted deletion of neutrophil elastase or MMP-12 protects from the development of cigarette smoke or gp91 deficiency-induced emphysema (CitationHautamaki et al 1997; CitationShapiro et al 2003; CitationKassim et al 2005). Furthermore, the structural alterations in ECM proteins may provoke an immune reaction, whereas degradation fragments generated during extensive tissue remodeling may lead to antigenic fragments also provoking an immune reaction. More specifically, exposure to reactive oxygen or nitrogen intermediates or aldehydes present in smoke or produced by inflammatory cells may lead to adduct formation of structural (lipo)proteins in the lung eg, as a result of lipid peroxidation or protein carbonyl formation (CitationRahman and Adcock 2006). In turn, this may result in the formation of neo-epitopes of ECM proteins (CitationKirkham et al 2004; CitationMorquette et al 2006). The induction of neo-antigens upon tobacco smoke exposure may lead to specific B and T cell clones that provoke autoimmune reactions. Alternatively, during embryogenesis specific B and T cells are being selected and generated by peptide fragments of extracellular matrix components secreted by lung epithelial cells. At that moment, the immune system is tolerant to these peptides. Upon smoke exposure, these B and/or T cells may be activated causing the inflammatory phenotype and specific lung parenchymal destruction as seen in emphysema. Some of the mechanisms are outlined in Figure . As COPD is detected in an advanced form only at later age, other models are needed to study these and other pathogenetic mechanisms of COPD.
Figure 1 Simplified summary of inflammatory and remodeling mechanisms in the airways in COPD. Exposure to cigarette smoke in susceptible individuals leads to an abnormal inflammation and tissue remodeling. This appears to be self-perpetuating and may be linked to infection. Tobacco smoke activates different cell types including macrophages, epithelial and smooth muscle cells to produce cytokines, growth factors or proteases. Reactive molecules in tobacco smoke stimulate airway macrophages to produce cytokines and reactive oxygen or nitrogen species. Activated macrophages and epithelial cells attract and activate inflammatory cells including monocytes, macrophages, neutrophils and T cells. Alternatively, reactive species may react with extracellular matrix (ECM), and lipid moieties causing cell damage, gene expression or oxidative stress in different cell types. Chemokines like CXCL-8 and CXCL-1 cause T cell and neutrophil chemotaxis and activation of neutrophils to degranulate proteases like elastase and MMPs, and produce reactive oxygen species like hydrogen peroxide or O2 •–. Radicals may activate proteases that in turn fragment ECM molecules and/or form ECM neo-epitopes. Oxygen radicals may also react with ECM leading to adducts or neo-epitopes. Altered or fragmented ECM molecules may stimulate inflammation and auto-immune-like reactions. Tobacco smoke may also activate smooth muscle cells and fibroblasts to produce pro-inflammatory cytokines and growth factors (GF) like VEGF, leading to Th1-mediated inflammation and vascular remodelling. Loss of epithelial cells due to direct toxicity of smoke, TNFα-induced apoptosis, or degradation of ECM, induces a repair process. Growth factors like EGF, FGF, TGFβ1 and VEGF stimulate tissue repair and vascular remodelling seen in COPD. Epithelial remodelling (squamous or mucous metaplasia, hyperplasia) may be due to excessive growth factor production or by TNFα resulting in a loss of lung clearance function and mucus hyperproduction. A-HNE, 4-hydroxy-2-nonenal; ROS, reactive oxygen species; RNS, reactive nitrogen species.
Cytokines and chemokines
Inflammation is one of the hallmarks of COPD. Inflammatory cells are known to migrate to chemotactic gradients, or to express and produce pro- or anti-inflammatory proteins, MMPs and other proteases, growth factors, antibacterial proteins like α-defensins, β-defensins and cathelicidins, and/or to degranulate by a plethora of molecules including cytokines, chemokines, growth factors, and arachidonic acid derivates (like prostaglandins, leukotrienes and thromboxanes) (CitationDe Boer 2002; CitationBals and Hiemstra 2006; CitationRahman and Adcock 2006; CitationRolin et al 2006). Classical cytokines include TNFα, interleukins (ILs) and interferons (IFNs). Chemokines can be subdivided into four subfamilies based on their structural homology around 4 cysteine residues: -C-, -CC-, -CXC-, and -CXXXC-, in which X substitutes for any amino acid. The major subfamilies are the -CXC- (or: α chemokines, and the -CC- chemokines (or: β-) chemokines) (CitationDe Boer 2002). Gene expression of cytokines and chemokines is regulated by transcription factors including AP-1 and Fos, and signal transduction kinases like mitogen activated protein kinase (MAPK) p38, Jun kinase (JNK), and NF-κB, and are synthesized by both structural cells (such as fibroblasts, epithelial, endothelial, and muscle cells) and inflammatory cells of the innate and the adaptive immune system. They act via specific membrane-bound receptors resulting in cell specific reactions. In patients with COPD, protein and/or mRNA levels of different cytokines and chemokines have been found to be altered compared with subjects without COPD (Table ).
Table 1 Molecules involved in inflammation in COPD
Among these, TNFα or TNFR levels, soluble IL-1 receptor antagonist (sIL-1Ra), CCL2 (monocyte chemoattractant protein 1, MCP-1) and its receptor CCR2, CCL3 (macrophage inflammatory protein 1α, MIP-1α) and CCL4 (MIP- 1β) and their receptor CCR5, CXCL8 (IL-8), and CXCL10 (interferon-inducible protein 10, IP-10) can be discerned as pro-inflammatory factors. In addition to the inflammatory effects, recent studies provided more evidence that cytokines and chemokines are also involved in tissue remodeling apart from growth factors, pointing to cytokine-driven effects of inflammatory cells on epithelial wound repair (CitationDe Boer et al 2007) (Figure ).
CCL2 is produced by a variety of cells including macrophages, endothelial and epithelial cells. It binds to CCR2, a receptor that is highly expressed on circulating blood DCs and monocytes, and thus primarily controls their recruitment from the blood vessels to the tissue. CCL2 is also involved in tissue remodelling. In vitro, CCL2 and its receptor CCR2 were demonstrated to be directly involved in endothelial and lung epithelial cell proliferation, migration and wound closure (CitationDe Boer et al 2007). In addition, CCL2 was found to stimulate collagen synthesis in rat lung fibroblasts via a TGFβ1-dependent pathway, hence potentially contributing to a fibrogenetic remodelling as seen in COPD. In turn, TGFβ1 was reported to induce CCL2 protein levels via downstream intracellular mechanisms including ROS, and MAPK p38 and p42/44 in mesangial cells (CitationCheng et al 2005). Results from studies in mice and cell lines suggest that oxidative stress activates MAPK p42/44 and p38 which stimulates the expression of TNFα, IL-1β, CCL2 and CXCL10 (CitationNishi et al 2005; CitationGuest et al 2006; CitationHuang et al 2006; CitationLoke et al 2006). Oxidative stress led to an influx of macrophages and increased expression of proteins like NADPH oxidase, thioredoxin- 1 and inducible nitric oxide synthase, via a CCR2- mediated mechanism (CitationHayasaki et al 2006). These effects were inhibitable by antioxidants including N-acetylcysteine, ascorbic acid, quercetin or overexpression of SOD1, deletion of CCR2, or by inhibition of MAPK p42/44 or p38 (CitationNishi et al 2005; CitationGuest et al 2006; CitationHayasaki et al 2006; CitationHuang et al 2006; CitationLoke et al 2006). Hence the CCL2-CCR2-TGFβ axis may potentially contribute to tissue remodeling and inflammation as seen in COPD.
CXCL1 (GROα) and CXCL8 are produced by both structural and inflammatory cells including macrophages. Both bind to their receptor CXCR2 whereas CXCL8 binds also to CXCR1. Both receptors are expressed on neutrophils while CXCR2 is also expressed on other inflammatory cells including a subset of CD8+ T cells, mast cells and macrophages. CXCL1 and CXCL8 are chemotactic and activate inflammatory cells while CXCL8 also induces neutrophils to degranulate and causes an oxidative burst. Recent studies demonstrated that cigarette smoke-conditioned medium induces human monocytes or macrophages in vitro to release CXCL8 (CitationKarimi et al 2006; CitationYang et al 2006). As inhibitors of Toll-like receptor (TLR)-4, NF-κB and IKK-2 impaired the CXCL8 expression this points to a TLR4 and NF-κB dependent mechanism. In addition, CXCL8 release was significantly inhibited by the anti-oxidant glutathione pointing to a regulatory effect of the chemokine expression by the redox status of the cells (CitationYang et al 2006). In contrast to monocytes, cigarette smoke conditioned medium did not affect CXCL8 or IL-6 expression nor NF-κB activation in human lung epithelial cells in vitro (CitationMoodie et al 2004). This suggests that oxidants exert differential effects on NF-κB signalling and chemokine expression depending on the cell type, and that inflammatory cells produce CXCL8 upon oxidant exposure. The study showed that also epithelial cells express CXCR2 suggesting a role for CXCL8/CXCR2 in wound repair (CitationDe Boer 2002). In vivo, impaired CXCR2 expression in mice resulted in delayed skin wound repair as compared to wild type mice, which cannot be restored by exogenous CXCL8 (CitationDevalaraja et al 2000; CitationMilatovic et al 2003). In contrast, topical application of CXCL1 or CXCL8 stimulated human skin wound repair in chimeric mice (CitationRennekampff et al 1997, Citation2000), and stimulated human colon, skin and lung epithelial proliferation and/or migration in vitro (CitationDe Boer et al 2007). Inhibition of CXCL8 or CXCL1 signaling or expression as a treatment target in COPD may hence inhibit inflammatory cell activation and tissue degradation, but may potentially delay wound repair in COPD.
Cigarette smoke has been shown in vivo to be a cause of increased adherence of leukocytes to vascular endothelium (CitationNoguera et al 1998). CitationShen and co-workers (1996) have shown that cigarette smoke condensate induces the expression of a subset of cell adhesion molecules, such as intercellular adhesion molecule (ICAM-1), endothelial leukocyte adhesion molecule 1 (ELAM-1), and vascular cell adhesion molecule (VCAM-1) in human umbilical vascular endothelial cells associated with an increase in the binding activity of NF-κB suggesting the increased transendothelial migration of monocytes by cigarette smoking. The release of proinflammatory mediators, such as IL-1β and soluble ICAM-1, was increased by cigarette smoke exposure in bronchial epithelial cells cultured from biopsy materials obtained from patients with COPD compared to smokers (CitationRusznak et al 2000). Moreover, CitationScott and coworkers (2000) demonstrated a clear dose-dependent relationship between smoke intake and sICAM-1 concentrations and sICAM-1 concentrations substantially reduced in those who stopped smoking for a year but remained elevated in continuing smokers. These results suggest that patients with COPD have a greater susceptibility to the effects of cigarette smoke.
Growth factors: VEGF and TGFβ1
Growth factors can be divided into different superfamilies based on structural and functional homology. These families include vascular endothelial growth factor (VEGF), TGF-β, epidermal growth factor (EGF)-like growth factors, fibroblast growth factor (FGF) and insulin-like growth factor (IGF) (CitationDe Boer et al 2007). With regard to COPD several studies suggest the involvement of these families in either pulmonary inflammation like for VEGF and TGFβ1 (CitationDe Boer et al 1998; CitationTakizawa et al 2001; CitationPostma and Timens 2006), vascular or tissue remodeling like for EGF-like growth factors, FGFs and VEGFs (CitationKranenburg et al 2002, Citation2005; CitationDe Boer et al 2006; CitationPostma and Timens 2006), or oxidative stress as with TGFβ1 or FGF-7 (CitationRahman et al 2000; CitationRahman et al 2002; CitationRay et al 2003) (Table ). A review on growth factors as a potential target for drug therapy is presented elsewhere (CitationDe Boer et al 2007).
VEGF receptor impairment, VEGF gene deletion or generation of antibodies against VEGF receptors all cause airspace enlargement in rodents without airway inflammation (CitationKasahara et al 2000). In addition, in murine models tobacco smoke exposure leads to decreased expression of VEGF and VEGF receptors as well as emphysematous lesions, as has also been observed in smokers with emphysema. Furthermore, blockade of VEGF receptors was shown to induce oxidative stress and alveolar cell apoptosis that was inhibited by exogenous administration of the SOD mimetic M40419 (CitationTuder et al 2003). These data link oxidative stress with development of emphysema and abrogated VEGF signaling rather than alveolar damage induced by inflammation alone. Tuder and coworkers proposed a disturbed balance between oxidative stress, proteinases, antiproteinases and apoptosis, and lung inflammation as a non-specific reaction secondary to alveolar tissue damage (CitationTuder et al 2006). However, these data may not be applied to COPD as a whole as VEGF and VEGFR expression was observed to be enhanced in relation to vascular remodeling in non-emphysematous patients making these patients less eligible for VEGF therapy (CitationKranenburg et al 2005; CitationDe Boer et al 2007).
