Abstract
While tobacco smoking is the main risk factor for chronic obstructive pulmonary disease (COPD) only a fraction of smokers go on to develop the disease. We investigated the relationship between the insertion (I) – deletion (D) polymorphisms in the Angiotensin converting enzyme (ACE) gene and the risk of developing COPD in smokers by determining the distribution of the ACE genotypes (DD, ID and II) in 151 life-long male smokers. 74 of the smokers had developed COPD (62 ± 2 years; FEV1 44 ± 6 % reference) whereas the rest retained normal lung function (56 ± 2 yrs; FEV1 95 ± 3 % reference). In addition, we genotyped 159 males recruited randomly from the general population. The prevalence of the DD genotype was highest (p = 0.01) in the smokers that developed COPD and its presence was associated with a 2-fold increase in the risk for COPD (OR 2.2; IC95% 1.1 to 5.5). Surprisingly, the 151 individuals in the smoking population did not demonstrate Hardy-Weinberg equilibrium unlike the 159 recruited from the general population. Our results suggest that ACE polymorphisms are associated with both the smoking history of an individual and their risk of developing COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) is now the sixth largest cause of death in the world and it’s prevalence is expected to increase in the future (CitationMurray and Lopez 1997). Tobacco smoking is the main risk factor for COPD but despite this only a fraction of smokers go on to develop the disease (CitationBarnes 2000). While it has been suggested that susceptibility to tobacco smoke may reflect a genetic component of the disease (CitationBarnes 1999) the only genetic factor identified so far in predisposing individuals to COPD is the α1-antitrypsin deficiency (CitationSilverman and Speizer 1996).
The Angiotensin-converting enzyme (ACE) is a zinc metallo-peptidase that is highly expressed in lungs, where it degrades bradykinin and catalyses the formation of the Angiotensin II (AII); a powerful vasoconstrictor, inflammatory modulator and cellular growth factor (CitationWoods et al 2000). ACE activity is increased in COPD (CitationBrice et al 1995; CitationUcar et al 1997) but it remains to be established whether this is a cause or consequence of the disease process.
The ACE gene is located in chromosome 17q23 and shows a functional polymorphism consisting of an insertion (I) or deletion (D) of a 287bp fragment that determines three genotypes (DD, ID and II) (CitationRigat et al 1992) where the presence of the D allele is associated with higher ACE activity (CitationRigat et al 1990; CitationBusjahn et al 1998; CitationChagani et al 1999; CitationMontgomery et al 1999). Given that previous studies show increased ACE activity in COPD patients (CitationBrice et al 1995; CitationUcar et al 1997) the aim of the present study was to test the hypothesis that the DD genotype is more prevalent in smokers that develop COPD.
Methods
Population and ethics
We genotyped 151 male life-long smokers, 74 of whom had developed moderate-severe COPD (FEV1 44 ± 6 %reference) and the rest retaining normal lung function (FEV1 95 ± 3 % reference). All COPD patients were in a stable phase of the disease (not having had an exacerbation for, at least, three months preceding their inclusion to the study. We exclude subjects with other chronic lung diseases (asthma, bronchiectasis, lung cancer and interstitial lung diseases) and/or chronic extrapulmonary disorders such as, diabetes, arterial hypertension, cancer, immune diseases and cardiac, hepatic or renal failure. In addition, we genotyped 159 healthy males recruited randomly from the general population. All individuals were from the Balearic Islands and belonged to the same Caucasian Mediterranean ethnic background. Participants were not relatives and all gave informed consent to the study that was approved by the ethical review board of the Hospital Universitari Son Dureta.
Lung function
Forced spirometry (GS, Warren E Collins, Braintree, MA, USA) was determined according to international guidelines (CitationSanchis et al 1989) and reference values were those for a Mediterranean population (CitationRoca et al 1986).
ACE genotyping
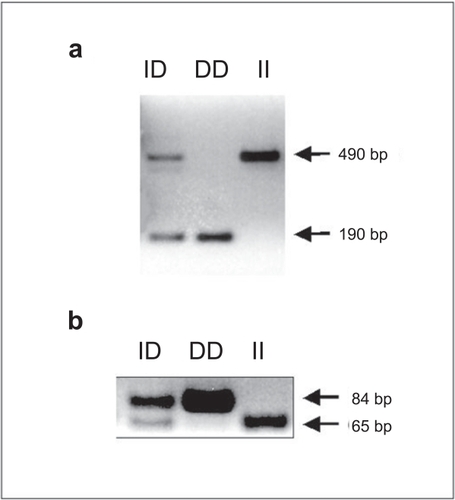
Genomic DNA was extracted from venous blood using standard methods (Promega, Madison, WI, USA) and stored at −80ºC for subsequent analysis. The ACE genotype was determined using the polymerase chain reaction (PCR).Since the D allele can be more effectively amplified than the longer I allele, increasing the potential for ID individuals to be genotyped as DD (CitationUeda et al 1996), we used two different genotyping methods. Initially, the genotype was determined using the method of CitationRigat et al (1992) where the PCR products were a 190 bp (allele D) and a 490 bp (allele I) (Figure , Panel a). Subsequently, the genotype was confirmed with the method described by CitationEvans et al (1994)where the amplified products were a 84 bp (allele D) and a 65 bp (allele I) (Figure , Panel b).
