Abstract
Many patients with chronic obstructive pulmonary disease (COPD) suffer from exercise intolerance. In about 40% of the patients exercise capacity is limited by alterations in skeletal muscle rather than pulmonary problems. Indeed, COPD is often associated with muscle wasting and a slow-to-fast shift in fiber type composition resulting in weakness and an earlier onset of muscle fatigue, respectively. Clearly, limiting muscle wasting during COPD benefits the patient by improving the quality of life and also the chance of survival. To successfully combat muscle wasting and remodeling during COPD a clear understanding of the causes and mechanisms is needed. Disuse, hypoxemia, malnutrition, oxidative stress and systemic inflammation may all cause muscle atrophy. Particularly when systemic inflammation is elevated muscle wasting becomes a serious complication. The muscle wasting may at least partly be due to an increased activity of the ubiquitin proteasome pathway and apoptosis. However, it might well be that an impaired regenerative potential of the muscle rather than the increased protein degradation is the crucial factor in the loss of muscle mass during COPD with a high degree of systemic inflammation. Finally, we briefly discuss the various treatments and rehabilitation strategies available to control muscle wasting and fatigue in patients with COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) is a major cause of morbidity and death throughout the world. The disease is mainly caused by smoking, but environmental pollution and α1–antritrypsin deficiency may also cause the development of COPD (CitationPetty 2006). In 2000 about 16 million people suffered from COPD in the USA alone (CitationMannino et al 2002) with the number of women suffering from this disorder increasing (CitationCasaburi 2001). The disease is progressive, but the severity and progress can be moderated by actions such as smoking cessation, careful management of infections and appropriate rehabilitation (CitationATS/ERS 1999; CitationFaulkner et al 2006).
One of the major problems of patients with COPD is exercise intolerance (CitationGosker et al 2000; CitationAliverti and Macklem 2001; CitationCasaburi 2001). Although the disease is characterized by reduced maximal expiratory flow (CitationATS/ERS 1999; CitationFaulkner et al 2006), FEV1 in COPD correlates poorly with exercise capacity (CitationKillian et al 1992; CitationGosselink et al 1996; CitationEngelen et al 2000; CitationGosker, Lencer et al 2003). Likewise, in single and double lung transplants, exercise capacity did improve after surgery, but was still lower than normal (CitationAmbrosino et al 1996) indicating that factors other than lung function alone limited exercise capacity (CitationEvans et al 1997). Also in patients with COPD who did not undergo a lung-transplantation, evidence that the exercise intolerance is not only related to a reduced lung function, but also to skeletal muscle dysfunction is growing (CitationSchols et al 1991; CitationGosselink et al 1996; CitationGosker, Lencer et al 2003). The importance of skeletal muscle dysfunction may increase over time as the deterioration in exercise capacity is uncoupled from the progression of airflow limitation (CitationOga et al 2005). It is important, therefore, to know how muscle function is affected, to identify the factors that contribute to the muscle dysfunction and the mechanism of muscle wasting in COPD so as to improve the management of the disease. Such knowledge may also have wider implications since COPD is one of a number of common disorders, including chronic heart failure (CitationGosker et al 2000) and cancer (CitationTisdale 2005), where muscle dysfunction and wasting are serious complications and may be brought about by similar underlying mechanisms. This review will focus on the changes in peripheral skeletal muscle structure, function and metabolism in COPD and discuss some of the potential underlying factors and mechanisms contributing to the observed muscle wasting and dysfunction.
The relation between muscle structure and function in COPD
Muscle weakness and loss of muscle mass
A loss of skeletal muscle mass is a common observation in patients with COPD and may not only lead to muscle weakness (CitationSchols et al 1993; CitationGosselink et al 1996; CitationBernard et al 1998; CitationEngelen et al 2000), but is also associated with an increased mortality of patients with COPD. Marquis and colleagues (CitationMarquis et al 2002) reported that 50% of their patients with a FEV1 predicted <25% and a mid-thigh crosssectional area (CSA) <70 cm2 died within 3 years, compared to only 12% of patients with a mid-thigh CSA >70 cm2. Schols and colleagues (CitationSchols et al 1993) found that about half of the patients with mild to severe COPD had a reduced body weight, which could be related to both a loss of muscle and adipose tissue. Since lean tissue depletion could even occur in overweight patients the prevalence of muscle wasting might be even higher (CitationDe Benedetto et al 2000).
Muscle atrophy occurs when the balance of protein synthesis and degradation shifts to net protein breakdown. Most of the research on muscle wasting in chronic diseases has focused on protein degradation pathways. For an extensive discussion of the molecular and cellular mechanisms involved in protein degradation, such as the ubiquitin-proteosome pathway, we refer the reader to several excellent reviews on this subject (CitationJagoe and Engelen 2003; CitationKandarian and Jackman 2006; CitationSaini et al 2006).
Besides an increased rate of protein degradation, also a decreased rate of protein synthesis contributes to the muscle wasting in many chronic diseases (CitationRennie et al 1983). This may be a consequence of systemic inflammation that often occurs in patients with COPD (CitationGan et al 2004). Indeed, recent studies indicate that the regenerative capacity of skeletal muscle is impaired in mice with elevated circulating tumor necrosis factor-α (TNF-α) levels (CitationGuttridge et al 2000; CitationLangen et al 2004, Citation2006). Furthermore, testosterone, an anabolic hormone, levels were lower in COPD patients (CitationCasaburi 1998; CitationCasaburi et al 2004; CitationVan Vliet et al 2005). The lower testosterone levels were associated with muscle weakness (CitationVan Vliet et al 2005). It has been speculated that chronic hypoxia (CitationAasebo et al 1993) and corticosteroid therapy (CitationKamischke et al 1998) contribute to low testosterone levels. It is equivocal whether insulin-like growth factor-I (IGF-1), which mediates muscle growth, is elevated or reduced in COPD patients (CitationCreutzberg and Casaburi 2003). Also, human studies of myostatin, a hormone that is produced in the muscle and suppresses muscle growth by inhibiting satellite cell activity, are scarce and its role in muscle wasting in COPD is unknown (CitationJespersen et al 2006). Clearly, more studies are necessary to assess whether protein synthesis rates are affected during COPD.
Besides muscle wasting also other factors, such as a decrease in maximal neural drive to the working muscles (CitationRutherford et al 1986) may contribute to muscle weakness during COPD. Indeed, a reduced neural drive may well explain the decline in force generating capacity per muscle cross-sectional area (specific tension) in vivo without a change in in-vitro specific tension of isolated bundles from the same muscle (CitationDebigare et al 2003). However, COPD patients who were matched for fat free mass index with control subjects did not show signs of muscle weakness or atrophy (CitationHeijdra et al 2003; CitationDegens et al 2005) indicating that the neural drive is maintained as long as fat free mass index is maintained.
Contractile properties and fiber type composition
During daily life most activities involve shortening contractions which require a sufficient power output from the muscle. Therefore, power output, which is the product of force and velocity, is more important during daily life than the ability of the muscle to generate isometric force. The loss of power in patients with COPD (CitationYquel et al 2006) as a result of a loss in muscle strength may be compensated for, to some extent, by the slow-to-fast transition in fiber type composition (CitationJobin et al 1998; CitationGosker, van Mameren et al 2002) and an increased proportion of hybrid fibers expressing more than one myosin heavy chain isoform (CitationGosker, van Mameren et al 2002). The slow-to-fast transition appears to be more marked during emphysema than in chronic bronchitis (CitationGosker, van Mameren et al 2002) and to be related to the severity of the disease in terms of FEV1 (CitationSatta et al 1997). Apart from a marked type IIX fiber atrophy and a slight increase in fibrosis and fat-cell replacement, which is not uncommon for skeletal muscle in the elderly, there are no myopathologic features in non-cachetic COPD patients (CitationGosker, Kubat et al 2003). Nevertheless, it is unlikely that the changes in fiber type composition during COPD, a disease that mostly becomes manifest after the age of 50, are just a reflection of the ageing process as normal ageing is, if anything, accompanied by a fast-to-slow rather than a slow-to-fast transition (CitationNarici et al 1991; CitationDegens and Alway 2006; CitationKorhonen et al 2006). Although the changes in fiber type composition during COPD may be too small to affect the rates of contraction and relaxation during electrically evoked isometric tetani (CitationDegens et al 2005), it remains to be established whether the fiber type transition is sufficient to cause a change in the shortening velocity during dynamic contractions. Nonetheless, as type II fibers are less efficient than type I fibers for force generation (CitationStienen et al 1996), the slow-to-fast-transition in fiber type composition may at least partly explain the reduced mechanical efficiency of COPD patients during one leg knee extensor exercise (CitationFranssen, Wouters, Baarends et al 2002; CitationRichardson et al 2004).