TGFβ1 has been associated with COPD either as a result of oxidative stress or an imbalance in proteinases and antiproteinases, but may also be related to an aberrant repair process and hence progression of COPD (CitationPostma and Timens 2006; CitationRahman and Adcock 2006). TGFβ1 expression was demonstrated to be enhanced in patients with COPD (CitationDe Boer et al 1998) but decreased in patients with emphysema (CitationZandvoort et al 2006). The expression of intracellular inhibitory signaling proteins of TGFβ1, Smad6 and Smad7, was observed to be decreased both in bronchial and alveolar tissue from patients with COPD, whereas in expression of stimulatory Smad molecules including Smad3 was unaltered (CitationSpringer et al 2004; CitationZandvoort et al 2006). Smad7 is involved in the regulation of expression of inflammatory proteins. In vivo wound healing study with mice demonstrated that overexpression of Smad7 inhibits TGFβ1, CCL2, VEGF, MMP-9 and TIMP-2 protein and mRNA expression (CitationSaika et al 2005). Reducing overexpression of Smad7 in patients with inflammatory bowel disease (IBD) using antisense Smad7 oligonucleotides caused a decreased production of proinflammatory cytokines IFNγ and TNFα upon treatment of intestinal tissue explants and cells with TGFβ1 (CitationMonteleone et al 2001). With regard to COPD, it is not known whether Smad7 downregulation is intrinsic or due to inflammation, oxidative stress, or other factors, and what the consequences are of differential expression of TGFβ1 in patients with COPD or emphysema alone. An alternative hypothesis is that tobacco smoke exposure causes excessive growth factor production resulting in tissue remodeling, independent of inflammation. Recent data from a murine study (CitationChurg et al 2006) provided support for this idea. Their study demonstrated that short-term smoke exposure for 2 hours stimulated early growth factor expression including TGFβ1 and type 1 procollagen synthesis before the onset of inflammation. Upon chronic smoke exposure for up to 6 months profibrotic growth factor expression continued as well as tissue remodeling characterized by enhanced collagen deposition, while other studies showed the development of airway inflammation and emphysema in rodents in this period. Taken together, the balance between TGFβ1 and Smad7 expression in pulmonary cells of patients with COPD seems to be delicate and may affect tissue remodeling and inflammation differently depending on the COPD phenotype. Targeting TGFβ1 as a therapy in COPD requires more studies on the precise role of these factors in the pathogenesis of COPD.
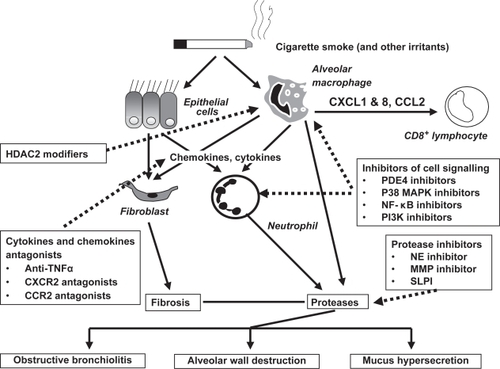
Figure outlines briefly the proposed remodeling and inflammatory mechanisms in COPD, whereas Figure summarizes potential intervention strategies. Based on this, specific anti-inflammatory therapies are being developed for COPD (CitationDe Boer 2005).
Figure 2 Emerging anti-inflammatory therapy. The chronic, persistent inflammation and tissue remodeling that ensues in COPD is thought to be responsible for both the symptoms of disease and also the progressive decline in lung function. The loss of airway function appears to be related to the destruction of alveoli resulting in a loss of elasticity linked to increased protease activity in emphysema, and/or obstruction and fibrosis of the (small) airways as a result of inflammation and mucus hypersecretion in chronic bronchitis. Emerging anti-inflammatory therapies under clinical investigation attack this chronic pulmonary inflammation via several strategies. Signaling pathway inhibitors such as PDE4 inhibitors, MAPK p38 inhibitors, NF-κB signaling inhibitors and PI3K inhibitors are in development. Reduction of pleiotropic inflammatory cytokines such as TNFα using monoclonal antibodies that target the ligands, or soluble receptors that bind and inactivate TNFα may also reduce the inflammatory burden in the lung. Targeting chemokines like CCL2 and CXCL8 may reduce the influx of inflammatory cells into the lungs from the circulation by reducing the chemotactic gradient. Inhibition of protease activity in the lung may attenuate lung tissue damage and reduces the numbers of lung neutrophils. Increased HDAC2 expression restores the sensitivity for steroids in the treatment of COPD. Reducing the severity of inflammation and tissue remodeling may improve lung function and slow the progression of COPD.
Current therapies
Therapies for COPD are mainly based on anti-inflammatory drugs for treating asthma, including corticosteroids or theophylline with or without bronchodilators like β2-agonists. Some studies reported reduction of the number of exacerbations, improved quality of life and an decline in FEV1 after short- or long-term treatment with inhaled corticosteroids, or no effect on lung function (CitationGartlehner et al 2006). Though some recent studies using higher doses or longer duration of treatment showed reduced airway inflammation, steroid treatment of patients with COPD is rather ineffective in reducing the decline in lung function (CitationBarnes and Stockley 2005; CitationGan et al 2005). Adverse effects of steroids include increased risk of hip fractures and osteoporosis, skin bruising and candidiasis (CitationGartlehner et al 2006), and the airway remodeling is not positively affected by the current treatment. Anti-oxidant therapy by mucolytics such as N-acetylcysteine is also being used as a treatment reducing acute exacerbation frequency, but generally fails to reduce airway inflammation or decline in lung function (CitationPoole and Black 2006; CitationSadowska et al 2007). Adverse effects of these mucolytic agents are rarely seen. The final part of this review focuses on the recent developments and advances in potential anti-oxidant and anti-cytokine treatment (Table ).
Table 2 Examples of potential cytokine and oxidant antagonist drugs for COPD
Development of antioxidant agents and anti-inflammatory therapies
Development of antioxidant therapies
Systemic and local antioxidant capacity and antioxidant vitamins
Smoking and exacerbations of COPD result in decreased antioxidant capacity in plasma in association with depleted protein sulphydryls in the plasma (CitationRahman et al 1996, Citation1997; CitationCorradi et al 2003). The decrease in antioxidant capacity in smokers occurs transiently during smoking and resolves rapidly after smoking cessation. In exacerbations of COPD, however, antioxidant capacity remains low for several days after the onset of the exacerbation, tending to return towards normal values at the time of recovery from the exacerbation (CitationRahman et al 1997). The depletion of antioxidant capacity could in part be explained by the increased release of ROS from peripheral blood neutrophils, as shown by a significant negative correlation between neutrophil superoxide anion release and plasma antioxidant capacity (CitationRahman et al 1996). Thus there is clear evidence that oxidants in cigarette smoke markedly decreases plasma antioxidants, which may play an important role in the pathogenesis of COPD. Furthermore, it is possible that inter-individual differences in antioxidant capacity may contribute to the differences in susceptibility to cigarette-smoke induced COPD. Thus, it is imperative to propose the rationale for antioxidant therapy ameliorating the increased oxidative stress and consequently the inflammatory response in COPD.
Depletion of total antioxidant capacity in smokers is associated with decreased levels of major plasma antioxidants in smokers (CitationPetruzzelli et al 1990; CitationBridges et al 1993; Citationvan Antwerpen et al 1993; CitationMezzetti et al 1995; CitationRahman and MacNee 1996a). These studies show depletion of ascorbic acid, vitamin E, β-carotene and selenium in the serum of chronic smokers and in patients with COPD (CitationPetruzzelli et al 1990; CitationBridges et al 1993; Citationvan Antwerpen et al 1993; CitationMezzetti et al 1995; CitationTug et al 2004). Moreover, decreased vitamin E and vitamin C levels were reported in leukocytes and BAL fluids from smokers. Ascorbate appears to be a particularly important antioxidant in the plasma. Cigarette smoke-induced lipid peroxidation of plasma in vitro is decreased by ascorbate (CitationCross et al 1994). Reduced levels of vitamin E and a marginal increase in vitamin C in the BAL fluid of smokers, compared to nonsmokers have been shown (CitationRahman and MacNee 1996a). Similarly, alveolar macrophages from smokers have both increased levels of ascorbic acid and augmented uptake of ascorbate, suggesting that these cells are trying to redress their antioxidant balance (CitationRahman and MacNee 1996a).
Dietary antioxidants supplementation is one of the simplest approaches to boost antioxidant defense systems. Supplementation of vitamin C, vitamin E and β-carotene has been attempted in cigarette smokers and patients with COPD (CitationCross et al 1993; CitationRautalahti et al 1997; CitationSteinberg and Chait 1998; CitationAghdassi et al 1999; CitationHabib et al 1999; CitationLykkesfeldt et al 2000; CitationUneri et al 2006). In the general population there is a positive association between dietary intake of antioxidant vitamins and lung function. Epidemiological studies have demonstrated negative associations of dietary antioxidant intake with pulmonary function and with obstructive airway disease (CitationGrievink et al 1998). Britton and co-workers (CitationBritton et al 1995) showed a positive association between dietary intake of the antioxidant vitamin E and lung function in a population of 2,633 subjects, supporting the hypothesis that this antioxidant may have a role in protecting against the development of COPD. Another study has suggested that antioxidant levels in the diet could be a possible explanation for differences in COPD mortality in different populations (CitationSargeant et al 2000). Dietary polyunsaturated fatty acids may also protect cigarette smokers against the development of COPD (CitationShahar et al 1999). These studies support the concept that dietary antioxidant supplementation including polyphenols may be a possible therapy to prevent or inhibit the oxidative stress and inflammatory responses, which are key features in the development of COPD. However, robust clinical trials using dietary antioxidant vitamins and polyphenols are urgently needed to address the beneficial effects of these antioxidants in COPD.
Directly increasing lung antioxidant capacity
The development and progress of COPD is associated with increased oxidative stress or decreased antioxidant resources (CitationBoots et al 2003). The most direct way to redress the oxidant/antioxidant imbalance in COPD would be to increase the pulmonary capacity by antioxidants (Table ). A variety of means by which to do this have been attempted with varying success.
Table 3 Examples of antioxidant compounds currently in clinical trials for COPD treatment
Glutathione and its biosynthesis
The thiol antioxidant glutathione (GSH) is concentrated in epithelial lining fluid compared with plasma and has an important protective role in the airspaces and intracellularly in epithelial cells. Several studies have suggested that GSH homeostasis may play a central role in the maintenance of the integrity of the lung airspace epithelial barrier. Decreasing the levels of GSH in epithelial cells leads to loss of barrier function and increased permeability (CitationMorrison et al 1999). Human studies have shown elevated levels of glutathione in epithelial lining fluid in chronic cigarette smokers compared with non-smokers (CitationMorrison et al 1999). However, this increase is not present immediately after acute cigarette smoking (CitationMorrison et al 1999). The two-fold increase in BALF GSH in chronic smokers may not be sufficient to deal with the excessive oxidant burden during smoking, when acute depletion of GSH may occur (CitationHarju et al 2002). In addition, the immunoreactivity of γ-glutamylcysteine synthetase (γ-GCS; now called as glutmate cysteine ligase, GCL), the rate limiting enzyme in GSH synthesis, was decreased in the airways of smokers compared to nonsmokers, suggesting that cigarette smoke predisposes lung cells to ongoing oxidant stress (CitationHarju et al 2002). Neurohr and colleagues recently showed that decreased GSH levels in BALF cells of chronic smokers were associated with a decreased expression of γ-GCS/GCL-light subunit without a change in γ-GCS/GCL-heavy subunit expression (CitationNeurohr et al 2003). Increasing the activity of γ-GCS/GCL, would be expected to increase cellular GSH levels. The induction of γ-GCS/GCL by molecular means to increase cellular GSH levels or γ-GCS/GCL gene therapy also holds great promise in protection against chronic inflammation and oxidant-mediated injury in COPD.
Direct increase of lung cellular levels of GSH would be a logical approach to enhance the antioxidant potential in the treatment of COPD. In fact, extracellular augmentation of GSH has been tried through intravenous administration of GSH, oral ingestion of GSH, and aerosol inhalation of nebulized GSH in an attempt to reduce inflammation in various lung diseases (CitationRahman and MacNee 1999, Citation2000a, Citation2000b). However, these routes of administration lead to undesirable effects suggesting that direct GSH therapy may not be an appropriate way of increasing GSH levels in lung epithelial lining fluid and cells in COPD. The bioavailability of GSH, pH, osmolility in the inflammatory micro-environment and the resultant formation of toxic products (GSSG and GSH-adducts) are further challenges for direct GSH administration. Alternative formulations may address bioavailability, such as liposomal delivery, but at present it seems that direct administration of GSH will not be successful in treating COPD.