Figure 1 Determination of DD, ID, and II genotypes by PCR. Panel a shows a representative experiment using the method described by CitationRigat et al (1992). PCR products are a 190 bp fragment (D genotype) and a 490 bp fragment (I genotype). Heterozygotes (ID) have both the 190 bp and 490 bp fragments. Panel b shows a representative experiment using the method described by CitationEvans et al (1994). PCR products are a 84 bp (D genotype) and a 65 bp (I genotype). Heterozygotes (ID) have both the 84 bp and 65 bp fragments.
Statistical analysis
Results are shown for continuous variables as mean and its standard error, and for discrete variables as percentages. Quantitative variables were compared between groups using ANOVA. Genotype distributions were compared between smokers with and without COPD using Pearson′s Chi-Square test, from which the odds ratio (OR) and the 95% confidence interval (95% CI) were calculated. Genotype frequencies were compared with values predicted by Hardy-Weinberg equilibrium equation (p2 + 2pq + q2 = 1) by the Chi-Square test, where p is frequency of dominant allele D and q is frequency of recessive allele d. A p value of less than 0.05 was considered significant.
Results
Age, smoking exposure and body mass index were similar in smokers with and without COPD (Table ) and, by definition, the smokers with COPD showed characteristics of airflow obstruction (Table ).
Table 1 Clinical and functional data of the two life-long smoker groups
The distribution of the ACE genotypes in smokers that developed COPD was significantly different (p = 0.023) from those with normal lung function (Table ). As hypothesized, the DD genotype was found to be more prevalent in patients with COPD (36.5% vs. 16.9%, p = 0.01) and the presence of the DD genotype in a smoker was calculated to give a 2-fold increase in the risk of developing COPD (OR 2.2; IC95% 1.1 to 5.5).
Table 2 Distribution of the ACE genotypes in the different groups studied
Surprisingly, we found Hardy-Weinberg disequilibrium in the smoking population (n = 151, p = 0.001). Deviations from this equilibrium can be caused by either genotyping errors and/or stratifications within the population being studied (CitationXu et al 2002) and we excluded the first possibility by using two different genotyping techniques. To address the second possibility, we further genotyped 159 randomly recruited individuals from the same population as our smokers and found their distribution of ACE genotypes to be in Hardy Weinberg equilibrium (Table ). This observation further supports the validity of our genotyping techniques but more importantly, suggests that smoking may be associated with the ACE genotype. In fact, an excess of the heterozygote (ID) was found in smokers (p = 0.05) irrespective of whether they had developed COPD (Table ).
Table 3 Allele/Genotype frequencies and test of Hardy-Weinberg (HW) equilibrium
Discussion
This study shows that the DD ACE genotype is associated with an increased risk of a smoker to developing COPD and, unexpectedly, that the ACE heterozygote is more prevalent in individuals that smoke.
ACE and COPD
Previous studies have shown an association between ACE genotype and several phenotypic characteristics of COPD, for example, the development of pulmonary vascular remodelling, right ventricular failure and skeletal muscle strength (Citationvan Suylen et al 1999; CitationKanazawa et al 2000; CitationHopkinson et al 2004). To our knowledge, however, this is the first study to show that the presence of the DD genotype increases the risk of a smoker developing chronic airflow obstruction. Plasma ACE activity is increased in COPD (CitationBrice et al 1995; CitationUcar et al 1997) and while we did not measure it here the D allele is associated with higher plasma ACE activity (CitationRigat et al 1990; CitationBusjahn et al 1998; CitationChagani et al 1999; CitationMontgomery et al 1999). The high prevalence of the DD genotype observed in the smokers that developed COPD is therefore consistent with increased plasma ACE activity in these patients (CitationBrice et al 1995; CitationUcar et al 1997). More importantly, our results suggest that the increased ACE activity reported in COPD (CitationBrice et al 1995; CitationUcar et al 1997) can be related to the genotype and is not a consequence of the disease (leading to translational or post-translational modification of the protein).