Metabolism and capillarization
Several studies have addressed the metabolic characteristics of muscles from COPD patients (CitationJakobsson et al 1990, Citation1995; CitationJobin et al 1998; CitationWhittom et al 1998; CitationGosker, van Mameren et al 2002). The results of these studies are equivocal. Part of the discrepancies in the literature can be ascribed to differences in disease severity, medication (see section medication) and whether locomotor or other muscles have been studied. While the oxidative capacity of the vastus lateralis muscle of patients with moderate-to-severe COPD was significantly reduced (CitationJakobsson et al 1995; CitationGosker, van Mameren et al 2002), the oxidative capacity in the musculature of the upper extremity was not affected (CitationGea, Pasto et al 2001). Also, the mechanical efficiency was lower in leg muscles, while arm mechanical efficiency was not significantly affected (CitationFranssen, Wouters, Baarends et al 2002). The different effect that COPD has on upper body and leg muscles was so marked that it is referred to as ‘the compartment theory’ (CitationGea, Orozco-Levi et al 2001). A simple explanation put forward for the differences between the two ‘compartments’ is the different degree of disuse they experience during COPD (CitationGosselink et al 2000; CitationGea, Orozco-Levi et al 2001). Nevertheless, the glycolytic capacity was elevated in both the muscles of the leg (CitationJakobsson et al 1995) and the upper body (CitationGea, Pasto et al 2001). In advanced stages of the disease, however, energy metabolism becomes increasingly compromised as reflected by lower levels of glycogen, ATP and PCr in the quadriceps femoris muscle of patients with respiratory failure, but not in those without respiratory failure (CitationJakobsson et al 1990).
Little work has yet been done on the capillarization of skeletal muscle in COPD (CitationJobin et al 1998; CitationWhittom et al 1998; CitationRichardson et al 2004). Although the number of capillaries per fiber might be reduced during COPD (CitationWhittom et al 1998), the capillary supply per fiber CSA and total numerical capillary density was maintained (CitationWhittom et al 1998). It seems that, at least anatomically, the microcirculation is intact in COPD patients (CitationRichardson et al 2004). This is not an unequivocal finding, however, as CitationJobin et al (1998) found an almost 50% decrease in capillary to fiber ratio and capillary density, suggesting that a disproportionate loss of capillaries may occur during COPD.
Skeletal muscle fatigue
Muscle fatigue can be defined as the inability of a muscle to maintain a certain force or power output. As has been mentioned above exercise intolerance, as reflected by a low peak oxygen uptake, is a major symptom in patients with COPD (CitationOga et al 2007). It is likely that the increased load and oxygen need of the respiratory muscles during COPD and reduced venous return compete with an impaired delivery of oxygen to the limb muscles (CitationAliverti and Macklem 2001). However, under circumstances where the cardio-respiratory system is unlikely to be the limiting factor, such as during one-leg exercise, or exercise of a single muscle or muscle group, the capillarization and oxidative capacity of a muscle are important determinants of muscle fatigue resistance (CitationDegens and Veerkamp 1994). Therefore, it is no surprise in light of these changes in the muscle as described above that an increased susceptibility to skeletal muscle fatigue has often been reported in COPD patients (CitationSerres et al 1998; CitationAllaire et al 2004; CitationCoronell et al 2004; CitationVan’t Hul et al 2004; CitationSaey et al 2005; CitationJanaudis-Ferreira et al 2006). Other studies, however, do report an unaltered fatigue resistance (CitationGosker, Lencer et al 2003; CitationDegens et al 2005; CitationFranssen et al 2005).
Besides changes in the muscle itself that may cause an earlier onset of muscle fatigue, changes in fatigue resistance could also be caused by an altered central drive (central fatigue) (CitationBigland-Ritchie et al 1978). To date, the central component in the development of muscle fatigue in COPD patients is poorly understood. However, it may play a role in the development of fatigue as systemic inflammation has been shown to cause feelings of tiredness (CitationSpath-Schwalbe et al 1998). However, muscle fatigability has been determined largely with series of voluntary contractions (CitationSerres et al 1998; CitationCoronell et al 2004; CitationVan’t Hul et al 2004; CitationJanaudis-Ferreira et al 2006), which makes it difficult to differentiate between central and peripheral factors. Using electrical or magnetic stimulation, however, one can exclude the contribution of central factors to the development of fatigue. As far as we know, only one study (CitationDegens et al 2005) has assessed peripheral muscle fatigue using electrical stimulation in patients with COPD. In that study, neither differences in contractile properties nor fatigability were found. This indicates that there were no apparent differences in motivation between patients and controls matched for fat free mass index and physical activity level (CitationDegens et al 2005). Although it is thus possible that the alterations in fatigue resistance observed in other studies may be related to a lower fat free mass index in patients with COPD and controls, the observation that muscle endurance was similar in wasted and non-wasted patients (CitationGosker, Lencer et al 2003; CitationFranssen et al 2005) argues against this. However, in several studies it has been explicitly stated that the COPD patients were significantly less active than the controls (CitationSerres et al 1998; CitationCoronell et al 2004). Clearly, at least part of the decline in fatigue resistance often observed during COPD is attributable to a reduced physical activity level, but also smoking itself may reduce muscle fatigue resistance (CitationWüst et al 2006).
Factors underlying muscle dysfunction and wasting
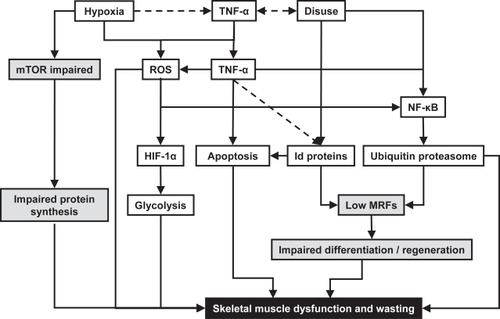
Many factors have been suggested to induce changes in skeletal muscle structure and function in COPD. Here we will summarize the possible role of airflow obstruction, disuse, hypoxemia, malnutrition, oxidative stress and systemic inflammation in the adaptations of skeletal muscle of patients with COPD. Figure summarizes how disuse, hypoxia and systemic inflammation may affect muscle wasting and dysfunction in COPD.
Figure 1 Pathways by which hypoxia, disuse and systemic inflammation contribute to muscle dysfunction and wasting during Chronic Obstructive Pulmonary Disease. Grey shaded boxes indicate impaired regeneration or protein synthesis; black shaded box represents the end result of the pathways in terms of muscle dysfunction and muscle wasting. Solid lines indicate observed relations; dotted lines indicate possible relations. TNF-α: tumor necrosis factor - α; mTOR: mammalian target of rapamycin; ROS: reactive oxygen species; HIF-1α: hypoxia-inducible factor - 1α; NF-κB: nuclear factor - κB; Id proteins: inhibitor of differentiation proteins; MRFs: myogenic regulatory factors (such as MyoD).