N-acetyl-L-cysteine (NAC)
NAC, a cysteine-donating reducing compound, acts as a cellular precursor of GSH and becomes de-acetylated in the gut to cysteine following oral administration (CitationCotgreave 1997; CitationRepine et al 1997). NAC may also reduce cystine to cysteine, which is an important mechanism for intracellular GSH elevation in vivo in lungs. It reduces disulphide bonds (a property of a good reducing agent), but also has the potential to interact directly with oxidants. NAC is also used as a mucolytic agent, to reduce mucus viscosity and to improve mucociliary clearance.
Pharmacological administration of NAC has been used in an attempt to enhance lung GSH in patients with COPD with varying success (CitationRasmussen and Glennow 1988; CitationBridgeman et al 1994). Schooten et al have reported that in a randomized, double-blind, placebo-controlled Phase II trial, a six month oral dose of 600 mg twice a day (b.i.d), reduced various plasma and BAL fluid oxidative biomarkers in smokers (CitationVan Schooten et al 2002). Similarly, it has been shown that treatment with NAC 600 mg once daily for 12 months also reduced the concentration of H2O2 in EBC compared to placebo in stable COPD patients (CitationKasielski and Nowak 2001). A more recent clinical trial also proves that oral administration of NAC 600 mg b.i.d. for 2 months rapidly reduces the oxidant burden in airways of stable COPD patients (CitationDe Benedetto et al 2005). This reduction in oxidative biomarkers results in clinical benefit such as reduction in bronchial hypersecretion, in addition to decline in FEV1 and in exacerbations (CitationStey et al 2000). Orally dosed NAC has been shown to increase phagocytic activity of BAL macrophages from healthy smokers, but similar results were not seen in COPD patients, possibly due to active concentrations of NAC not reaching the lung, as in vitro analysis of cells support an induction of phagocytosis by NAC. It has also been reported recently that orally dosed NAC increased the quadriceps endurance time of severe COPD patients (CitationKoechlin et al 2004), thus suggesting that NAC administration may have beneficial effects on the systemic oxidative stress associated with COPD. However, a multi-centre study using NAC delivered by metered dose inhalers in patients with chronic cough failed to show a positive effect on well being, sensation of dyspnoea, cough or lung function (CitationDueholm et al 1992).
Whilst there is a body of evidence that the administration of NAC provides benefit for COPD patients, it is not clear whether this represents a maintenance therapy (CitationDecramer et al 2001, Citation2005). A phase III multicentre Bronchitis Randomized on NAC Cost-Utility Study (BRONCUS) has recently been completed, with the aim of addressing this question and determining whether the effectiveness of NAC as an “antioxidant” results in an alteration in the rate of decline in FEV1, exacerbation rate and quality of life in patients with moderate to severe COPD and hence support NAC administration as a maintenance therapy for COPD (CitationGerrits et al 2003; CitationDekhuijzen 2004). The results of the phase III BRONCUS trials showed no effect on decline in FEV1 but a reduction in hyperinflation in patients with severe COPD and exacerbation rate in patients who are not treated with inhaled glucocorticoids (CitationDecramer et al 2001, Citation2005).
N-acystelyn (NAL)
NAL is a lysine salt of N-acetyl-L-cysteine, and is a mucolytic and antioxidant (reducing) thiol compound. The advantage of NAL over NAC is that it has a neutral pH in solution, whereas NAC is acidic. NAL can be aerosolized into the lung without causing significant side effects (CitationGillissen et al 1997). Gillissen and co-workers compared the effect of NAL and NAC and found that both drugs enhance intracellular glutathione in alveolar epithelial cells and inhibited hydrogen peroxide and O2 • −released from human blood-derived polymorphonuclear cells (PMN) from smokers with COPD (CitationGillissen et al 1997). NAL also inhibited ROS generation induced by serum-opsonised zymosan by human neutrophils. This inhibitory response was comparable to the effects of NAC (CitationGillissen et al 1997). Antonicelli and colleagues have shown that NAL inhibited oxidant-mediated CXCL8 release in alveolar epithelial A549 cells suggesting an anti-inflammatory effect of NAL (CitationAntonicelli et al 2002). Therefore, NAL may represent an interesting alternative approach to augment the antioxidant screen and thereby inhibiting inflammatory responses in the lungs and has the advantage over other antioxidant agents in that it may be administered by inhalation. The clinical trials using NAL in the treatment of COPD are in progress.
N-isobutyrylcysteine (NIC)
Because NAC becomes hydrolyzed in biological systems, the measured bioavailability of the drug is low. Thus, it was speculated that a drug might be synthesized that possessed greater bioavailability than NAC, and could be used as a more effective treatment for chronic bronchitis. N-isobutyrylcysteine (NIC) is a NAC-like thiol compound that does not undergo effective first-pass hydrolysis and hence has a higher oral bioavailability than NAC (CitationEkberg-Jansson et al 1999). The oral bioavailability can be as high as 80%, dependent on food intake. However, when evaluated as a therapy for exacerbations of chronic bronchitis, NIC performed no better than placebo drugs, and not as well as NAC (CitationGillissen et al 1997). Furthermore, a study of NIC also failed to reduce exacerbation rates in patients with COPD (CitationEkberg-Jansson et al 1999).
Erdosteine
Erdosteine is a new thiol compound that also acts as an anti-oxidant, but in addition has mucoactive properties and reduces bacterial adhesiveness. In the “Equalife” randomized placebo controlled clinical study, erdosteine was dosed orally 300 mg twice a day (b.i.d) for a period of 8 months (CitationMoretti et al 2004). Patients receiving erdosteine had significantly fewer exacerbations and spent less days in hospital than the placebo group. Moreover, patients receiving erdosteine showed no reduction in lung function over this period and showed a significant improvement in health related quality of life. It is not clear whether this clinical benefit is due to antioxidant effects of erdosteine. The mucolytic effect of erdosteine is perhaps due to the presence of a sulphydryl group. It may be possible that erdosteine may reduce bacterial colonization through a direct effect on adhesion.
Procysteine
Procysteine (L-2-oxothiazolidine-4-carboxylate), is a cysteine donating compound which increases the cysteine levels of the cells and has a greater bioavailability than NAC. This thiol compound is well tolerated is has been shown to increase mitochondrial levels of GSH in alveolar type II cells (CitationGuidot and Brown 2000). Glutathione esters, particularly GSH monoethyl esters can increase the GSH levels of the cells by cleavage of ester bond (an ethyl group esterified to glycine). GSH esters have been shown to increase GSH levels in the lungs of rats, however, this compound can be cytotoxic and variation in the uptake levels of GSH has been shown in various cellular models (CitationButterworth et al 1993).
Antioxidant enzyme mimetics and spin traps
Increased activity of antioxidant enzymes (superoxide dismutase and catalase) in alveolar macrophages from young smokers have been reported (CitationMcCusker and Hoidal 1990). However, Kondo and co-workers (CitationKondo et al 1994) found that the increased superoxide generation by alveolar macrophages in elderly smokers was associated with decreased antioxidant enzyme activities when compared with non-smokers. The activities of CuZnSOD, glutathione-S-transferase and glutathione peroxidase (GP) are all decreased in alveolar macrophages from elderly smokers (CitationGilks et al 1998).
The activities of SOD and glutathione peroxidase have been shown to be higher in the lungs of rats exposed to cigarette smoke. CitationMcCusker and Hoidal (1990) have also demonstrated enhanced antioxidant enzyme activity in alveolar macrophages from hamsters following cigarette smoke exposure, which resulted in reduced mortality when the animals were subsequently exposed to >95% oxygen. They speculated that alveolar macrophages undergo an adaptive response to chronic oxidant exposure that ameliorates potential damage to lung cells from further oxidant stress. The mechanism(s) for the induction of antioxidant enzymes in erythrocytes, alveolar macrophages, and lungs, by cigarette smoke exposure are currently unknown.
Spin traps such as α-phenyl-N-tert-butyl nitrone react directly with reactive oxygen and reactive nitrogen species at the site of inflammation (CitationChabrier et al 1999). In a recent study, Smith and colleagues have shown that intratracheal instillation of a catalytic antioxidant, manganese (III) mesotetrakis (N,N'-diethyl-1,3-imidazolium-2-yl) porphyrin (AEOL 10150 and AEOL 10113) inhibited the cigarette smoke-induced inflammatory response (decreased number of neutrophils and macrophages) in rats after 2 d or 8 weeks (6 hours/day, 3 days/week) exposure (CitationSmith et al 2002). These compounds also mimic extracellular SOD and catalase, scavenging both lipid peroxides and peroxynitrite, and have been shown to be effective in a number of animal models of lung disease.
It has been shown that SOD mimetic M40419 blocked the development of emphysema and significantly reduced lung markers of oxidative stress in an animal model (CitationTuder et al 2003). Animal studies have shown that recombinant SOD treatment can prevent the neutrophil influx to the airspaces and CXCL8 release induced by cigarette smoking through a mechanism involving down regulation of NF-κB (CitationNishikawa et al 1999). This further substantiate the idea that generation of compounds with anti-oxidant enzyme properties may be able to act as novel anti-inflammatory drugs by regulating the molecular events in COPD.
Development of anti-inflammatory therapies
NF-κB inhibitors
Studies with IκBα mutants (CitationBaldwin 1996; CitationGhosh et al 1998) gave the first evidence that NF-κB pathway could be specifically inhibited. Signal-induced phosphorylation and degradation of cytoplasmic IκBα is required for NF-κB pathway activation. However, an IκBα protein with mutations at serine-32 and 36 is not phosphorylated by IKK (IκBα kinase) and therefore not degraded by the proteasome. This IκBα mutant or super-repressor exerts its negative effect by sequestering NF-κB in the cytoplasm and thus prevents the induction of specific NF-κB target genes.
Another novel way whereby NF-κB activity may be regulated is by the use of inhibitors of proteasome function, which can reduce the degradation of IκB and thus prevent NF-κB activation (CitationBaldwin 1996; CitationGhosh et al 1998). A series of peptide aldehydes such as MG101, MG132, and MG115, make up a family of agents that inhibit the protease activity of the proteasome. Lactacystin, another class of proteasome inhibitor, blocks proteolytic activity by acylating a threonine residue in one of the key proteasome subunits. Furthermore, a group of boronic acid peptides, including PS-341, are extremely potent inhibitors of proteasome function (CitationAdams et al 1999), thus inhibiting activation of the NF-κB pathway. It is also possible that inhibitors of the ubiquitin ligase that mediates IκB ubiquitination may be a useful target in preventing proteasome degradation of IκB. Thus, a wide variety of potential inhibitors of proteasome function may have a therapeutic role in anti- NF-κB pathway dependent strategies.
Certain natural antioxidants/products such as flavonoids/polyphenols quercetin, curcumin, resveratrol, and myricetin are also known to mediate their anti-inflammatory properties through down-regulation of the NF-κB pathway (CitationTsai et al 1999; CitationHolmes-McNary and Baldwin 2000). For example, resveratrol, which is found in red wine, can inhibit NF-κB activity and induce apoptosis in transformed cells, which may reduce mortality from coronary heart diseases, certain cancers and inflammatory diseases (CitationHolmes-McNary and Baldwin 2000). Resveratrol has strong inhibitory effects on iNOS expression and NO generation in activated macrophages (CitationTsai et al 1999). Since treatment of macrophages with resveratrol blocks LPS-induced phosphorylation and degradation of IκBα to decrease NF-κB DNA binding activity, is suggestive of the fact that its anti-inflammatory effects may be due at least in part to the inhibition of NF-κB-dependent NO synthesis (CitationTsai et al 1999). Thus several of the biological activities of flavonoids may be mediated by their inhibition of the NF-κB pathway.
Thus it is evident that there are several possible approaches to inhibition of NF-kB, including gene transfer of IκB, inhibitors of IκB kinases (IKK), NF-κB-inducing kinase and IκB ubiquitin ligase, which regulate the activity of NF-κB, and inhibit the degradation of IκB (CitationDelhase et al 2000). The most promising approach however, may be the inhibition of IKK-2 by small molecule inhibitors (CitationCastro et al 2003) (Table ), which suppress the release of inflammatory cytokines and chemokines from alveolar macrophages (CitationJazrawi et al 2003). This in particular might be more effective in COPD, particularly since alveolar macrophages are resistant to the anti-inflammatory actions of corticosteroids (see HDACs modifiers). It is however, of concern that long-term inhibition of NF-κB, with effective inhibitors may result in immune suppression and therefore impair host defenses. This concern is validated from a study that mice lacking NF-κB genes succumb to septicemia. However, alternative modulation of pathways of NF-κB activation via kinases other than IKK might be a more safer approach in inflammatory disease and would have less potential effect on innate and adaptive immune responses (CitationNasuhara et al 1999).