Our study was not designed to investigate the potential mechanisms linking the ACE genotype to the pathogenesis of COPD. However, several possibilities can be conceived to explain a higher risk of COPD among smokers who carry the DD genotype. As discussed earlier, the presence of the D allele will increase ACE activity (CitationRigat et al 1990; CitationBusjahn et al 1998; CitationChagani et al 1999; CitationMontgomery et al 1999) and, in turn, cause higher Angiotensin II (AII) levels. Increased AII levels will contribute to the inflammatory response that characterizes COPD (CitationBarnes 1999, Citation2000) by (1) up-regulating expression of the endothelial adhesion molecules ICAM-1 and VCAM-1 (CitationGuba et al 2000) and facilitating neutrophil retention within the lungs (CitationSelby et al 1991; CitationKeatings and Barnes 1997), (2) activating the neutrophil respiratory burst and contributing to oxidative stress (CitationZafari et al 1998), (3) enhancing airway hyper-responsiveness (CitationMyou et al 2000), (4) regulating vascular smooth-muscle cell growth and migration (CitationHatakeyama et al 1994), that may contribute to pulmonary hypertension in COPD and (5) modulating tissue repair and fibrosis (CitationWoods et al 2000).
ACE and smoking
We found that our smoking population was in Hardy-Weinberg disequilibrium and are confident that this did not result from genotyping errors or population stratification due to the reasons given previously.
There are a number of assumptions required to evaluate whether the genotype distribution within a population fulfils the requirements for Hardy – Weinberg equilibrium, namely that the gene is located in a somatic chromosome, the organism has discrete generations, there is random mating within a single population and an infinite population size (or sufficiently large so as to minimize the effect of genetic drift). Finally, there should be experiences of no selection, no mutation, and no migration (gene flow). We believe all apply in the populations studied here; however, replication by different investigators in different populations would be welcome to identify any causal link between the ACE gene and COPD. Departure from Hardy-Weinberg equilibrium also occurs when the selection criteria are based on disease-susceptibility genotypes rather than on independently selected alleles. In fact, marker-disease association can be detected by testing for Hardy-Weinberg disequilibrium at a marker locus (CitationNielsen et al 1998). Therefore, to explain the excess ID genotype observed among smokers (Table ), we propose the existence of linkage disequilibrium between the trait of being a life-long smoker and the ACE locus. This interpretation is supported by a recent study showing that the ACE ID polymorphism influences the amount of cigarettes smoked per week (CitationHubacek et al 2001).
Molecular heterosis occurs when heterozygous subjects for a given polymorphism show a significantly greater or lesser effect for a trait than in homozygous subjects for either allele. We found molecular heterosis in smokers, where a significant proportion of individuals carried the ID genotype (Table ). It is worth noting here that molecular heterosis (and Hardy-Weinberg disequilibrium) has also been found in studies of the dopamine D2 receptor gene (DRD2) in smoking males (CitationLee et al 2002; CitationLee 2003).
The biological basis of nicotine dependence is not fully understood, but it is well established that nicotine stimulates the central dopamine reward pathway (CitationPontieri et al 1996; CitationPontieri et al 1998; CitationAlcantara et al 2003). Like nicotine (CitationZhou et al 2001), Angiotensin II also modulates dopamine release in the striatum (CitationJenkins et al 1997; CitationZhou et al 2001) and it is likely that the ACE gene is involved in the regulation of the dopamine reward pathway. This provides a mechanistic basis for the interpretation of our observation of Hardy-Weinberg disequilibrium and molecular heterosis in smokers.
Conclusion
Our study shows, first, that the presence of the DD ACE genotype increases the risk of smokers going on to develop COPD and, second, that smoking is linked to a Hardy-Weinberg disequilibrium in the allelic distribution of the ACE gene consistent with it playing a role in smoking addiction.
Acknowledgements
Supported, in part, by FIS 00/0437, FIS 02/3008and RTIC C03/11 (Red Respira); CICYT PB98-1084, SEPAR and ABEMAR.
Notes
XB and NGM equally contributed to this work.