Airflow obstruction
In the GOLD classification the severity of COPD is determined as the degree of airflow obstruction as indicated by the percentage of the predicted FEV1. Despite the fact that a low FEV1 indicates a severe case of COPD no correlations between FEV1 and skeletal muscle strength or fatigue have been observed (CitationGosker, Kubat et al 2003; CitationGosker, Lencer et al 2003; CitationDegens et al 2005). In terms of muscle fatigability, this is not surprising, as during muscle fatigue tests a relatively small muscle mass is recruited of which the oxygen requirement is well within the limits that can be provided by the affected lung. Although, the increased cost of breathing as a result of the obstructive airflow may well be a cause of exercise intolerance during COPD (CitationAliverti and Macklem 2001) and may cause structural changes in the respiratory muscles due to the continuous overload (CitationOrozco-Levi et al 2001), it seems unlikely that the airflow obstruction per se will affect peripheral skeletal muscle structure or function.
Disuse
The physical activity level of patients with COPD is lower than that of the average population (CitationPitta et al 2005, Citation2006b) and during and after a period of exacerbations patients become even less active (CitationPitta et al 2006a). This is thought to be a consequence of the so-called dyspnea spiral: patients do not exert themselves too much in order to avoid the occurrence of dyspnea, which in turn causes a decline in fitness and an earlier occurrence of dyspnea and so on (CitationSerres et al 1998). It is therefore not surprising that disuse contributes significantly to the alterations in skeletal muscle structure and function during COPD (CitationDegens and Alway 2006). In fact, in a patient group compared with a physical activity level matched control group, no differences in muscle strength, fatigue resistance and contractile properties were detected (CitationDegens et al 2005). However, disuse alone is inadequate to explain all the changes occurring in skeletal muscle structure and function. For instance, CitationGosker et al (Gosker, Engelen et al 2002) showed that atrophy mainly occurred in type IIX fibers, whereas disuse would cause atrophy of each fiber type, with type I fibers being affected the most (CitationDegens and Alway 2006). Also, a 12-weeks physical-rehabilitation program did not entirely reverse the effects of COPD in terms of capillarization and fiber type distribution (CitationWhittom et al 1998).
Hypoxemia
Due to the difficulties with breathing and impaired oxygen uptake, patients with severe COPD may have a decreased hemoglobin oxygen saturation level (hypoxemia), which may result in local tissue hypoxia. The abundance of the transcription factor hypoxia-inducible factor-1α (HIF-1α) increases during hypoxia (CitationRaguso et al 2004) and may induce a down-regulation of oxidative enzymes and an upregulation of glycolytic enzymes (CitationHoppeler et al 2003). In addition, it has been shown in cardiomyocytes that hypoxia inactivates the transcription factor peroxisome proliferator-activated receptor α (PPARα) and thereby decreases the expression of genes involved in fatty acid oxidation (CitationHuss et al 2001). These changes in transcriptional regulation of the expression of metabolic genes during hypoxia may result in an increased glycolytic and a reduced oxidative capacity similar to what is observed during COPD (CitationHoppeler et al 2003; CitationRaguso et al 2004)
Chronic hypoxia may be linked with muscle wasting and weakness. Just 8 weeks at altitudes greater than 5000 m has been shown to cause as much as a 10% reduction in muscle mass and peak power (CitationFerretti et al 1990; CitationHoppeler et al 1990). Although a decrease in fiber CSA is associated with exposure to hypoxia (CitationHoppeler et al 1990; CitationMacDougall et al 1991), other confounding factors such as a decreased food intake, due to an hypoxia-induced expression of leptin, together with detraining may contribute to muscle wasting during hypoxia (CitationWesterterp and Kayser 2006).
Hypoxia has been shown to impair the mTOR pathway, which is involved in transcription of DNA and translation of mRNA into protein (CitationProud 2004b) and may, as a consequence, contribute to muscle wasting during COPD. In addition, it has been reported in cell culture studies that hypoxia inhibits myoblast differentiation by degradation of MyoD, a myogenic transcription factor, via the ubiquitin proteasome pathway (CitationDi Carlo et al 2004). Clearly, such an effect in vivo will have a negative impact on the regenerative potential of skeletal muscle. Moreover, hypoxia may also induce inflammation (CitationOrth et al 2005), causing muscle atrophy through inflammatory pathways (see below and Figure ).
In addition to these relatively long-term effects, there is evidence that hypoxia might acutely affect the contractile apparatus. Indeed, single fibers isolated from bundles exposed to hypoxia for 30 min exhibited a marked force loss which was attributable to a reduced fraction of strongly attached cross bridges, while reoxygenation completely reversed the contractile dysfunction (CitationOttenheijm, Heunks, Geraedts et al 2006). In addition to muscle weakening, hypoxia has also been shown to reduce the maximum shortening velocity, power output, force frequency relation and endurance in muscle bundles (CitationMachiels et al 2001; CitationZhu et al 2005), possibly through nitrotyrosylation of proteins (CitationOttenheijm, Heunks, Geraedts et al 2006) and the presence of reactive oxygen species (ROS). Although in vitro there seems to be a clear effect of acute hypoxia on skeletal muscle function this is not necessarily the case in vivo. In the electrically stimulated human quadriceps muscle no change in contractile properties, strength or fatigue resistance could be detected during acute exposure to hypoxia (CitationDegens et al 2006). The discrepancy between in vitro and in vivo observations might be caused by absence of oxidative stress in vivo (CitationDousset et al 2002). It is possible that hemoglobin and myoglobin, absent in the in vitro situation, scavenge nitric oxygen (NO) and ROS, so that the detrimental effects of these substances are attenuated, at least for a limited period (CitationOrdway and Garry 2004). Indeed, in chronic hypoxemic COPD patients oxidative stress is enhanced (CitationKoechlin et al 2005).
Hypoxia itself, however, can not fully account for all the observed changes in skeletal muscle as many patients with COPD suffering from muscle wasting and dysfunction do not exhibit hypoxemia. In addition, hypoxia may even protect the nucleus from apoptosis (CitationRiva et al 2001), whereas an elevated occurrence of apoptosis in skeletal muscle has been observed in depleted patients with COPD (CitationAgusti et al 2002; Degens et al submitted).
Malnutrition
Many patients with COPD suffer from semi-starvation, possibly caused by elevated levels of circulating leptin, which negatively affects dietary intake and consequently muscle mass and function (CitationEngelen et al 1994; CitationCasaburi 2001; CitationFranssen, Wouters, Schols 2002; CitationSchols 2003a). Moreover, the basal metabolism in COPD is increased as a consequence of extra work required for breathing and/or the presence of systemic inflammation. Hypermetabolism in combination with a decreased appetite often leads to a negative nutrition balance and ultimately weight loss (CitationSchols 2003b).
Oxidative stress
Reative Oxygen Species (ROS) and free radicals are elevated in patients with COPD both during rest and exercise (CitationCouillard et al 2002, Citation2003; CitationGosker, Bast et al 2005; Citationvan Helvoort, Heijdra, Thijs 2006). The mitochondrial electron transport chain and neutrophils are an important source of ROS (CitationZhang et al 1990). Also hypoxia, cigarette smoke, sepsis and an increased cost of breathing (CitationHeunks and Dekhuijzen 2000) cause an increased generation of ROS and reactive nitrogen species in the lungs and respiratory muscles, spilling into the circulation. There is evidence that an abnormal oxidative stress response to submaximal and maximal exercise may be more severe in muscle-wasted than non-muscle-wasted patients with COPD (Citationvan Helvoort, Heijdra, Thijs 2006). Oxidative stress may acutely affect skeletal muscle function via inhibition of the activity of the sodium/potassium pump (CitationComellas et al 2006), sarcoplasmic reticulum function, myosin ATPase and mitochondrial respiration (CitationZhang et al 1990), and in the long run may also cause muscle wasting and dysfunction in both respiratory and peripheral muscle (CitationLangen et al 2003; CitationKoechlin et al 2004, Citation2005 CitationOttenheijm, Heunks, Geraedts et al 2006).