PDE4 inhibitors
Phosphodiesterase 4 (PDE4) is the predominant PDE isoenzyme in most inflammatory cells thought to have a role in the pathogenesis of COPD (Figure ). Its activity is elevated in lung macrophages from COPD patients (CitationBarber et al 2004). In contrast to steroids that have a limited anti-inflammatory efficacy in cigarette smoke models both in the mouse and guinea pig, there are increasing numbers of studies documenting the in vivo efficacy of PDE4 inhibitor in animal models of COPD. There are at least currently five oral PDE4 inhibitors in clinical development for COPD, one of which is suspended (C1393 in phase II, from Merck) (see Table ). A major hurdle in their development has been to overcome their side effects which include nausea, emesis, and headache.
In 24 weeks Phase multi-center III trails in COPD patients (RECORD trial), oral administration of roflumilast or cilomilast improved pre- and post-bronchodilator FEV1 (CitationRabe et al 2005; CitationRennard et al 2006). The health-related quality of life (SGRQ) was also improved when compared with the placebo control. In addition, exacerbation frequency was lower in drugs group than in the placebo group.
The relationship between these improvements in clinical outcome and potential anti-inflammatory activity has been investigated in a single study (CitationGamble et al 2003; CitationGrootendorst et al 2005). After a 4-week treatment with roflumilast post-bronchodilator FEV1 improved by 68.7 ml compared with placebo. Treatment with roflumilast significantly reduced the absolute numbers of neutrophils and eosinophils of sputum. These were paralleled with by a reduction in CXCL8 and neutrophil elastase. Although a 12 weeks treatment with cilomilast had no effect on sputum neutrophils, macrophages, elastase, CXCL8 or lung funtion, bronchial biopsies demonstrated that cilomilast treatment was associated with significant reductions in CD8+ T lymphocyte and CD68+ cells. The results showed that related outcomes observed in longer term trials could be due, at least in part, to anti-inflammatory activity of drugs.
In an attempt to reduce the potential for systemic side effects and to administer relatively higher doses to the lung, inhaled PDE4 inhibitors are being developed. GSK842470 (AWD-12-281) was licensed from Elbion and reached Phase II for asthma and COPD but there have been unconfirmed reports that it had no advantage over oral PDE4 inhibitors. This compound no longer appears on GSK’s pipeline but remains in development for rhinitis by Elbion. Currently, GSK (SB256066, Phase I) and Pfizer (Phase II) are reported to have inhaled PDE4 inhibitors in clinical development for COPD.
Experimental data suggest that PDE4D inhibition is one likely cause of the side effects of the orally-delivered compounds, while PDE4B is a therapeutically relevant target. Therefore, PDE4 subtype inhibitors eg, PDE4B for treatment of COPD is being studied by Plexxikon.
MAPK p38 inhibitors
MAPKs play a key role in chronic inflammation and several complex enzyme cascades have now been defined (CitationJohnson and Lapadat 2002). One of these, the p38 MAPK pathway, is activated by cellular stress and regulates the expression of a wide variety of inflammatory cytokines that include CXCL8, TNFα and MMPs (CitationMeja et al 2000). Small molecule inhibitors of MAP kinase p38, such as SB 203580, SB 239063 and RWJ 67657 having a broad range of anti-inflammatory effects have been developed (CitationKumar et al 2003) (Table ). Administration of SB203580 has beneficial effects in animal disease models such as collagen-induced arthritis and endotoxin-induced septic shock (CitationLee et al 1999). p38 has also been shown to upregulate cytokine production by several independent mechanisms, including direct phosphorylation of transcription factors, and direct or indirect (through downstream kinases such as MAPKAPK2) stabilization and increased translation of mRNAs containing 3% untranslated region adenylate/uridylate-rich elements (AREs) by phosphorylation of AREbinding proteins (CitationDean et al 2004; CitationBriata et al 2005; CitationHitti et al 2006). These observations have attracted interest in p38 as a molecular target in the treatment of inflammatory human diseases. MAPK p38 has 4 isozymes. Each inhibitor has its own specificity towards one of more of these isozymes, causing differential effects Studies in healthy volunteers given p38α/p38β inhibitors found reductions in pro-inflammatory cytokine secretion from ex-vivo LPS-stimulated peripheral-blood mononuclear cells (PBMCs) (CitationParasrampuria et al 2003), and decreased LPS-induced pro-inflammatory cytokine production, neutrophil and endothelial-cell activation in vivo. SB239063 on the other hand reduces neutrophil infiltration and the concentrations of IL-6 and MMP-9 in BALF of rats after endotoxin inhalation, suggesting its potential as an antiinflammatory agent in COPD (CitationUnderwood et al 2000).
The potential therapeutic utility of p38 MAPK inhibition in respiratory disease has been supported by data generated in a range of pulmonary inflammatory models in vivo including LPS induced pulmonary neutrophilia (CitationHaddad et al 2001), bleomycin induced fibrosis (CitationMatsuoka et al 2002), and antigen induced eosinophilia (CitationUnderwood et al 2000). A recent study demonstrated the efficacy of p38α MAPK inhibitor, SD282, in mouse COPD models (CitationFitzgerald et al 2006). In this model, SD-282 inhibited cigarette smoke induced pulmonary neutrophilia and macrophage recruitment. Although a number of oral p38 MAPK inhibitors are in clinical development for arthritis and cancer only two compounds are currently in development for COPD. GSK-681323 is currently in a 4 week Phase II trial where the efficacy outcome measures include lung function, sputum and serum biomarkers, including CRP. GSK-85633 is in Phase I. Although it is likely that such a broad spectrum anti-inflammatory drug might ensue some toxicity or impair natural immune responses, but inhalation route might be a feasible therapeutic approach.
Elastase inhibitor
Neutrophil elastase is a serine protease that is synthesized in the neutrophils and secreted following neutrophils activation. One of the major actions of neutrophil elastase is its ability to degrade matrix proteins and appears to be a key enzyme in the development of hereditary due to α-antitrypsin deficiency. Although its role in the development of non hereditary emphysema is currently unclear it also has a broad range of actions that are consistent with it having a pro-inflammatory role in COPD pathogenesis (CitationShapiro et al 2003; CitationChughtai and O’Riordan 2004). Neutrophil elastase can increase MMPs activity by directly activating MMPs such as MMP-9 (CitationFerry et al 1997) and by inactivating the endogenous MMP inhibitor, TIMP (CitationOkada et al 1988), thus potentially enhancing the role of MMPs in COPD. Neutrophil elastase has also been reported to stimulate mucin secretion and modulate apoptosis of human lung epithelial cells. Therefore, neutrophil elastase may play a role in emphysema and lung remodeling through matrix degradation and by inducing apoptosis.
In cigarette smoke models, neutrophil elastase knockout mice are protected from emphysema development and this effect is accompanied by an inhibition of both neutrophils and macrophages recruitment (CitationShapiro et al 2003). Treatment with α1-antitrypsin inhibited emphysema development and reduced recruitment of pulmonary neutrophils and macrophages (CitationChurg et al 2003; CitationPemberton et al 2006). Similarly, a small molecule neutrophil elastase inhibitor, ZD0892, reduced cigarette smoke induced emphysema and pulmonary cells recruitment in a guinea pig model (CitationWright et al 2002). These studies suggest inhibition of neutrophil elastase can be anti-inflammatory in addition to preventing emphysema development.
The search for potent, safe oral inhibitors of neutrophil elastase has been going on for over 20 years and many of the compounds that progressed into clinical development failed because of poor pharmacokinetics and a low therapeutic index. Subsequently, tripeptidyl trifluoromethyl ketones were developed that had a better oral profile, although these molecules were not fully optimized for oral delivery (CitationEdwards et al 1997). Also monocyclic b-lactam neutrophil elastase inhibitors were identified (CitationVincent et al 1997). More recently, both GSK (GSK-311366) and ONO Pharmaceuticals (ONO6818) have had oral compounds in Phase I/II. However, they failed in early clinical trials and thus far no oral elastase inhibitors have been evaluated fully in Phase II COPD trials. The AstraZeneca protease inhibitor AZD3342 (thought to be an elastase inhibitor) is scheduled to progress to Phase II studies in the second half of 2006 and Bayer GmbH has an inhibitor in Phase I but the outcomes of these trials are still awaited.
Protein inhibitors of neutrophil elastase administered via nebulisation are also being pursued and supportive data has been obtained for inhaled recombinant α1-antitrypsin in mouse cigarette smoke models. The probability of inhaled combination products consisting of elastase inhibitors with bronchodilators is low. However, the opportunity for safe long acting inhaled elastase inhibitors with low systemic bioavailability that are suitable for combination with bronchodilators is being pursued by Argenta Discovery.
HDACs modifiers
NF-κB being a pro-inflammatory transcription factor, on activation binds to specific recognition sequence motifs in DNA and subsequently interacts with a multitude of co-activator molecules, such as cAMP-response-element-binding protein (CREB), CREB-binding protein (CBP), p300 and p300/CBP-associated factor (pCAF). These co-activators act as molecular switches of transcription and acetylate histones by their intrinsic histone acetyltransferase (HAT) activity (CitationOgryzko et al 1996; CitationRoth et al 2001). Acetylation of core histones allow DNA unwinding and thus access to wide variety of transcription factors (CitationRoth et al 2001). The transcriptional process can be reversibly switched off by deacetylation of acetylated histones, thus rewinding the exposed DNA, consequently leading to gene silencing. The deacetylation of acetylated histones is brought about by histone deacetylases (HDACs), which act as co-repressors, in association with other co-repressor proteins that are concomitantly recruited. Since all inflammatory processes involve acetylation and deacetylation of histones (chromatin remodeling) (CitationBarnes 2006), HDACs therefore are attractive targets for anti-inflammatory therapies.
Ito and co-workers have shown a role for histone acetylation and deacetylation in IL-1β-induced TNF-α release in alveolar macrophages derived from cigarette smokers (CitationIto et al 2001). They have also suggested that oxidants may play an important role in the modulation of HDAC and inflammatory cytokine gene transcription. Furthermore, we have shown that both cigarette smoke/H2O2 and TNF-α caused an increase in histone acetylation (HAT activity) leading to CXCL8 expression in monocytes and alveolar epithelial cells both in vitro and in vivo in rat lungs (CitationRahman et al 2002; CitationMarwick et al 2004; CitationMoodie et al 2004).
Glucocorticoid suppression of inflammatory genes requires recruitment of HDAC2 to the transcription activation complex by the glucocorticoid receptor (CitationIto et al 2001; CitationRahman et al 2004). This results in deacetylation of histones and a decrease in inflammatory gene transcription. A reduced level of HDAC2 was associated with increased proinflammatory response and reduced responsiveness to glucocorticoids in alveolar macrophages obtained from smokers (CitationIto et al 2001; CitationRahman et al 2002; CitationMarwick et al 2004; CitationMoodie et al 2004; CitationRahman et al 2004). Culpitt and co-workers have shown that cigarette smoke solution stimulated release of CXCL8 and GM-CSF, which was not inhibited by dexamethasone, in alveolar macrophages obtained from patients with COPD compared to that of smokers (CitationCulpitt et al 2003). They suggested that the lack of efficacy of corticosteroids in COPD might be due to steroid insensitivity of macrophages in the respiratory tract. Thus, the cigarette smoke/oxidant-mediated reduction in HDAC2 levels in alveolar epithelial cells and macrophages will not only increase inflammatory gene expression but will also cause a decrease in glucocorticoid function in patients with COPD. HDAC2 activity has also been measured in bronchial biopsies and alveolar macrophages from COPD patients and smoking control, demonstrating a significant decrease in HDAC2 activity, the magnitude of which increased with severity of disease (CitationIto et al 2005) (Table ). Moreover, protein expression of HDAC2 was decreased in a similar manner in COPD patients.
Consequently, a potential means by which to treat COPD would be to increase HDAC2 expression and activity such that steroids regain their anti-inflammatory activity. We have shown that co-incubation of cells with NAC and H2O2 protects HDAC2 from down-regulation and reduction of specific activity (CitationMoodie et al 2004). In addition, it has been reported that theophylline has a similar effect in lung macrophage cells, increasing HDAC2 expression and re-sensitising the cells to steroids (CitationCosio et al 2004). Similar data were obtained for curcumin, a dietary polyphenols, in restoration of steroid efficacy (unpublished data). Alternative means of upregulating HDAC2 activity would be of great interest for potential combination therapies of the future.