References
- AlcantaraAAChenVHerringBE2003Localization of dopamine D2 receptors on cholinergic interneurons of the dorsal striatum and nucleus accumbens of the ratBrain Res98622912965226
- BarnesPJ1999Molecular genetics of chronic obstructive pulmonary diseaseThorax542455210325902
- BarnesPJ2000Chronic obstructive pulmonary diseaseN Engl J Med3432688010911009
- BriceEAFriedlanderWBatemanED1995Serum angiotensin-converting enzyme activity, concentration, and specific activity in granulomatous interstitial lung disease, tuberculosis, and COPDChest107706107874941
- BusjahnAVossAKnoblauchH1998Angiotensin-converting enzyme and angiotensinogen gene polymorphisms and heart rate variability in twinsAm J Cardiol81755609527087
- CelliBRMacNeeW2004Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paperEur Respir J239324615219010
- ChaganiTParePDZhuS1999Prevalence of tumor necrosis factor-alpha and angiotensin converting enzyme polymorphisms in mild/moderate and fatal/near-fatal asthmaAm J Respir Crit Care Med1602788210390412
- EvansAEPoirierOKeeF1994Polymorphisms of the angiotensin-converting-enzyme gene in subjects who die from coronary heart diseaseQ J Med87211148208911
- GubaMSteinbauerMBuchnerM2000Differential effects of short-term ace- and AT1-receptor inhibition on postischemic injury and leukocyte adherence in vivo and in vitroShock13190610718375
- HatakeyamaHMiyamoriIFujitaT1994Vascular aldosterone. Biosynthesis and a link to angiotensin II-induced hypertrophy of vascular smooth muscle cellsJ Biol Chem26924316207929089
- HopkinsonNSNickolAHPayneJ2004Angiotensin converting enzyme genotype and strength in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med170395915117739
- HubacekJAPithaJSkodovaZ2001Angiotensin converting enzyme gene–a candidate gene for addiction to smoking?Atherosclerosis159237811689227
- JenkinsTAMendelsohnFAChaiSY1997Angiotensin-converting enzyme modulates dopamine turnover in the striatumJ Neurochem681304119048778
- KanazawaHOkamotoTHirataK2000Deletion polymorphisms in the angiotensin converting enzyme gene are associated with pulmonary hypertension evoked by exercise challenge in patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1621235811029323
- KeatingsVMBarnesPJ1997Granulocyte activation markers in induced sputum: comparison between chronic obstructive pulmonary disease, asthma, and normal subjectsAm J Respir Crit Care Med155449539032177
- LeeHS2003Gender-specific molecular heterosis and association studies: dopamine D2 receptor gene and smokingAm J Med Genet B Neuropsychiatr Genet11855912627467
- LeeHSKimSHLee2002Gender-specific molecular heterosis of dopamine D2 receptor gene (DRD2) for smoking in schizophreniaAm J Med Genet114593712210271
- MontgomeryHClarksonPBarnardM1999Angiotensin-converting-enzyme gene insertion/deletion polymorphism and response to physical trainingLancet353541510028982
- MurrayCJLopezAD1997Mortality by cause for eight regions of the world: Global Burden of Disease StudyLancet3491269769142060
- MyouSFujimuraMKurashimaK2000Type 1 angiotensin II receptor antagonism reduces antigen-induced airway reactionsAm J Respir Crit Care Med16245910903218
- NielsenDMEhmMGWeirBS1998Detecting marker-disease association by testing for Hardy-Weinberg disequilibrium at a marker locusAm J Hum Genet631531409867708
- PontieriFEPassarelliFCaloL1998Functional correlates of nicotine administration: similarity with drugs of abuseJ Mol Med761932019535552
- PontieriFETandaGOrziF1996Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugsNature38225578717040
- RigatBHubertCAlhenc-GelasF1990An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levelsJ Clin Invest86134361976655
- RigatBHubertCCorvolP1992PCR detection of the insertion/deletion polymorphism of the human angiotensin converting enzyme gene (DCP1) (dipeptidyl carboxypeptidase 1)Nucleic Acids Res2014331313972
- RocaJSanchisJAgusti-VidalA1986Spirometric reference values from a Mediterranean populationBull Eur Physiopathol Respir22217243730638
- SanchisJCasanPCastilloJ1989Normativa para la práctica de la espirometría forzadaArch Bronconeumol2513242
- SelbyCDrostELannanS1991Neutrophil retention in the lungs of patients with chronic obstructive pulmonary diseaseAm Rev Respir Dis1431359642048825
- SilvermanEKSpeizerFE1996Risk factors for the development of chronic obstructive pulmonary diseaseMed Clin North Am80501228637301
- UcarGYildirimZAtaolE1997Serum angiotensin converting enzyme activity in pulmonary diseases: correlation with lung function parametersLife Sci611075829307053
- UedaSHeeleyRPLeesKR1996Mistyping of the human angiotensin-converting enzyme gene polymorphism: frequency, causes and possible methods to avoid errors in typingJ Mol Endocrinol1727308863184
- van SuylenRJWoutersEFPenningsHJ1999The DD genotype of the angiotensin converting enzyme gene is negatively associated with right ventricular hypertrophy in male patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1591791510351920
- WoodsDRHumphriesSEMontgomeryHE2000The ACE I/D polymorphism and human physical performanceTrends Endocrinol Metab114162011091119
- XuJTurnerALittleJ2002Positive results in association studies are associated with departure from Hardy-Weinberg equilibrium: hint for genotyping error?Hum Genet111573412516594
- ZafariAMUshio-FukaiMAkersM1998Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophyHypertension32488959740615
- ZhouFMLiangYDaniJA2001Endogenous nicotinic cholinergic activity regulates dopamine release in the striatumNat Neurosci41224911713470