Systemic inflammation
A common feature in many chronic diseases including COPD is the presence of systemic and/or local inflammation (CitationGan et al 2004). In patients with COPD the lung is thought to be the main source of inflammatory cytokines. It has been shown that resistive breathing may cause the respiratory muscles also to produce inflammatory cytokines and thereby to contribute to the development of cachexia in patients with COPD (CitationVassilakopoulos et al 2004). The negative correlation between muscle strength and a marker of systemic inflammation during an exacerbation (CitationSpruit et al 2003) suggests that inflammation is indeed an important factor in muscle adaptations during COPD (CitationDegens and Alway 2006).
Inflammatory cytokines may also have central actions leading to “sickness behavior”, a loss of motivation and thus contribute to tiredness and the downward spiral of inactivity (CitationSpath-Schwalbe et al 1998). For instance, acute administration of IL-6 to otherwise healthy trained runners seriously impaired exercise performance (CitationRobson-Ansley et al 2004), but the effect may be mediated by an altered (serotonergic) activity in the brain increasing the sensation of generalized fatigue (the central fatigue hypothesis), rather than peripheral factors (CitationPolkey 2003).
In the remainder of this section we will concentrate mainly on the effects of TNF-α on skeletal muscle tissue, as most research has focused on this cytokine. Cachectin, or TNF-α, has long been known to induce muscle wasting (CitationBeutler and Cerami 1986). In a transgenic mouse model that over-expresses TNF-α in the lung, not only circulating TNF-α levels, but also the expression of TNF-α in the muscle were increased; the latter was thought to be the result of TNF-α inducing its own expression via a positive feed back loop (CitationLangen et al 2006). One can imagine, that in particular during exacerbations, when the inflammation is aggravated (CitationPapi et al 2006), this is a serious complication.
TNF-α may also have acute effects on skeletal muscle function. For instance, systemic administration of TNF-α to dogs caused diaphragm weakness after only three hours (CitationWilcox et al 1994). Since then, it has been shown in vitro that exposure of single muscle fibers to TNF-α decreased the force generating capacity (CitationLi et al 2000; CitationReid et al 2002), possibly through the generation of ROS or reactive nitrogen species. It should be noted, however, that in these studies supraphysiological doses of TNF-α were applied and it remains to be determined whether physiological levels of TNF-α have similar effects.
Chronically elevated systemic inflammation increases the activity of the ubiquitin proteasome pathway via activation of nuclear factor – κB (NF-κB), a factor that plays an important role in muscle atrophy (CitationDebigare et al 2001; CitationKandarian and Jackman 2006). The ubiquitin proteasome pathway is an ATP-dependent protein degradation pathway where proteins are labeled by ubiquitin for subsequent degradation in the proteasome. Although the activation of the ubiquitin-proteasome pathway is apparent in many other chronic diseases, including cancer (CitationKhal et al 2005), it is not known whether activation of the ubiquitin-proteasome pathway also plays a role in peripheral skeletal muscle wasting during COPD. Only recently it has been shown that the ubiquitin-proteasome pathway is activated in the diaphragm in patients with mild-to-moderate COPD (CitationOttenheijm, Heunks, Li et al 2006) leading to a loss of myosin, and thereby force generating capacity (CitationOttenheijm et al 2005).
Besides activating the ubiquitin proteaseome pathway TNF-α induces apoptosis, or programmed cell death, in skeletal muscle cells and myoblasts (CitationBazzoni and Beutler 1996; CitationStewart et al 2004). The loss of myonuclei may cause an increase of the myonuclear domain, the volume of cytoplasm associated with a single myonucleus, beyond a sustainable size and consequently be followed by muscle fiber atrophy (CitationAllen et al 1999). Apoptosis may indeed play a role in muscle wasting during COPD in particular in patients with a low body mass index (CitationAgusti et al 2002; CitationPlataki et al 2006). In addition, TNF-α induces expression of Id proteins in astrocytes (CitationTzeng et al 1999) and Id proteins in turn can induce apoptosis both via alteration of gene transcription and binding to regulators of apoptosis (CitationFlorio et al 1998). Although a link between TNF-α and Id protein expression has not been investigated in other cell types, Id proteins themselves have been shown to induce apoptosis in many cell types, including neonatal cardiomyocytes and myoblasts (CitationYokota and Mori 2002). It is therefore tempting to speculate that altered Id expression also plays a role in the development of apoptosis during COPD and impaired regenerative capacity. This has not yet been investigated in patients with COPD, but both the elevated expression of Id2 protein and apoptotic factors in the diaphragm and soleus of the emphysematous hamster (CitationAlway et al 2004) hint to this possibility.
As mentioned above, it is possible that the primary problem of muscle atrophy during COPD is a decreased regenerative capacity. Myogenic regulatory factors, such as MyoD, play an important role in satellite cell differentiation and hence regenerative capacity of the muscle (Charge et al 2004). Both in transgenic mice overexpressing TNF-α in the lung (CitationLangen et al 2006) and in mice treated with TNF-α (CitationGuttridge et al 2000; CitationLangen et al 2004), the regeneration of skeletal muscle from disuse atrophy is delayed. This decline in regenerating capacity was possibly related to an accelerated breakdown of MyoD by the ubiquitin proteasome pathway (CitationLangen et al 2004). Interestingly, MyoD becomes more prone to degradation by the ubiquitin proteasome pathway when it forms dimers with inhibitors of differentiation (Id) proteins (CitationReid 2005). Elevation of these proteins may thus contribute to the reduced abundance of MyoD protein but not mRNA in the soleus and diaphragm of emphysematous hamsters (CitationDegens et al 2004), indicating MyoD breakdown rather than reduced transcription. No studies have so far investigated whether alterations in MyoD and Id expression also occur in skeletal muscle from patients with COPD and whether they also suffer from an impaired skeletal muscle regenerative capacity.
Rehabilitation and medication
Here we only briefly discuss several of the more common potential treatments that target skeletal muscle wasting during COPD. For a more extensive discussion we refer the reader to other reviews that specifically address this issue (CitationSpruit et al 2004; CitationHansen et al 2006).
Exercise training
As disuse is considered an important factor contributing to muscle wasting and dysfunction it is not surprising that many studies have addressed the efficacy of exercise training on skeletal muscle structure and function in patients with COPD (CitationSerres et al 1998; CitationFranssen et al 2005; CitationGosker, Schrauwen et al 2005).
Endurance training improves exercise tolerance in patients with moderate and severe COPD, with the effect being largely influenced by the intensity of the training; a low intensity produces less of an effect than high intensity training sessions (CitationCasaburi 2001). This improvement in exercise tolerance is accompanied by at least a reduction of the percentage of type II fibers, muscle hypertrophy and an increase in oxidative capacity (CitationWhittom et al 1998; CitationCasaburi 2001; CitationVogiatzis et al 2005).
Strength training is effective in increasing muscle mass and strength and is associated with an improved quality of life in patients with COPD (CitationCasaburi 2001). Combined strength/endurance training results in the benefits that each of the programs separately would achieve, ie, not only an increase in submaximal exercise capacity, but also improvements in lean body mass and strength (CitationOrtega et al 2002). Moreover, interval training might also be preferred above constant-load exercise as it minimizes leg discomfort and ratings of dyspnea, without compromising the benefits of the endurance training program (CitationVogiatzis et al 2005).