Cytokine and chemokine antagonists
Several recent reviews point to the development of novel antagonists of cytokines, chemokines or their receptors (CitationDe Boer 2005; CitationChung 2006; CitationDe Boer et al 2007). These molecules may reduce gene expression, impair production or secretion of mature proteins, antagonize binding of cytokines and chemokines to their receptors or inhibit receptor signal transduction (Table ). Antibodies and solubilized receptors like TNFR often scavenge solubilized cytokines and chemokines, or prevent binding of these proteins to their receptors. Small molecules 1) prevent binding of cytokines and chemokines to their receptors by non-activating mimicking of cytokines or chemokines, or 2) prevent intracellular signal transduction activation, or 3) interfere with gene expression and translation by direct inhibition of transcription factors (like IKK2 inhibition) or mRNA binding via small interference (si) RNA or antisense mRNA.
TNFα and receptors antagonists
As a major proinflammatory cytokine TNFα and its receptors TNFR1 (or: TNFR p55) and TNFR2 (or: TNFR p75) seem to play an important role in many chronic diseases including COPD and asthma. Therefore, several drugs have been developed to reduce TNFα levels, of which some have been approved by eg, the Federal Drug Administration (FDA) for treatment of RA, ankylosing spondylitis, Crohn’s disease, or psoriasis. These approved drugs include etanercept (soluble human TNFR2), infliximab (chimeric human/mouse IgG1 antibody against TNFα), and adalumimab (human IgG1 antibody against TNFα). Many others are being developed in order to enhance efficacy, reduce side effects due to frequent subcutaneous injection, increase bioavailability or protection to proteolytic degradation by coupling to polyethylene glycol (PEG) chains, or reduce immunogenicity by humanization of antibodies or designing small molecules. Some examples are given in Table . Recent clinical trial phase II studies demonstrated that patients with moderate to severe asthma may profit from treatment with either of these drugs. Three infusions of infliximab over 6 weeks reduced the number of exacerbations as well as sputum levels of TNFα, IL-6, CXCL8 and CXCL10 but not peak expiratory flow (PEF) or inflammatory cell count in sputum of patients with moderate asthma (CitationErin et al 2006). Other studies demonstrated that twice-weekly treatment with etanercept during 10 to 12 weeks improved the bronchial hyperresponsiveness (BHR, expressed as PC20), post-bronchodilator FEV1 and the quality of life of patients with refractory, severe asthmatic patients (CitationHowarth et al 2005; CitationBerry et al 2006). Treatment of asthmatics with Marimastat, an inhibitor of TNFα and MMP activation, also reduced BHR but failed to significantly reduce sputum inflammatory cell numbers, asthma symptoms, FEV1 or bronchodilator use (CitationBruce and Thomas 2005). In contrast to asthma, 2 studies showed that treatment of COPD patients with 3 infusions of infliximab over 6 to 24 weeks did not result in any significant improvement of lung function, airway inflammation, or quality of life (CitationAbdelhady et al 2005; Citationvan der Vaart et al 2005; CitationRennard et al 2007). Treatment may have some beneficial effect on physical endurance seen by the 6 minute walking distance test (CitationRennard et al 2007). This ineffectiveness may be due to a rather short treatment period, the choice of infliximab over other drugs, or to the complex or less significant role of TNFα in progressed COPD. Future studies should sort out whether long-term treatment or treatment with different TNFα antagonists are beneficial to all COPD patients or only a specific population of COPD patients (CitationRennard et al 2007).
CXCL1, CXCL8, and receptors antagonists
As previously mentioned (CitationDe Boer 2005), several CXCR2 and CXCL8 antagonists are available, some of which were in clinical trial for COPD. Updated information shows that either the testing of these drugs is discontinued (like the antibody ABX-IL-8 against human CXCL8) or is not to be found in the public domain. Hence, little is known yet on treatment of patients with COPD with CXCL8 or CXCR2 antagonists. The small molecule CXCR2 antagonist SB-656933 (by GSK) has recently been demonstrated to inhibit the CXCL8-induced expression of CD11b molecules on peripheral blood neutrophils from COPD patients (CitationNicholson et al 2007). The antagonist was mentioned to enter clinical trial studies for COPD in 2005, but is not so in GSK’s pipeline of 2006. AZD-8309 is a pyrimidine derivate currently in phase I clinical trial for COPD and phase II for RA. Data from these studies have not yet been published. SB-265610 is a small molecule inhibiting CXCR2. Studies demonstrated that hyperoxia in newborn rats led to pulmonary inflammation by neutrophils and the formation of ROS and RNS mediating impaired lung development and lipid peroxidation (CitationAuten et al 2001; CitationLiao et al 2006). Treatment with SB-265610 reduced airway neutrophilia, radical formation, lipid peroxidation and protein nitration, as well as improved conservation of lung development and lung function. This points to the importance of reducing neutrophilia in order to reduce reactive species formation, peroxidation or nitration and tissue destruction or alterations. Data from other studies supported the effectiveness of CXCL8 or CXCR2 antagonists in reducing neutrophilia in vivo in rodents and inhibition of neutrophil activation and degranulation in vitro (CitationDe Boer 2002, Citation2005). These data point to the potential need for development of novel antagonists of CXCR1, CXCR2 or their ligands CXCL1 and CXCL8. Recent studies showed that novel thiazolopyrimidine, cyclobutenedione (eg, SCH 527123), or imidazolylpyrimidine CXCR2 antagonists had a good oral bioavailability in rats with reasonable pharmacokinetics (half life of at least 1.2h) (CitationBaxter et al 2006; CitationDwyer et al 2006; CitationHo et al 2006), and inhibition of CXCL1- or CXCL8-induced chemotaxis of cells (CitationBaxter et al 2006; CitationDwyer et al 2006).
CCL2 and CCR2 antagonists
The humanized monoclonal antibody MLN1202 (by Millennium) against CCR2 is currently in phase II for treatment of chronic inflammatory diseases like multiple sclerosis and artherosclerosis. Clinical data from these studies have not yet been published. Other inhibitors in clinical trial include the small molecule CCR2 inhibitors CCX915 (by ChemoCentryx) for multiple sclerosis (phase I), INCB3284 (by Incyte/Pfizer) which is in phase IIa for treatment of RA, and the monoclonal antibody ABN912 against CCL2 (by Novartis) for COPD (phase I) and RA (phase II). Recently, patients with RA did not clinically and immunohistologically respond to a 2-week treatment with ABN912 (CitationHaringman et al 2006). Serum levels of CCL2 increased clearly after treatment (doses higher than 1 mg/kg bw), and chemotactic complexes of the antibody with CCL2 were formed. This may hamper the treatment of patients with COPD, however these results have to be awaited. Several other compounds based on butyramides or γ-aminoamides were recently developed as specific CCR2 antagonists with a good oral bioavailablity in rats (CitationButora et al 2006; CitationPasternak et al 2006). Others are in preclinical phase, including INCB8696, JNJ-27553292, SKL-2841 and INCB3344. INCB3344 is a rodent CCR2 small molecule antagonist (CitationBrodmerkel et al 2005). The compound was shown to inhibit macrophage influx in a mouse model for delayed hypersensitivity.
Other anti-inflammatory agents for COPD include ion channel blocker (P2X7, Phase II, from AstraZeneca), monoclonal antibody to IL-1β (Phase I, from Novartis), inhibitors of 5-lipoxygenase (PEP03 inhaled, Phase II, from PharmaEngine) and iNOS inhibitors (GW-274150, Phase II, from GSK).
Target growth factors
Several studies pointed to the involvement of VEGF/VEGFR in pulmonary and vascular remodeling and inflammation. Therefore, VEGF inhibitors may be of potential use for treatment of vascularization, as seen in asthma or in specific subtypes of COPD, such as chronic bronchitis, but not emphysema (CitationMarwick et al 2006; CitationDe Boer et al 2007). Several drugs have been developed to reduce tumor growth and metastasis by impairing the neovascularization. Antagonists include those in clinical trials and in preclinical investigations. Among those in clinical trial are VEGF-Trap, bevacizumab (or: Avastin), CEP-7055, VEGF Trap, and PTK787 (CitationFerrara et al 2003). However, little is known yet of side effects or their efficacy in the treatment of COPD. In addition, in patients with emphysema for example, inhibitors of apoptosis are needed whereas in contrast to chronic bronchitis, expression of hypoxia-inducible factor, VEGF, and VEGFR2 may be stimulated to overcome endothelial and epithelial cell death and improve vascular restoration (CitationVoelkel and Coll 2003). Hence, treatment with VEGF/VEGFR inhibitors should be further investigated and may be restricted to specific subtypes of chronic lung diseases.
As discussed aforementioned, TGFβ1 expression was increased in COPD as compared to smokers without COPD, inhibition of TGFβ-mediated pathway may hence be a target for therapeutic intervention. However, almost all of TGFβ1 antagonists are still in preclinical development. Hence, little can be discussed yet on clinical applicability.
Concluding remarks
In order to reverse continuing damage and remodeling needs stopping exposure to tobacco smoke by many patients with COPD. However, after smoking cessation inflammation goes on which may hamper tissue repair. The ongoing inflammation may be due to an autoimmune component and/or epigenetics alterations. However, not all patients with COPD are smokers but do suffer from tissue damage and/or remodeling. Hence, an effective treatment of patients with COPD not only needs stopping exposure to toxic compounds like smoke, but also needs reduction of excessive inflammation, oxidative status, and stimulation of parenchymal and/or airway repair (CitationLeff 2006). This needs further development and testing of therapeutic drugs antagonizing a low antioxidant status, inflammation, and excessive remodeling. There are several small molecule compounds in clinical trials that target oxidant signaling or quench oxidants derived from inflammatory cells and cigarette smoke. Regarding inhibition of inflammation several drugs have been used successfully for treating different chronic inflammatory diseases, but not yet for COPD. As pointed out in this review, success may be hindered by the complexity of actions of eg, chemokines and antioxidants, or by a low efficacy and bioavailability of the drugs. Whereas more insight is needed in the pathogenesis of COPD or its subtypes, individualized combination therapy with such novel and specific antiinflammatory and anti-oxidant drugs may be advantageous to patients with specific phenotypes of the disease.