This response to training represents a normal response of the muscle to increased use (CitationSalmons and Henriksson 1981). The beneficial effects of exercise may, at least partly, be brought about by an increase in the expression of myogenin (CitationSiu, Donley et al 2004), a reduction of the occurrence of apoptosis (CitationSiu, Bryner et al 2004), and suppression of the muscle specific ubiquitin ligase atrogin (Leger et al 2006). Furthermore, endurance training may attenuate systemic inflammation (CitationGarrod et al 2007) and it is thus possible that the beneficial effects of training are partly mediated via a reduction in inflammation. So far, it is not clear whether all patients would benefit from exercise programs as for instance in some elderly people the hypertrophic response is attenuated, indicating a reduced plasticity at old age (CitationWelle et al 1996; CitationDegens and Alway 2006). This attenuated response has been shown to be related to elevated baseline levels of soluble TNF-receptors in the elderly (CitationBruunsgaard et al 2004). This would imply that chronic patients with a significantly elevated systemic inflammation may have reduced improvements in response to exercise training, particularly when one considers that the inflammatory and oxidative stress response is augmented in muscle-wasted patients (Citationvan Helvoort, Heijdra, Dekhuijzen 2006). Supporting the notion that exercise may loose its effectiveness when systemic inflammation is present, is the observation that the cellular protein breakdown in patients with a low fat free mass does not decline after an exercise training regime (CitationBolton et al 2006).
Nutrition
Nutritional support has been shown to result in functional improvements and decreased mortality in depleted patients (CitationSchols 2003b). One of the benefits of nutritional support is an increase in muscle strength (CitationEfthimiou et al 1988), which will inevitably improve the quality of life of the patient. Combination of nutritional support and exercise training may be the best approach to obtain functional improvements in patients with COPD (CitationSchols 2003a). For detailed information about the effects of nutrition of muscle performance in patients with COPD and other chronic diseases, the reader is referred to some excellent reviews on this subject (CitationSchols 2003b; CitationEngelen et al 2005).
Oxygen therapy
Oxygen supplementation is often used to reduce dyspnea experienced by severe hypoxemic COPD patients. Oxygen supplementation may not only increase the exercise capacity (CitationAliverti and Macklem 2001), but also reduce the normal exercise-induced elevation in systemic inflammation and oxidative stress in muscle-wasted patients with COPD (Citationvan Helvoort, Heijdra, Heunks 2006). An additional benefit of supplemental oxygen during exercise is that patients can exercise at higher work loads (CitationEmtner et al 2003) and thereby augment the improvement in exercise tolerance.
Long-term oxygen supplementation only benefits hypoxemic, but not normoxemic patients with COPD by increasing survival and attenuating the progression of pulmonary hypertension (CitationZielinski 1999). However, to our knowledge no studies have addressed the direct or long-term effects of oxygen therapy on skeletal muscle adaptations in COPD patients.
Medication
Anabolic hormone, such as testosterone, supplementation has been suggested to treat muscle wasting and dysfunction during COPD (CitationCasaburi et al 2004; CitationHansen et al 2006). Combination of strength training and testosterone supplementation appeared to give additive effects on lean body mass and strength in patients with COPD (CitationCasaburi et al 2004). However, the long-term (side and beneficial) effects of hormone replacement remain to be established. The observation that IGF-I is only positively related to muscle strength when IL-6 levels are low but not when IL-6 levels are high (CitationBarbieri et al 2003) suggests that the effectiveness of testosterone replacement (which probably acts in part through an effect on IGF-1) may be attenuated in patients with high levels of systemic inflammation.
Since systemic inflammation plays an important role in the progression of the disease anti-inflammatory drugs have been used to attenuate the disease progression and the concomitant observed muscle wasting (CitationHansen et al 2006). Corticosteroids are often prescribed to minimize the inflammatory reaction, in particular following exacerbations. Unfortunately, corticosteroids also induce the activity of the ubiquitin proteasome pathway (CitationTisdale 2005) and prolonged treatment with corticosteroids results in a ‘steroid myopathy’, characterized by a preferential type II fiber atrophy (CitationDecramer et al 1996). The vitamin D analogue α-calcidol, already widely used for the treatment of osteoporosis, has proved successful in reducing circulating TNF-α levels and the release of cytokines by macrophages and improving muscle power in patients with rheumatoid arthritis (CitationScharla et al 2005). No studies have so far addressed the efficacy of α-calcidol in the treatment of muscle weakness (and/or osteoporosis) in COPD.
One could also consider to provide supplementation with IGF-I, or inhibitors of TNF-α, but IGF-I may increase the chance of getting cancer and TNF-α inhibitors increase the risk of sepsis and infection (CitationHansen et al 2006). Therefore, factors downstream, such as the ubiquitin proteasome pathway, or factors involved in satellite cell differentiation, may be better targets. In this context it is worthwhile to note that Id proteins may be an interesting target as they play a role in muscle wasting and their inhibition is also a potential target in the treatment of cancer (CitationBenezra et al 2001).
Drugs that inhibit NF-κB, which is downstream of the TNF-receptor and an upstream regulator of MuRF1, a ubiquitin ligase, appear to be effective in attenuating muscle wasting during cachexia (CitationTisdale 2005). Indeed, there are clear indications for further drug developments such as with inhibitors of the ubiquitin-proteasome pathway and apoptosis (CitationLibera and Vescovo 2004; CitationTisdale 2005; CitationHansen et al 2006). As suggested by CitationTisdale (2005), it is advisable to combine inhibitors of protein degradation with stimulators of protein synthesis by, for instance, an increased intake of amino acids, such as leucine, that stimulate the mTOR pathway (CitationProud 2004a).
In clinical settings, it seems that a combination of multiple strategies results in the best functional improvements. Clearly, however, more studies are needed to determine the best combination of therapies in terms of cost-effectiveness, benefit and patient compliance (CitationSchols 2003a).
Summary and conclusion
In this article, we present an overview of the changes in muscle structure and function in COPD and the factors contributing to these changes are discussed. Muscle wasting should be considered as a serious complication in COPD and has important implications for survival. One notable feature of the literature is the great diversity of muscle symptoms in patients with apparently similar degrees of lung disease. Whether this indicates a difference in the nature of the disease or of the genetic susceptibility to muscle complications remains an important question. There is no doubt, however, that continued research into the question of muscle wasting and decreased skeletal muscle fatigue resistance in COPD will eventually lead to the development of specific treatments for cachexia, such as targeting the ubiquitin proteasome pathway, cytokine inhibition, administration of anabolic factors as well as life style changes, such as exercise and nutrition. The clinical approach to cachexia in COPD and other chronic diseases should change dramatically in the near future.
References
- AaseboUGyltnesABremnesRM1993Reversal of sexual impotence in male patients with chronic obstructive pulmonary disease and hypoxemia with long-term oxygen therapyJ Steroid Biochem Mol Biol467998038274414
- AgustiAGSauledaJMirallesC2002Skeletal muscle apoptosis and weight loss in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med166485912186825
- AlivertiAMacklemPT2001How and why exercise is impaired in COPDRespiration682293911416240
- AllaireJMaltaisFDoyonJF2004Peripheral muscle endurance and the oxidative profile of the quadriceps in patients with COPDThorax59673815282387
- AllenDLRoyRREdgertonVR1999Myonuclear domains in muscle adaptation and diseaseMuscle Nerve2213506010487900
- AlwaySESwisherASiuPM2004Increased expression of apoptotic genes in the diaphragm and soleus muscle of the emphysematous hamsterFaseb J18A358
- AmbrosinoNBruschiCCallegariG1996Time course of exercise capacity, skeletal and respiratory muscle performance after heart-lung transplantationEur Respir J91508148836667
- ATS/ERS1999Skeletal muscle dysfunction in chronic obstructive pulmonary disease. A statement of the American Thoracic Society and European Respiratory SocietyAm J Respir Crit Care Med159S14010194189
- BarbieriMFerrucciLRagnoE2003Chronic inflammation and the effect of IGF-I on muscle strength and power in older personsAm J Physiol Endocrinol Metab284E481712419777
- BazzoniFBeutlerB1996The tumor necrosis factor ligand and receptor familiesN Engl J Med3341717258637518
- BenezraRRafiiSLydenD2001The Id proteins and angiogenesisOncogene2083344111840326
- BernardSLeBlancPWhittomF1998Peripheral muscle weakness in patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med158629349700144
- BeutlerBCeramiA1986Cachectin and tumour necrosis factor as two sides of the same biological coinNature32058483010124
- Bigland-RitchieBJonesDAHoskingGP1978Central and peripheral fatigue in sustained maximum voluntary contractions of human quadriceps muscleClin Sci Mol Med5460914657729
- BoltonCEBroekhuizenRIonescuAA2006Cellular protein breakdown and systemic inflammation are unaffected by pulmonary rehabilitation in COPDThorax in press.