References
- AbdelhadyHFloresRTanH2005Infliximab treatment for chronic obstructive pulmonary disease (COPD) – a pilot studyProc Am Thorac Soc2A133
- AdamsJPalombellaVJSausvilleEAProteasome inhibitors1999a novel class of potent and effective antitumor agentsCancer Res5926152210363983
- AghdassiERoyallDAllardJP1999Oxidative stress in smokers supplemented with vitamin CInt J Vitam Nutr Res69455110052021
- AgustiAGNogueraASauledaJ2003Systemic effects of chronic obstructive pulmonary diseaseEur Respir J213476012608452
- AntonicelliFParmentierMDrostEM2002Nacystelyn inhibits oxidant-mediated interleukin-8 expression and NF-kappaB nuclear binding in alveolar epithelial cellsFree Radic Biol Med3249250211958950
- AutenRLRichardsonRMWhiteJR2001Nonpeptide CXCR2 antagonist prevents neutrophil accumulation in hyperoxia-exposed newborn ratsJ Pharmacol Exp Ther29990511561067
- BaldwinASJr1996The NF-kappa B and I kappa B proteins: new discoveries and insightsAnnu Rev Immunol14649838717528
- BalsRHiemstraPS2006Antimicrobial peptides in COPD – basic biology and therapeutic applicationsCurr Drug Targets77435016787176
- BarberRBaillieGSBergmannR2004Differential expression of PDE4 cAMP phosphodiesterase isoforms in inflammatory cells of smokers with COPD, smokers without COPD, and nonsmokersAm J Physiol Lung Cell Mol Physiol287L3324315047569
- BarnesPJShapiroSDPauwelsRA2003Chronic obstructive pulmonary disease: molecular and cellular mechanismsEur Respir J226728814582923
- BarnesPJStockleyRA2005COPD: current therapeutic interventions and future approachesEur Respir J25108410615929966
- BarnesPJ2006Corticosteroid effects on cell signallingEur Respir J274132616452600
- Basu-ModakSLuscherPTyrrellRM1996Lipid metabolite involvement in the activation of the human heme oxygenase-1 geneFree Radic Biol Med20887978743975
- BaxterACooperAKinchinE2006Hit-to-Lead studies: the discovery of potent, orally bioavailable thiazolopyrimidine CXCR2 receptor antagonistsBioorg Med Chem Lett16960316297626
- BerryMAHargadonBShelleyM2006Evidence of a role of tumor necrosis factor alpha in refractory asthmaN Engl J Med35469770816481637
- BootsAWHaenenGRBastA2003Oxidant metabolism in chronic obstructive pulmonary diseaseEur Respir J Suppl4614s27s14621103
- BrackeKRD’HulstAIMaesT2006Cigarette smoke-induced pulmonary inflammation and emphysema are attenuated in CCR6-deficient miceJ Immunol1774350916982869
- BriataPForcalesSVPonassiM2005p38-dependent phosphorylation of the mRNA decay-promoting factor KSRP controls the stability of select myogenic transcriptsMol Cell2089190316364914
- BridgemanMMMarsdenMSelbyC1994Effect of N-acetyl cysteine on the concentrations of thiols in plasma, bronchoalveolar lavage fluid, and lung tissueThorax4967058066561
- BridgesABScottNAParryGJ1993Age, sex, cigarette smoking and indices of free radical activity in healthy humansEur J Med220588261071
- BrightlingCEMcKennaSHargadonB2005Sputum eosinophilia and the short term response to inhaled mometasone in chronic obstructive pulmonary diseaseThorax60193815741434
- BrittonJRPavordIDRichardsKA1995Dietary antioxidant vitamin intake and lung function in the general populationAm J Respir Crit Care Med151138377735589
- BrodmerkelCMHuberRCovingtonM2005Discovery and pharmacological characterization of a novel rodent-active CCR2 antagonist, INCB3344J Immunol1755370816210643
- BruceCThomasPS2005The effect of marimastat, a metalloprotease inhibitor, on allergen-induced asthmatic hyper-reactivityToxicol Appl Pharmacol2051263215893540
- ButoraGMorrielloGJKothandaramanS20064-Amino-2-alkylbutyramides as small molecule CCR2 antagonists with favorable pharmacokinetic propertiesBioorg Med Chem Lett1647152216870431
- ButterworthMUpshallDGHobbsM1993Elevation of cysteine and replenishment of glutathione in rat lung slices by cysteine isopropylester and other cysteine precursorsBiochem Pharmacol451769748494535
- CantorJOCerretaJMArmandG1997Further investigation of the use of intratracheally administered hyaluronic acid to ameliorate elastase-induced emphysemaExp Lung Res23229449184790
- CantorJOShteyngartBCerretaJM2000The effect of hyaluronan on elastic fiber injury in vitro and elastase-induced airspace enlargement in vivoProc Soc Exp Biol Med225657110998200
- CantorJOCerretaJMOchoaM2005Aerosolized hyaluronan limits airspace enlargement in a mouse model of cigarette smoke-induced pulmonary emphysemaExp Lung Res314173016025922
- CastroACDangLCSoucyF2003Novel IKK inhibitors: betacarbolinesBioorg Med Chem Lett1324192212824047
- CeledonJCLangeCRabyBA2004The transforming growth factorbeta1 (TGFB1) gene is associated with chronic obstructive pulmonary disease (COPD)Hum Mol Genet1316495615175276
- ChabrierPEAuguetMSpinnewynB1999BN 80933, a dual inhibitor of neuronal nitric oxide synthase and lipid peroxidation: a promising neuroprotective strategyProc Natl Acad Sci U S A9610824910485910
- ChanezPVignolaAMO’ShaugnessyT1997Corticosteroid reversibility in COPD is related to features of asthmaAm J Respir Crit Care Med1551529349154853
- ChengJDiaz EncarnacionMMWarnerGM2005TGF-beta1 stimulates monocyte chemoattractant protein-1 expression in mesangial cells through a phosphodiesterase isoenzyme 4-dependent processAm J Physiol Cell Physiol289C9597015930146
- ChoiAMElbonCLBruceSA1994Messenger RNA levels of lung extracellular matrix proteins during ozone exposureLung17215308295431
- ChughtaiBO’RiordanTG2004Potential role of inhibitors of neutrophil elastase in treating diseases of the airwayJ Aerosol Med172899815684729
- ChungKF2006Cytokines as targets in chronic obstructive pulmonary diseaseCurr Drug Targets76758116787167
- ChurgAWangRDXieC2003alpha-1-Antitrypsin ameliorates cigarette smoke-induced emphysema in the mouseAm J Respir Crit Care Med16819920712689849
- ChurgATaiHCoulthardT2006Cigarette smoke drives small airway remodeling by induction of growth factors in the airway wallAm J Respir Crit Care Med17413273417008639
- CorradiMRubinsteinIAndreoliR2003Aldehydes in exhaled breath condensate of patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1671380612522029
- CosioBGTsaprouniLItoK2004Theophylline restores histone deacetylase activity and steroid responses in COPD macrophagesJ Exp Med2006899515337792
- CotgreaveIA1997N-acetylcysteine: pharmacological considerations and experimental and clinical applicationsAdv Pharmacol38205278895810
- CrossCEO’NeillCAReznickAZ1993Cigarette smoke oxidation of human plasma constituentsAnn N Y Acad Sci6867289discussion 89–90.8512263
- CrossCEvan der VlietAO’NeillCA1994Oxidants, antioxidants, and respiratory tract lining fluidsEnviron Health Perspect102Suppl 10185917705296
- CulpittSVRogersDFShahP2003Impaired inhibition by dexamethasone of cytokine release by alveolar macrophages from patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med167243112406856
- CulpittSVRogersDFTravesSL2005Sputum matrix metalloproteases: comparison between chronic obstructive pulmonary disease and asthmaRespir Med997031015878486
- De BenedettoFAcetoADraganiB2005Long-term oral n-acetylcysteine reduces exhaled hydrogen peroxide in stable COPDPulm Pharmacol Ther1841715607126
- De BoerWIvan SchadewijkASontJK1998Transforming growth factor beta1 and recruitment of macrophages and mast cells in airways in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med158195179847291
- De BoerWI2002Cytokines and therapy in COPD: a promising combination?Chest121209S218S12010854
- de BoerWI2005Perspectives for cytokine antagonist therapy in COPDDrug Discov Today109310615718158
- De BoerWIHauCMvan SchadewijkA2006Expression of epidermal growth factors and their receptors in the bronchial epithelium of subjects with chronic obstructive pulmonary diseaseAm J Clin Pathol1251849216393673
- De BoerWIAlagappanVKSharmaHS2007Molecular mechanisms in chronic obstructive pulmonary disease: potential targets for therapyCell Biochem Biophys471314817406066
- DeanJLSullyGClarkAR2004The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisationCell Signal1611132115240006
- DecramerMDekhuijzenPNTroostersT2001The bronchitis randomized on NAC cost-utility study (BRONCUS): hypothesis and design BRONCUS-trial CommitteeEur Respir J173293611405507
- DecramerMRutten-van MolkenMDekhuijzenPN2005Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (Bronchitis Randomized on NAC Cost-Utility Study, BRONCUS): a randomised placebo-controlled trialLancet36515526015866309
- DekhuijzenPN2004Antioxidant properties of N-acetylcysteine: their relevance in relation to chronic obstructive pulmonary diseaseEur Respir J236293615083766
- DelhaseMLiNKarinM2000Kinase regulation in inflammatory responseNature406367810935625
- DemedtsIKMorel-MonteroALebecqueS2006Elevated MMP-12 protein levels in induced sputum from patients with COPDThorax6119620116308335
- DentenerMAVernooyJHHendriksS2005Enhanced levels of hyaluronan in lungs of patients with COPD: relationship with lung function and local inflammationThorax601141915681498
- DentenerMALouisRClootsRH2006Differences in local versus systemic TNFalpha production in COPD: inhibitory effect of hyaluronan on LPS induced blood cell TNFalpha releaseThorax614788416517575
- DevalarajaRMNanneyLBDuJ2000Delayed wound healing in CXCR2 knockout miceJ Invest Dermatol1152344410951241
- Di StefanoACapelliALusuardiM2001Decreased T lymphocyte infiltration in bronchial biopsies of subjects with severe chronic obstructive pulmonary diseaseClin Exp Allergy3189390211422154
- DillonTJWalshRLScicchitanoR1992Plasma elastin-derived peptide levels in normal adults, children, and emphysematous subjects. Physiologic and computed tomographic scan correlatesAm Rev Respir Dis146114381443863
- DueholmMNielsenCThorshaugeH1992N-acetylcysteine by metered dose inhaler in the treatment of chronic bronchitis: a multicentre studyRespir Med8689921615189
- DwyerMPYuYChaoJ2006Discovery of 2-Hydroxy-N, N-dimethyl-3-{2-[[(R)-1-(5- methylfuran-2-yl)propyl]amino]-3,4-dioxocyclobut-1-enylamino}benzamide (SCH 527123): A Potent, Orally Bioavailable CXCR2/CXCR1 Receptor AntagonistJ Med Chem497603617181143
- EdwardsPDAndisikDWBryantCA1997Discovery and biological activity of orally active peptidyl trifluoromethyl ketone inhibitors of human neutrophil elastaseJ Med Chem401876859191965
- Ekberg-JanssonALarsonMMacNeeW1999N-isobutyrylcysteine, a donor of systemic thiols, does not reduce the exacerbation rate in chronic bronchitisEur Respir J138293410362048
- ErinEMLeakerBRNicholsonGC2006The effects of a monoclonal antibody directed against tumor necrosis factor-alpha in asthmaAm J Respir Crit Care Med1747536216840747
- FerraraNGerberHPLeCouterJ2003The biology of VEGF and its receptorsNat Med96697612778165
- FerryGLonchamptMPennelL1997Activation of MMP-9 by neutrophil elastase in an in vivo model of acute lung injuryFEBS Lett402111159037177
- FitzgeraldMFSpicerDMaJY2006Efficacy of the p38 MAP Kinase Inhibitor, SD-282, in an Inflammatory Model of COPD in A/J MiceProc Am Thorac Soc3A850
- GambleEGrootendorstDCBrightlingCE2003Antiinflammatory effects of the phosphodiesterase-4 inhibitor cilomilast (Ariflo) in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1689768212816740
- GanWQManSFSinDD2005Effects of inhaled corticosteroids on sputum cell counts in stable chronic obstructive pulmonary disease: a systematic review and a meta-analysisBMC Pulm Med5315707484
- GartlehnerGHansenRACarsonSS2006Efficacy and safety of inhaled corticosteroids in patients with COPD: a systematic review and meta-analysis of health outcomesAnn Fam Med42536216735528
- GerritsCMHeringsRMLeufkensHG2003N-acetylcysteine reduces the risk of re-hospitalisation among patients with chronic obstructive pulmonary diseaseEur Respir J21795812765423
- GhoshSMayMJKoppEB1998NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responsesAnnu Rev Immunol16225609597130
- GilksCBPriceKWrightJL1998Antioxidant gene expression in rat lung after exposure to cigarette smokeAm J Pathol152269789422544
- GillissenAJaworskaMOrthM1997Nacystelyn, a novel lysine salt of N-acetylcysteine, to augment cellular antioxidant defence in vitroRespir Med91159689135855
- GosmanMMBoezenHMvan DiemenCC2006A disintegrin and metalloproteinase 33 and chronic obstructive pulmonary disease pathophysiologyThorax62242717090574
- GosmanMMWillemseBWJansenDF2006Increased number of B-cells in bronchial biopsies in COPDEur Respir J2760416387936
- GrashoffWFSontJKSterkPJ1997Chronic obstructive pulmonary disease: role of bronchiolar mast cells and macrophagesAm J Pathol1511785909403729
- GrievinkLSmitHAOckeMC1998Dietary intake of antioxidant (pro)-vitamins, respiratory symptoms and pulmonary function: the MORGEN studyThorax53166719659349
- GrootendorstDCGauwSASterkPJ2005Treatment with PDE4 inhibitor roflumilast reduces sputum neutrophil and eosinophil numbers in patients with COPDProc Am Thoracic Soc2A543
- GuestTMVlastosGAlameddineFM2006Mechanoregulation of monocyte chemoattractant protein-1 expression in rat vascular smooth muscle cellsAntioxid Redox Signal814617116987003
- GuidotDMBrownLA2000Mitochondrial glutathione replacement restores surfactant synthesis and secretion in alveolar epithelial cells of ethanol-fed ratsAlcohol Clin Exp Res241070610924012
- HabibMPTankLJLaneLC1999Effect of vitamin E on exhaled ethane in cigarette smokersChest1156849010084476
- HaddadEBBirrellMMcCluskieK2001Role of p38 MAP kinase in LPSinduced airway inflammation in the ratBr J Pharmacol13217152411309243
- HaringmanJJGerlagDMSmeetsTJ2006A randomized controlled trial with an anti-CCL2 (anti-monocyte chemotactic protein 1) monoclonal antibody in patients with rheumatoid arthritisArthritis Rheum5423879216869001
- HarjuTKaarteenaho-WiikRSoiniY2002Diminished immunoreactivity of gamma-glutamylcysteine synthetase in the airways of smokers’ lungAm J Respir Crit Care Med166754912204877
- HarrisonDJCantlayAMRaeF1997Frequency of glutathione S-transferase M1 deletion in smokers with emphysema and lung cancerHum Exp Toxicol16356609257159
- HautamakiRDKobayashiDKSeniorRM1997Requirement for macrophage elastase for cigarette smoke-induced emphysema in miceScience277200249302297
- HayasakiTKaikitaKOkumaT2006CC chemokine receptor-2 deficiency attenuates oxidative stress and infarct size caused by myocardial ischemia-reperfusion in miceCirc J703425116501303
- HittiEIakovlevaTBrookM2006Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich elementMol Cell Biol26239940716508014
- HoKKAuldDSBohnstedtAC2006Imidazolylpyrimidine based CXCR2 chemokine receptor antagonistsBioorg Med Chem Lett162724816540318
- HoggJC2004Pathophysiology of airflow limitation in chronic obstructive pulmonary diseaseLancet3647092115325838
- Holmes-McNaryMBaldwinASJr2000Chemopreventive properties of trans-resveratrol are associated with inhibition of activation of the IkappaB kinaseCancer Res6034778310910059
- HoogendoornMFeenstraTLRutten-van MolkenMP2006[Projections of future resource use and the costs of asthma and COPD in the Netherlands]Ned Tijdschr Geneeskd15012435016796176
- HowarthPHBabuKSArshadHS2005Tumour necrosis factor (TNFalpha) as a novel therapeutic target in symptomatic corticosteroid dependent asthmaThorax6010121816166100
- HuangSMWuCHYenGC2006Effects of flavonoids on the expression of the pro-inflammatory response in human monocytes induced by ligation of the receptor for AGEsMol Nutr Food Res5011293917103373
- HynninenKMBreitveMHWiborgAB2005Psychological characteristics of patients with chronic obstructive pulmonary disease: a reviewJ Psychosom Res594294316310027
- ItoKLimSCaramoriG2001Cigarette smoking reduces histone deacetylase 2 expression, enhances cytokine expression, and inhibits glucocorticoid actions in alveolar macrophagesFaseb J1511101211292684
- ItoKItoMElliottWM2005Decreased histone deacetylase activity in chronic obstructive pulmonary diseaseN Engl J Med35219677615888697
- JazrawiECosioBGBarnesPJ2003Inhibition of IKK2 and JNK differentially regulates GM-CSF and IL-8 release in epithelial cells and alveolar macrophagesAm J Respir Crit Care Med167A798
- JohnsonGLLapadatR2002Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinasesScience29819111212471242
- JonesPW2006Health status: what does it mean for payers and patients?Proc Am Thorac Soc3222616636089
- JoosLHeJQShepherdsonMB2002The role of matrix metalloproteinase polymorphisms in the rate of decline in lung functionHum Mol Genet115697611875051
- KarimiKSarirHMortazE2006Toll-like receptor-4 mediates cigarette smoke-induced cytokine production by human macrophagesRespir Res76616620395
- KasaharaYTuderRMTaraseviciene-StewartL2000Inhibition of VEGF receptors causes lung cell apoptosis and emphysemaJ Clin Invest10613111911104784
- KasielskiMNowakD2001Long-term administration of N-acetylcysteine decreases hydrogen peroxide exhalation in subjects with chronic obstructive pulmonary diseaseRespir Med954485611421501
- KassimSYFuXLilesWC2005NADPH oxidase restrains the matrix metalloproteinase activity of macrophagesJ Biol Chem28030201515983040
- KeatingsVMCaveSJHenryMJ2000A polymorphism in the tumor necrosis factor-alpha gene promoter region may predispose to a poor prognosis in COPDChest118971511035665
- KirkhamPASpoonerGRahmanI2004Macrophage phagocytosis of apoptotic neutrophils is compromised by matrix proteins modified by cigarette smoke and lipid peroxidation productsBiochem Biophys Res Commun31832715110749
- KoechlinCCouillardACristolJP2004Does systemic inflammation trigger local exercise-induced oxidative stress in COPD?Eur Respir J235384415083751
- KoetheSMKuhnmuenchJRBeckerCG2000Neutrophil priming by cigarette smoke condensate and a tobacco anti-idiotypic antibodyAm J Pathol15717354311073832
- KondoTTagamiSYoshiokaA1994Current smoking of elderly men reduces antioxidants in alveolar macrophagesAm J Respir Crit Care Med149178828111579
- KranenburgARDe BoerWIVan KriekenJH2002Enhanced expression of fibroblast growth factors and receptor FGFR-1 during vascular remodeling in chronic obstructive pulmonary diseaseAm J Respir Cell Mol Biol275172512397010
- KranenburgARde BoerWIAlagappanVK2005Enhanced bronchial expression of vascular endothelial growth factor and receptors (Flk-1 and Flt-1) in patients with chronic obstructive pulmonary diseaseThorax601061315681497
- KranenburgARWillems-WidyastutiAMooriWJ2006Enhanced bronchial expression of extracellular matrix proteins in chronic obstructive pulmonary diseaseAm J Clin Pathol1267253517111536
- KucukaycanMVan KrugtenMPenningsHJ2002Tumor necrosis factor-alpha +489G/A gene polymorphism is associated with chronic obstructive pulmonary diseaseRespir Res32912537602
- KumagaiTMatsukawaNKanekoY2004A lipid peroxidation-derived inflammatory mediator: identification of 4-hydroxy-2-nonenal as a potential inducer of cyclooxygenase-2 in macrophagesJ Biol Chem279483899615355999
- KumarSBoehmJLeeJC2003p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseasesNat Rev Drug Discov27172612951578
- LangMRFiauxGWGilloolyM1994Collagen content of alveolar wall tissue in emphysematous and non-emphysematous lungsThorax49319268202900
- LangenRCKornSHWoutersEF2003ROS in the local and systemic pathogenesis of COPDFree Radic Biol Med352263512885585
- LeeJCKassisSKumarS1999p38 mitogen-activated protein kinase inhibitors – mechanisms and therapeutic potentialsPharmacol Ther823899710454214
- LeffAR2006Tissue remodeling and repair mechanisms in chronic obstructive pulmonary diseaseProc Am Thorac Soc37346
- LiaoLNingQLiY2006CXCR2 blockade reduces radical formation in hyperoxia-exposed newborn rat lungPediatr Res6029930316923948
- LindbergABjerg-BacklundARonmarkE2006Prevalence and underdiagnosis of COPD by disease severity and the attributable fraction of smoking Report from the Obstructive Lung Disease in Northern Sweden StudiesRespir Med1002647215975774
- LindholtJSJorgensenBShiGP2003Relationships between activators and inhibitors of plasminogen, and the progression of small abdominal aortic aneurysmsEur J Vasc Endovasc Surg255465112787697
- LokeWMProudfootJMMcKinleyAJ2006Augmentation of monocyte intracellular ascorbate in vitro protects cells from oxidative damage and inflammatory responsesBiochem Biophys Res Commun34510394316712788
- LykkesfeldtJChristenSWallockLM2000Ascorbate is depleted by smoking and repleted by moderate supplementation: a study in male smokers and nonsmokers with matched dietary antioxidant intakesAm J Clin Nutr71530610648268
- MaenoTHoughtonAMQuinteroPA2007CD8+ T cells are required for inflammation and destruction in cigarette smoke-induced emphysema in miceJ Immunol1788090617548647
- Martin-MosqueroCPeces-BarbaGRubioML2006Increased collagen deposition correlated with lung destruction in human emphysemaHistol Histopathol21823816691534
- MarwickJAKirkhamPAStevensonCS2004Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungsAm J Respir Cell Mol Biol316334215333327
- MarwickJAStevensonCSGiddingsJ2006Cigarette smoke disrupts VEGF165-VEGFR-2 receptor signaling complex in rat lungs and patients with COPD: morphological impact of VEGFR-2 inhibitionAm J Physiol Lung Cell Mol Physiol290L89790816361360
- MatsuokaHAraiTMoriM2002A p38 MAPK inhibitor, FR-167653, ameliorates murine bleomycin-induced pulmonary fibrosisAm J Physiol Lung Cell Mol Physiol283L1031212060566
- McCuskerKHoidalJ1990Selective increase of antioxidant enzyme activity in the alveolar macrophages from cigarette smokers and smoke-exposed hamstersAm Rev Respir Dis141678822310098
- MejaKKSeldonPMNasuharaY2000p38 MAP kinase and MKK-1 co-operate in the generation of GM-CSF from LPS-stimulated human monocytes by an NF-kappa B-independent mechanismBr J Pharmacol13111435311082122
- MenezesAMPerez-PadillaRJardimJR2005Chronic obstructive pulmonary disease in five Latin American cities (the PLATINO study): a prevalence studyLancet36618758116310554
- MezzettiALapennaDPierdomenicoSD1995Vitamins E, C and lipid peroxidation in plasma and arterial tissue of smokers and nonsmokersAtherosclerosis1129197772072
- MilatovicSNanneyLBYuY2003Impaired healing of nitrogen mustard wounds in CXCR2 null miceWound Repair Regen112131912753603
- MonteleoneGKumberovaACroftNM2001Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel diseaseJ Clin Invest108601911518734
- MoodieFMMarwickJAAndersonCS2004Oxidative stress and cigarette smoke alter chromatin remodeling but differentially regulate NF-kappaB activation and proinflammatory cytokine release in alveolar epithelial cellsFaseb J181897915456740
- MorettiMBottrighiPDallariR2004The effect of long-term treatment with erdosteine on chronic obstructive pulmonary disease: the EQUALIFE StudyDrugs Exp Clin Res301435215553660
- MorquetteBShiQLavigneP2006Production of lipid peroxidation products in osteoarthritic tissues: new evidence linking 4-hydroxynonenal to cartilage degradationArthritis Rheum542718116385544
- MorrisonDRahmanILannanS1999Epithelial permeability, inflammation, and oxidant stress in the air spaces of smokersAm J Respir Crit Care Med159473799927360
- MullerKHardwickSJMarchantCE1996Cytotoxic and chemotactic potencies of several aldehydic components of oxidised low density lipoprotein for human monocyte-macrophagesFEBS Lett38816588690078
- NasuharaYAdcockIMCatleyM1999Differential IkappaB kinase activation and IkappaBalpha degradation by interleukin-1beta and tumor necrosis factor-alpha in human U937 monocytic cells Evidence for additional regulatory steps in kappaB-dependent transcriptionJ Biol Chem274199657210391945
- NeurohrCLenzAGDingI2003Glutamate-cysteine ligase modulatory subunit in BAL alveolar macrophages of healthy smokersEur Respir J2282712882455
- NicholsonGCTennantRCCarpenterDC2007A novel flow cytometric assay of human whole blood neutrophil and monocyte CD11b levels: upregulation by chemokines is related to receptor expression, comparison with neutrophil shape change, and effects of a chemokine receptor (CXCR2) antagonistPulm Pharmacol Ther2052916406722
- NishiTMaierCMHayashiT2005Superoxide dismutase 1 overexpression reduces MCP-1 and MIP-1 alpha expression after transient focal cerebral ischemiaJ Cereb Blood Flow Metab2513122415829914
- NishikawaMKakemizuNItoT1999Superoxide mediates cigarette smoke-induced infiltration of neutrophils into the airways through nuclear factor-kappaB activation and IL-8 mRNA expression in guinea pigs in vivoAm J Respir Cell Mol Biol20189989922209
- NogueraABusquetsXSauledaJ1998Expression of adhesion molecules and G proteins in circulating neutrophils in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med158166489817722
- OgryzkoVVSchiltzRLRussanovaV1996The transcriptional coactivators p300 and CBP are histone acetyltransferasesCell8795398945521
- OkadaYWatanabeSNakanishiI1988Inactivation of tissue inhibitor of metalloproteinases by neutrophil elastase and other serine proteinasesFEBS Lett229157603162216