- BruunsgaardHBjerregaardESchrollM2004Muscle strength after resistance training is inversely correlated with baseline levels of soluble tumor necrosis factor receptors in the oldest oldJ Am Geriatr Soc522374114728633
- CasaburiR1998Rationale for anabolic therapy to facilitate rehabilitation in chronic obstructive pulmonary diseaseBaillieres Clin Endocrinol Metab124071810332562
- CasaburiR2001Skeletal muscle dysfunction in chronic obstructive pulmonary diseaseMed Sci Sports Exerc33S6627011462075
- CasaburiRBhasinSCosentinoL2004Effects of testosterone and resistance training in men with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med170870815271690
- ComellasAPDadaLALecuonaE2006Hypoxia-mediated degradation of Na,K-ATPase via mitochondrial reactive oxygen species and the ubiquitin-conjugating systemCirc Res9813142216614303
- CoronellCOrozco-LeviMMendezR2004Relevance of assessing quadriceps endurance in patients with COPDEur Respir J241293615293615
- CouillardAKoechlinCCristolJP2002Evidence of local exerciseinduced systemic oxidative stress in chronic obstructive pulmonary disease patientsEur Respir J201123912449164
- CouillardAMaltaisFSaeyD2003Exercise-induced quadriceps oxidative stress and peripheral muscle dysfunction in patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1671664912672647
- CreutzbergECCasaburiR2003Endocrinological disturbances in chronic obstructive pulmonary diseaseEur Respir J Suppl4676s80s14621109
- De BenedettoFDel PonteAMarinariS2000In COPD patients, body weight excess can mask lean tissue depletion: a simple method of estimationMonaldi Arch Chest Dis55273811057077
- DebigareRCoteCHHouldFS2003In vitro and in vivo contractile properties of the vastus lateralis muscle in males with COPDEur Respir J21273812608441
- DebigareRCoteCHMaltaisF2001Peripheral muscle wasting in chronic obstructive pulmonary disease. Clinical relevance and mechanismsAm J Respir Crit Care Med1641712711719314
- DecramerMde BockVDomR1996Functional and histologic picture of steroid-induced myopathy in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1531958648665061
- DegensHAlwaySE2006Control of muscle size during disuse, disease, and agingInt J Sports Med2794916475053
- DegensHOttenheijmCAHeunksLM2004Altered MyoD and Id2 protein expression in the diaphragm and soleus muscle of the emphysematous hamsterAm J Respir Crit Care Med169A857
- DegensHSanchez HornerosJMHeijdraYF2005Skeletal muscle contractility is preserved in COPD patients with normal fat-free massActa Physiol Scand1842354215954991
- DegensHSanchez HornerosJMHopmanMT2006Acute hypoxia limits endurance but does not affect muscle contractile propertiesMuscle Nerve33532716372323
- DegensHSwisherAKHeijdraYFSubmitted. Apoptosis and Id2 expression in diaphragm and soleus muscle from the emphysematous hamsterAm J Physiol Regul Integr Comp Physiol
- DegensHVeerkampJH1994Changes in oxidative capacity and fatigue resistance in skeletal muscleInt J Biochem2687188063011
- Di CarloADe MoriRMartelliF2004Hypoxia inhibits myogenic differentiation through accelerated MyoD degradationJ Biol Chem27916332814754880
- DoussetESteinbergJGFaucherM2002Acute hypoxemia does not increase the oxidative stress in resting and contracting muscle in humansFree Radic Res36701412184222
- EfthimiouJFlemingJGomesC1988The effect of supplementary oral nutrition in poorly nourished patients with chronic obstructive pulmonary diseaseAm Rev Respir Dis1371075823057956
- EmtnerMPorszaszJBurnsM2003Benefits of supplemental oxygen in exercise training in nonhypoxemic chronic obstructive pulmonary disease patientsAm J Respir Crit Care Med16810344212869359
- EngelenMPRuttenEPDe CastroCL2005Altered interorgan response to feeding in patients with chronic obstructive pulmonary diseaseAm J Clin Nutr823667216087980
- EngelenMPScholsAMBakenWC1994Nutritional depletion in relation to respiratory and peripheral skeletal muscle function in outpatients with COPDEur Respir J7179377828687
- EngelenMPScholsAMDoesJD2000Skeletal muscle weakness is associated with wasting of extremity fat-free mass but not with airflow obstruction in patients with chronic obstructive pulmonary diseaseAm J Clin Nutr71733810702166
- EvansABAl-HimyaryAJHrovatMI1997Abnormal skeletal muscle oxidative capacity after lung transplantation by 31P-MRSAm J Respir Crit Care Med155615219032203
- FaulknerMALenzTLStadingJA2006Cost-effictiveness of smoking cessation and the implications for COPDInt J of COPD127987
- FerrettiGHauserHdi PramperoPE1990Maximal muscular power before and after exposure to chronic hypoxiaInt J Sports Med11Suppl 1S3142323862
- FlorioMHernandezMCYangH1998Id2 promotes apoptosis by a novel mechanism independent of dimerization to basic helix-loop-helix factorsMol Cell Biol185435449710627
- FranssenFMBroekhuizenRJanssenPP2005Limb muscle dysfunction in COPD: effects of muscle wasting and exercise trainingMed Sci Sports Exerc372915632660
- FranssenFMWoutersEFBaarendsEM2002Arm mechanical efficiency and arm exercise capacity are relatively preserved in chronic obstructive pulmonary diseaseMed Sci Sports Exerc341570612370557
- FranssenFMWoutersEFScholsAM2002The contribution of starvation, deconditioning and ageing to the observed alterations in peripheral skeletal muscle in chronic organ diseasesClin Nutr2111411884007
- GanWQManSFSenthilselvanA2004Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysisThorax595748015223864
- GarrodRAnsleyPCanavanJ2007Exercise and the inflammatory response in chronic obstructive pulmonary disease (COPD)-Does training confer anti-inflammatory properties in COPD?Med Hypotheses68291817010529
- GeaJOrozco-LeviMBarreiroE2001Structural and functional changes in the skeletal muscles of COPD patients: the compartments theoryMonaldi Arch Chest Dis562142411665501
- GeaJGPastoMCarmonaMA2001Metabolic characteristics of the deltoid muscle in patients with chronic obstructive pulmonary diseaseEur Respir J179394511488330
- GoskerHRBastAHaenenGR2005Altered antioxidant status in peripheral skeletal muscle of patients with COPDRespir Med991182515672860
- GoskerHREngelenMPvan MamerenH2002Muscle fiber type IIX atrophy is involved in the loss of fat-free mass in chronic obstructive pulmonary diseaseAm J Clin Nutr76113912081824
- GoskerHRKubatBSchaartG2003Myopathological features in skeletal muscle of patients with chronic obstructive pulmonary diseaseEur Respir J22280512952261
- GoskerHRLencerNHFranssenFM2003Striking similarities in systemic factors contributing to decreased exercise capacity in patients with severe chronic heart failure or COPDChest12314162412740256
- GoskerHRSchrauwenPBroekhuizenR2005Exercise training restores uncoupling protein-3 content in limb muscles of patients with chronic obstructive pulmonary diseaseAm J Physiol Endocrinol Metab
- GoskerHRvan MamerenHvan DijkPJ2002Skeletal muscle fibre-type shifting and metabolic profile in patients with chronic obstructive pulmonary diseaseEur Respir J196172511998989
- GoskerHRWoutersEFvan der VusseGJ2000Skeletal muscle dysfunction in chronic obstructive pulmonary disease and chronic heart failure: underlying mechanisms and therapy perspectivesAm J Clin Nutr7110334710799364