- PapiARomagnoliMBaraldoS2000Partial reversibility of airflow limitation and increased exhaled NO and sputum eosinophilia in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1621773711069811
- ParasrampuriaDAde BoerPDesai-KriegerD2003Single-dose pharmacokinetics and pharmacodynamics of RWJ 67657, a specific p38 mitogen-activated protein kinase inhibitor: a first-in-human studyJ Clin Pharmacol434061312723461
- ParolaMPinzaniMCasiniA1993Stimulation of lipid peroxidation or 4-hydroxynonenal treatment increases procollagen alpha 1 (I) gene expression in human liver fat-storing cellsBiochem Biophys Res Commun1941044508352762
- PasternakAMarinoDVicarioPP2006Novel, orally bioavailable gamma-aminoamide CC chemokine receptor 2 (CCR2) antagonistsJ Med Chem494801416884289
- PauwelsRARabeKF2004Burden and clinical features of chronic obstructive pulmonary disease (COPD)Lancet3646132015313363
- PembertonPAKobayashiDWilkBJ2006Inhaled recombinant alpha 1-antitrypsin ameliorates cigarette smoke-induced emphysema in the mouseCopd3101817175673
- PetruzzelliSHietanenEBartschH1990Pulmonary lipid peroxidation in cigarette smokers and lung cancer patientsChest9893052209151
- PizzichiniEPizzichiniMMGibsonP1998Sputum eosinophilia predicts benefit from prednisone in smokers with chronic obstructive bronchitisAm J Respir Crit Care Med1581511179817701
- PoolePJBlackPN2006Mucolytic agents for chronic bronchitis or chronic obstructive pulmonary diseaseCochrane Database Syst Rev3CD00128716855965
- PostmaDSTimensW2006Remodeling in asthma and chronic obstructive pulmonary diseaseProc Am Thorac Soc3434916799088
- RabeKFBatemanEDO’DonnellD2005Roflumilast – an oral anti-inflammatory treatment for chronic obstructive pulmonary disease: a randomised controlled trialLancet3665637116099292
- RahmanIAdcockIM2006Oxidative stress and redox regulation of lung inflammation in COPDEur Respir J282194216816350
- RahmanIMacNeeW1996aOxidant/antioxidant imbalance in smokers and chronic obstructive pulmonary diseaseThorax51348508733482
- RahmanIMacNeeW1996bRole of oxidants/antioxidants in smoking-induced lung diseasesFree Radic Biol Med21669818891669
- RahmanIMacNeeW1998Role of transcription factors in inflammatory lung diseasesThorax53601129797762
- RahmanIMacNeeW1999Lung glutathione and oxidative stress: implications in cigarette smoke-induced airway diseaseAm J Physiol277L10678810600876
- RahmanIMacNeeW2000aOxidative stress and regulation of glutathione in lung inflammationEur Respir J165345411028671
- RahmanIMacNeeW2000bRegulation of redox glutathione levels and gene transcription in lung inflammation: therapeutic approachesFree Radic Biol Med2814052010924859
- RahmanIMorrisonDDonaldsonK1996Systemic oxidative stress in asthma, COPD, and smokersAm J Respir Crit Care Med1541055608887607
- RahmanISkwarskaEMacNeeW1997Attenuation of oxidant/antioxidant imbalance during treatment of exacerbations of chronic obstructive pulmonary diseaseThorax5256589227727
- RahmanIGilmourPSJimenezLA2002Oxidative stress and TNFalpha induce histone acetylation and NF-kappaB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammationMol Cell Biochem234523948
- RahmanIvan SchadewijkAACrowtherAJ20024-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med166490512186826
- RahmanIMarwickJKirkhamP2004Redox modulation of chromatin remodeling: impact on histone acetylation and deacetylation, NF-kappaB and pro-inflammatory gene expressionBiochem Pharmacol6812556715313424
- RajagopalanSMengXPRamasamyS1996Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stabilityJ Clin Invest98257298958220
- RasmussenJBGlennowC1988Reduction in days of illness after long-term treatment with N-acetylcysteine controlled-release tablets in patients with chronic bronchitisEur Respir J135153294038
- RautalahtiMVirtamoJHaukkaJ1997The effect of alpha-tocopherol and beta-carotene supplementation on COPD symptomsAm J Respir Crit Care Med1561447529372659
- RayPDevauxYStolzDB2003Inducible expression of keratinocyte growth factor (KGF) in mice inhibits lung epithelial cell death induced by hyperoxiaProc Natl Acad Sci U S A100609810312732722
- RennardSIFogartyCKelsenSfor the COPD Investigators2007The safety and efficacy of infliximab in moderate to severe chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1759263417290043
- RennardSISchachterNStrekM2006Cilomilast for COPD: results of a 6-month, placebo-controlled study of a potent, selective inhibitor of phosphodiesterase 4Chest129566616424413
- RennekampffHOHansbroughJFWoodsVJr1997Role of melanoma growth stimulatory activity (MGSA/gro) on keratinocyte function in wound healingArch Dermatol Res289204129143736
- RennekampffHOHansbroughJFKiessigV2000Bioactive interleukin-8 is expressed in wounds and enhances wound healingJ Surg Res93415410945942
- RepineJEBastALankhorstI1997Oxidative stress in chronic obstructive pulmonary disease. Oxidative Stress Study GroupAm J Respir Crit Care Med156341579279209
- RolinSMasereelBDogneJM2006Prostanoids as pharmacological targets in COPD and asthmaEur J Pharmacol5338910016458293
- RothSYDenuJMAllisCD2001Histone acetyltransferasesAnnu Rev Biochem708112011395403
- RussellRECulpittSVDeMatosC2002Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary diseaseAm J Respir Cell Mol Biol26602911970913
- RusznakCMillsPRDevaliaJL2000Effect of cigarette smoke on the permeability and IL-1beta and sICAM-1 release from cultured human bronchial epithelial cells of never-smokers, smokers, and patients with chronic obstructive pulmonary diseaseAm J Respir Cell Mol Biol23530611017919
- SadowskaAMManuelYKBDe BackerWA2007Antioxidant and antiinflammatory efficacy of NAC in the treatment of COPD: discordant in vitro and in vivo dose-effects: a reviewPulm Pharmacol Ther2092216458553
- SaettaMBaraldoSCorbinoL1999CD8+ve cells in the lungs of smokers with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1607111710430750
- SaikaSIkedaKYamanakaO2005Expression of Smad7 in mouse eyes accelerates healing of corneal tissue after exposure to alkaliAm J Pathol16614051815855641
- SandfordAJChaganiTWeirTD2001Susceptibility genes for rapid decline of lung function in the lung health studyAm J Respir Crit Care Med1634697311179124
- SargeantLAJaeckelAWarehamNJ2000Interaction of vitamin C with the relation between smoking and obstructive airways disease in EPIC Norfolk. European Prospective Investigation into Cancer and NutritionEur Respir J1639740311028650
- SchaurRJDussingGKinkE1994The lipid peroxidation product 4-hydroxynonenal is formed by – and is able to attract – rat neutrophils in vivoFree Radic Res20365738081452
- ScottDAStapletonJAWilsonRF2000Dramatic decline in circulating intercellular adhesion molecule-1 concentration on quitting tobacco smokingBlood Cells Mol Dis26255810950946
- ShaharEBolandLLFolsomAR1999Docosahexaenoic acid and smoking-related chronic obstructive pulmonary disease. The Atherosclerosis Risk in Communities Study InvestigatorsAm J Respir Crit Care Med1591780510351918
- ShapiroSDGoldsteinNMHoughtonAM2003Neutrophil elastase contributes to cigarette smoke-induced emphysema in miceAm J Pathol16323293514633606
- ShenYRattanVSultanaC1996Cigarette smoke condensate-induced adhesion molecule expression and transendothelial migration of monocytesAm J Physiol270H1624338928867
- SinDDAnthonisenNRSorianoJB2006Mortality in COPD: role of comorbiditiesEur Respir J2812455717138679
- SmithKRUyeminamiDLKodavantiUP2002Inhibition of tobacco smoke-induced lung inflammation by a catalytic antioxidantFree Radic Biol Med3311061412374622
- SniderGL1989Chronic obstructive pulmonary disease: risk factors, pathophysiology and pathogenesisAnnu Rev Med40411292658758
- SpringerJScholzFRPeiserC2004SMAD-signaling in chronic obstructive pulmonary disease: transcriptional down-regulation of inhibitory SMAD 6 and 7 by cigarette smokeBiol Chem3856495315318814
- SpycherSETabataba-VakiliSO’DonnellVB1997Aldose reductase induction: a novel response to oxidative stress of smooth muscle cellsFaseb J1118189039961
- SteinbergFMChaitA1998Antioxidant vitamin supplementation and lipid peroxidation in smokersAm J Clin Nutr68319279701189
- SteinerovaARacekJStozickyF2001Antibodies against oxidized LDL – theory and clinical usePhysiol Res501314111522041
- SteyCSteurerJBachmannS2000The effect of oral N-acetylcysteine in chronic bronchitis: a quantitative systematic reviewEur Respir J162536210968500
- StonePJGottliebDJO’ConnorGT1995Elastin and collagen degradation products in urine of smokers with and without chronic obstructive pulmonary diseaseAm J Respir Crit Care Med15195297697272
- SullivanAKSimonianPLFaltaMT2005Oligoclonal CD4+ T cells in the lungs of patients with severe emphysemaAm J Respir Crit Care Med172590615937291
- TakizawaHTanakaMTakamiK2001Increased expression of transforming growth factor-beta1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD)Am J Respir Crit Care Med16314768311371421
- Taraseviciene-StewartLScerbaviciusRChoeKH2005An animal model of autoimmune emphysemaAm J Respir Crit Care Med1717344215563631
- TjalkensRBCookLWPetersenDR1999Formation and export of the glutathione conjugate of 4-hydroxy-2, 3-E-nonenal (4-HNE) in hepatoma cellsArch Biochem Biophys361113199882435
- TsaiSHLin-ShiauSYLinJK1999Suppression of nitric oxide synthase and the down-regulation of the activation of NFkappaB in macrophages by resveratrolBr J Pharmacol1266738010188978
- TuderRMZhenLChoCY2003Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockadeAm J Respir Cell Mol Biol29889712600822
- TuderRMYoshidaTArapW2006State of the art. Cellular and molecular mechanisms of alveolar destruction in emphysema: an evolutionary perspectiveProc Am Thorac Soc35031016921129
- TugTKaratasFTerziSM2004Antioxidant vitamins (A, C and E) and malondialdehyde levels in acute exacerbation and stable periods of patients with chronic obstructive pulmonary diseaseClin Invest Med27123815305803
- UnderwoodDCOsbornRRBochnowiczS2000SB 239063, a p38 MAPK inhibitor, reduces neutrophilia, inflammatory cytokines, MMP-9, and fibrosis in lungAm J Physiol Lung Cell Mol Physiol279L89590211053025
- UnderwoodDCOsbornRRKotzerCJ2000SB 239063, a potent p38 MAP kinase inhibitor, reduces inflammatory cytokine production, airways eosinophil infiltration, and persistenceJ Pharmacol Exp Ther293281810734180
- UneriCSariMBaglamT2006Effects of vitamin E on cigarette smoke induced oxidative damage in larynx and lungLaryngoscope1169710016481818
- van AntwerpenLTheronAJMyerMS1993Cigarette smoke-mediated oxidant stress, phagocytes, vitamin C, vitamin E, and tissue injuryAnn N Y Acad Sci68653658512261
- van der StrateBWPostmaDSBrandsmaCA2006Cigarette smoke-induced emphysema: A role for the B cell?Am J Respir Crit Care Med173751816399994
- van der VaartHKoeterGHPostmaDS2005First study of infliximab treatment in patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med172465915937294
- Van SchootenFJBesaratiniaADe FloraS2002Effects of oral administration of N-acetyl-L-cysteine: a multi-biomarker study in smokersCancer Epidemiol Biomarkers Prev111677511867504
- VincentSHPainterSKLuffer-AtlasD1997Orally active inhibitors of human leukocyte elastase. II. Disposition of L-694,458 in rats and rhesus monkeysDrug Metab Dispos2593299280401
- VoelkelNFCollCD2003Pulmonary vascular involvement in chronic obstructive pulmonary diseaseEur Respir J Suppl4628s32s14621104
- WangRDTaiHXieC2003Cigarette smoke produces airway wall remodeling in rat tracheal explantsAm J Respir Crit Care Med1681232612958058
- WeathingtonNMvan HouwelingenAHNoeragerBD2006A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammationNat Med123172316474398
- WrightJLFarmerSGChurgA2002Synthetic serine elastase inhibitor reduces cigarette smoke-induced emphysema in guinea pigsAm J Respir Crit Care Med1669546012359653
- WrightJLChurgA2006Advances in the pathology of COPDHistopathology491916842241
- YangSRChidaASBauterMR2006Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophagesAm J Physiol Lung Cell Mol Physiol291L465716473865
- YoshidaMKorfhagenTRWhitsettJA2001Surfactant protein D regulates NF-kappa B and matrix metalloproteinase production in alveolar macrophages via oxidant-sensitive pathwaysJ Immunol16675141911390505
- YoungRPHopkinsRBlackPN2006Functional variants of antioxidant genes in smokers with COPD and in those with normal lung functionThorax61394916467073
- ZandvoortAPostmaDSJonkerMR2006Altered expression of the Smad signalling pathway: implications for COPD pathogenesisEur Respir J285334116707515