- GosselinkRTroostersTDecramerM1996Peripheral muscle weakness contributes to exercise limitation in COPDAm J Respir Crit Care Med153976808630582
- GosselinkRTroostersTDecramerM2000Distribution of muscle weakness in patients with stable chronic obstructive pulmonary diseaseJ Cardiopulm Rehabil203536011144041
- GuttridgeDCMayoMWMadridLV2000NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexiaScience2892363611009425
- HansenMJGualanoRCBozinovskiS2006Therapeutic prospects to treat skeletal muscle wasting in COPD (chronic obstructive lung disease)Pharmacol Ther1091627216154635
- HeijdraYFPinto-PlataVFrantsR2003Muscle strength and exercise kinetics in COPD patients with a normal fat-free mass index are comparable to control subjectsChest124758212853505
- HeunksLMDekhuijzenPN2000Respiratory muscle function and free radicals: from cell to COPDThorax557041610899251
- HoppelerHKleinertESchlegelC1990Morphological adaptations of human skeletal muscle to chronic hypoxiaInt J Sports Med11Suppl 1S392323861
- HoppelerHVogtMWeibelER2003Response of skeletal muscle mitochondria to hypoxiaExp Physiol881091912525860
- HussJMLevyFHKellyDP2001Hypoxia inhibits the peroxisome proliferator-activated receptor alpha/retinoid X receptor gene regulatory pathway in cardiac myocytes: a mechanism for O2-dependent modulation of mitochondrial fatty acid oxidationJ Biol Chem276276051211371554
- JagoeRTEngelenMP2003Muscle wasting and changes in muscle protein metabolism in chronic obstructive pulmonary diseaseEur Respir J Suppl4652s63s14621107
- JakobssonPJorfeldtLBrundinA1990Skeletal muscle metabolites and fibre types in patients with advanced chronic obstructive pulmonary disease (COPD), with and without chronic respiratory failureEur Respir J319262311744
- JakobssonPJorfeldtLHenrikssonJ1995Metabolic enzyme activity in the quadriceps femoris muscle in patients with severe chronic obstructive pulmonary diseaseAm J Respir Crit Care Med15137477842194
- Janaudis-FerreiraTWadellKSundelinG2006Thigh muscle strength and endurance in patients with COPD compared with healthy controlsRespir Med1001451716337114
- JespersenJKjaerMSchjerlingP2006The possible role of myostatin in skeletal muscle atrophy and cachexiaScand J Med Sci Sports16748216533345
- JobinJMaltaisFDoyonJF1998Chronic obstructive pulmonary disease: capillarity and fiber-type characteristics of skeletal muscleJ Cardiopulm Rehabil1843279857275
- KamischkeAKemperDECastelMA1998Testosterone levels in men with chronic obstructive pulmonary disease with or without glucocorticoid therapyEur Respir J114159543268
- KandarianSCJackmanRW2006Intracellular signaling during skeletal muscle atrophyMuscle Nerve331556516228971
- KhalJHineAVFearonKC2005Increased expression of proteasome subunits in skeletal muscle of cancer patients with weight lossInt J Biochem Cell Biol37219620616125116
- KillianKJLeblancPMartinDH1992Exercise capacity and ventilatory, circulatory, and symptom limitation in patients with chronic airflow limitationAm Rev Respir Dis146935401416421
- KoechlinCCouillardASimarD2004Does oxidative stress alter quadriceps endurance in chronic obstructive pulmonary disease?Am J Respir Crit Care Med1691022715001462
- KoechlinCMaltaisFSaeyD2005Hypoxaemia enhances peripheral muscle oxidative stress in chronic obstructive pulmonary diseaseThorax608344115964914
- KorhonenMTCristeaAAlenM2006Aging, muscle fiber type, and contractile function in sprint-trained athletesJ Appl Physiol1019061716690791
- LangenRCKornSHWoutersEF2003ROS in the local and systemic pathogenesis of COPDFree Radic Biol Med352263512885585
- LangenRCScholsAMKeldersMC2006Muscle Wasting and Impaired Muscle Regeneration in a Murine Model of Chronic Pulmonary InflammationAm J Respir Cell Mol Biol
- LangenRCVan Der VeldenJLScholsAM2004Tumor necrosis factor-alpha inhibits myogenic differentiation through MyoD protein destabilizationFaseb J182273714769817
- LiXMoodyMREngelD2000Cardiac-specific overexpression of tumor necrosis factor-alpha causes oxidative stress and contractile dysfunction in mouse diaphragmCirculation1021690611015349
- LiberaLDVescovoG2004Muscle wastage in chronic heart failure, between apoptosis, catabolism and altered anabolism: a chimaeric view of inflammation?Curr Opin Clin Nutr Metab Care74354115192447
- MacDougallJDGreenHJSuttonJR1991Operation Everest II: structural adaptations in skeletal muscle in response to extreme simulated altitudeActa Physiol Scand14242171927554
- MachielsHAvan der HeijdenHFHeunksLM2001The effect of hypoxia on shortening contractions in rat diaphragm muscleActa Physiol Scand1733132111736693
- ManninoDMHomaDMAkinbamiLJ2002Chronic obstructive pulmonary disease surveillance – United States, 1971–2000MMWR Surveill Summ51116
- MarquisKDebigareRLacasseY2002Midthigh muscle crosssectional area is a better predictor of mortality than body mass index in patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1668091312231489
- NariciMVBordiniMCerretelliP1991Effect of aging on human adductor pollicis muscle functionJ Appl Physiol711277811757349
- OgaTNishimuraKTsukinoM2005Exercise capacity deterioration in patients with COPD: longitudinal evaluation over 5 yearsChest12862916002917
- OgaTNishimuraKTsukinoM2007Longitudinal deteriorations in patient reported outcomes in patients with COPDRespir Med1011465316713225
- OrdwayGAGarryDJ2004Myoglobin: an essential hemoprotein in striated muscleJ Exp Biol2073441615339940
- Orozco-LeviMLloretaJMinguellaJ2001Injury of the human diaphragm associated with exertion and chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1641734911719318
- OrtegaFToralJCejudoP2002Comparison of effects of strength and endurance training in patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1666697412204863
- OrthTAAllenJAWoodJG2005Exercise training prevents the inflammatory response to hypoxia in cremaster venulesJ Appl Physiol982113815705731
- OttenheijmCAHeunksLMGeraedtsMC2006Hypoxia-induced skeletal muscle fiber dysfunction: role for reactive nitrogen speciesAm J Physiol Lung Cell Mol Physiol290L1273516113049
- OttenheijmCAHeunksLMLiYP2006Activation of ubiquitinproteasome pathway in the diaphragm in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med
- OttenheijmCAHeunksLMSieckGC2005Diaphragm dysfunction in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med172200515849324
- PapiALuppiFFrancoF2006Pathophysiology of exacerbations of chronic obstructive pulmonary diseaseProc Am Thorac Soc32455116636093
- PettyTL2006The history of COPDInternational Journal of COPD131418046898
- PittaFTroostersTProbstVS2006aPhysical activity and hospitalization for exacerbation of COPDChest1295364416537849
- PittaFTroostersTProbstVS2006bQuantifying physical activity in daily life with questionnaires and motion sensors in COPDEur Respir J2710405516707399
- PittaFTroostersTSpruitMA2005Activity monitoring for assessment of physical activities in daily life in patients with chronic obstructive pulmonary diseaseArch Phys Med Rehabil8619798516213242
- PlatakiMTzortzakiERytilaP2006Apoptotic mechanisms in the pathogenesis of COPDInt J of COPD116171
- PolkeyMI2003Peripheral muscle weakness in COPD: where does it come from?Thorax58741212947126
- ProudCG2004amTOR-mediated regulation of translation factors by amino acidsBiochem Biophys Res Commun3134293614684180
- ProudCG2004bThe multifaceted role of mTOR in cellular stress responsesDNA Repair (Amst)39273415279778
- RagusoCAGuinotSLJanssensJP2004Chronic hypoxia: common traits between chronic obstructive pulmonary disease and altitudeCurr Opin Clin Nutr Metab Care7411715192444
- ReidMB2005Response of the ubiquitin-proteasome pathway to changes in muscle activityAm J Physiol Regul Integr Comp Physiol288R14233115886351
- ReidMBLannergrenJWesterbladH2002Respiratory and limb muscle weakness induced by tumor necrosis factor-alpha: involvement of muscle myofilamentsAm J Respir Crit Care Med1664798412186824
- RennieMJEdwardsRHEmeryPW1983Depressed protein synthesis is the dominant characteristic of muscle wasting and cachexiaClin Physiol3387986357592
- RichardsonRSLeekBTGavinTP2004Reduced mechanical efficiency in chronic obstructive pulmonary disease but normal peak VO2 with small muscle mass exerciseAm J Respir Crit Care Med169899614500263
- RivaCChevrierCPasqualN2001Bcl-2/Bax protein expression in heart, slow-twitch and fast-twitch muscles in young rats growing under chronic hypoxia conditionsMol Cell Biochem22691611768244
- Robson-AnsleyPJde MilanderLCollinsM2004Acute interleukin-6 administration impairs athletic performance in healthy, trained male runnersCan J Appl Physiol29411815317982
- RutherfordOMJonesDANewhamDJ1986Clinical and experimental application of the percutaneous twitch superimposition technique for the study of human muscle activationJ Neurol Neurosurg Psychiatry491288913794735
- SaeyDMichaudACouillardA2005Contractile fatigue, muscle morphometry, and blood lactate in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med17111091515735055
- SainiANasserASStewartCE2006Waste management – cytokines, growth factors and cachexiaCytokine Growth Factor Rev174758617118696
- SalmonsSHenrikssonJ1981The adaptive response of skeletal muscle to increased useMuscle Nerve4941057010156
- SattaAMiglioriGBSpanevelloA1997Fibre types in skeletal muscles of chronic obstructive pulmonary disease patients related to respiratory function and exercise toleranceEur Respir J102853609493673
- ScharlaSHSchachtELempertUG2005Alfacalcidol versus plain vitamin D in inflammation induced bone lossJ Rheumatol Suppl76263216142848
- ScholsA2003aNutritional modulation as part of the integrated management of chronic obstructive pulmonary diseaseProc Nutr Soc627839115018476
- ScholsAM2003bNutritional and metabolic modulation in chronic obstructive pulmonary disease managementEur Respir J Suppl4681s86s14621110
- ScholsAMMostertRSoetersPB1991Body composition and exercise performance in patients with chronic obstructive pulmonary diseaseThorax4669591750015
- ScholsAMSoetersPBDingemansAM1993Prevalence and characteristics of nutritional depletion in patients with stable COPD eligible for pulmonary rehabilitationAm Rev Respir Dis147115168484624
- SerresIGautierVVarrayA1998Impaired skeletal muscle endurance related to physical inactivity and altered lung function in COPD patientsChest11390059554623
- SiuPMBrynerRWMartynJK2004Apoptotic adaptations from exercise training in skeletal and cardiac musclesFaseb J181150215132982
- SiuPMDonleyDABrynerRW2004Myogenin and oxidative enzyme gene expression levels are elevated in rat soleus muscles after endurance trainingJ Appl Physiol972778515033961
- Spath-SchwalbeEHansenKSchmidtF1998Acute effects of recombinant human interleukin-6 on endocrine and central nervous sleep functions in healthy menJ Clin Endocrinol Metab83157399589658
- SpruitMAGosselinkRTroostersT2003Muscle force during an acute exacerbation in hospitalised patients with COPD and its relationship with CXCL8 and IGF-IThorax58752612947130
- SpruitMATroostersTTrappenburgJC2004Exercise training during rehabilitation of patients with COPD: a current perspectivePatient Educ Couns52243814998593
- StewartCENewcombPVHollyJM2004Multifaceted roles of TNF-alpha in myoblast destruction: a multitude of signal transduction pathwaysJ Cell Physiol1982374714603526
- StienenGJKiersJLBottinelliR1996Myofibrillar ATPase activity in skinned human skeletal muscle fibres: fibre type and temperature dependenceJ Physiol493(Pt 2):2993078782097
- TisdaleMJ2005Molecular pathways leading to cancer cachexiaPhysiology (Bethesda)20340816174873
- TzengSFKahnMLivaS1999Tumor necrosis factor-alpha regulation of the Id gene family in astrocytes and microglia during CNS inflammatory injuryGlia261395210384879
- Van’t HulAHarlaarJGosselinkR2004Quadriceps muscle endurance in patients with chronic obstructive pulmonary diseaseMuscle Nerve292677414755493
- van HelvoortHAHeijdraYFDekhuijzenPN2006Systemic immunological response to exercise in patients with chronic obstructive pulmonary disease: what does it mean?Respiration732556416432297
- van HelvoortHAHeijdraYFHeunksLM2006Supplemental oxygen prevents exercise-induced oxidative stress in muscle-wasted patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1731122916514109
- Van HelvoortHAHeijdraYFThijsHM2006Exercise-induced systemic effects in muscle-wasted patients with COPDMed Sci Sports Exerc3815435216960513
- Van VlietMSpruitMAVerledenG2005Hypogonadism, quadriceps weakness, and exercise intolerance in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med17211051116100014
- VassilakopoulosTRoussosCZakynthinosS2004The immune response to resistive breathingEur Respir J2410334315572550
- VogiatzisITerzisGNanasS2005Skeletal muscle adaptations to interval training in patients with advanced COPDChest12838384516354852
- WelleSTottermanSThorntonC1996Effect of age on muscle hypertrophy induced by resistance trainingJ Gerontol A Biol Sci Med Sci51M27058914498
- WesterterpKRKayserB2006Body mass regulation at altitudeEur J Gastroenterol Hepatol181316357611
- WhittomFJobinJSimardPM1998Histochemical and morphological characteristics of the vastus lateralis muscle in patients with chronic obstructive pulmonary diseaseMed Sci Sports Exerc301467749789845
- WilcoxPGWakaiYWalleyKR1994Tumor necrosis factor alpha decreases in vivo diaphragm contractility in dogsAm J Respir Crit Care Med1501368737952566
- WüstRCIMorseCIJonesDA2006The effects of smoking on contractile properties and fatigue resistance of human quadriceps muscleProc Physiol Soc1PC24
- YokotaYMoriS2002Role of Id family proteins in growth controlJ Cell Physiol19021811807807
- YquelRJTessonneauFPoirierM2006Peak anaerobic power in patients with COPD: gender related differencesEur J Appl Physiol973071516770466
- ZhangYMarcillatOGiuliviC1990The oxidative inactivation of mitochondrial electron transport chain components and ATPaseJ Biol Chem2651633062168888
- ZhuXHeunksLMVersteegEM2005Hypoxia-induced dysfunction of rat diaphragm: role of peroxynitriteAm J Physiol Lung Cell Mol Physiol288L162615361360
- ZielinskiJ1999Effects of long-term oxygen therapy in patients with chronic obstructive pulmonary diseaseCurr Opin Pulm Med581710813256