Abstract
Chronic obstructive pulmonary disease (COPD) is a complex disease with multifactorial background, based on the interaction of environmental and genetic factors. Environmental factors are clearly related to the development of the disease. However, family and twin studies suggested genetics factors to be one of the important determinants for the development of COPD. Different approaches have been used to identify genes of interest. Genomewide linkage analysis found areas of interest on different chromosomes, with some genes located in this regions being identified and replicated as susceptibility genes. Numerous of candidate genes that could be linked to disease pathogenesis have been implicated in COPD genetics. However, the candidate gene approach is often limited by inconsistent results in other study populations. Recently, a combination of different methods is used giving more evidence for some candidate genes, including TGFß-1, Surfactant, SERPINE2 and microsomal epoxide hydrolase. In the future ongoing exact phenotype definition, combination of several approaches, genome-wide association studies and animal model genetics will lead to new insights into the genetics of COPD, with epigenetic factors needs to be further investigated and considered in concert with genetic findings.
Introduction
Chronic obstructive pulmonary disease (COPD) is a major health problem as it causes increasing mortality and morbidity in all industrial countries (CitationPauwels et al 2001; CitationHurd 2005). Progressive irreversible airflow limitation is the central pathogenetic aspect in COPD. However, the disease is not only determined by pulmonary function, but by airway tissue inflammation, airway remodeling and systemic consequences.
The most important risk factor in the etiopathogenesis of COPD is cigarette smoking. Nevertheless, only 10%–20% of all heavy cigarette smokers develop COPD (CitationFletcher 1976; CitationRedline et al 1987; CitationSherrill 1990; CitationGlobal Initiative for Chronic Obstuctive Lung Disease 2001; CitationMannino 2002, Citation2003; CitationMannino et al 2003). There is strong evidence for a relationship between decline in lung function and smoking behavior. However, smoking account only for about 15% of lung function variability (CitationBeck et al 1981). These observations demonstrate that next to smoking other factors seems to be of importance for the development of the disease. Epidemiological data suggest genetics to be one of those factors, as COPD is known to aggregate in families (CitationLarson et al 1970; CitationTager et al 1976; CitationLebowitz et al 1984), with a stronger correlation between parents and children or siblings than between spouses (CitationHiggins and Keller 1975; CitationTager et al 1976; CitationKauffmann et al 1989). Twin (CitationRedline 1987, Citation1990) and segregation studies (CitationGivelber 1998) provide evidence that genetic predisposition plays a role in COPD (CitationHubert et al 1982; CitationAstemborski et al 1985; CitationRedline et al 1987; CitationRybicki et al 1990; CitationChen et al 1996; CitationGivelber et al 1998) and had indicated that the genetic background of COPD is composed of several genes with small effects, rather than a single major gene. Despite the general fact that genetic studies in COPD are difficult to conduct due to the variating phenotypes subsume under the disease definition, there are different possible approaches to analyze genetic factors of importance. Basic genetic studies include family and twin studies, as well as segregation studies. In linkage and positional cloning studies susceptibility genes could be identified on the basis of their chromosomal position. The genetic bases of COPD might also be investigated, similar to other diseases with complex and multifactorial etiology, by the analysis of candidate genes – genes that are postulated to play an important role in disease pathogenesis. This review will focus on the candidate gene approach after giving an short overview about the results of linkage studies.
Linkage studies
The genetic bases of lung function and/or COPD had been investigated using linkage or positional cloning studies. Linkage studies investigate haplotypes, or short segments of the genome, conserved between generations by virtue of their size. If a haplotype is found that is passed down along with a disease through a family, it could be speculated that there is a gene within or close to that may have an effect on the disease. To perform linkage analysis phenotypic data and DNA from affected families are needed at least from two generations. From each family all members typed for genetic markers throughout the whole genome, following by determination whether any of these markers are inherited with the disease more often than predicted by chance. In case of a positive correlation the disease is “linked” to that marker on a distinct chromosome. This method provides the possibility to identify novel genes, because linkage of the disease to a genetic marker depends only on the close proximity of that marker with the susceptibility gene, independent of a potential functional correlation. But these studies are not easy to conduct in COPD since the disease has a late onset in life and family members that are needed for analysis may no longer be alive.
Genomewide linkage analysis exploring genetic linkage to lung function was performed in several studies. The genetic determinants of FEV1, FVC and FEV1/FVC ratio had been studied in healthy subjects and patients with COPD. Two population- and one disease-based linkage analysis found strongest evidence of linkage to FEV1/FVC ratio (chromosome 4 [logarithm of the odds favoring genetic linkage (LOD) 3,5], chromosome 2q [LOD 4,12], and chromosome 2 [LOD 4,12]) (CitationJoost et al 2002; CitationSilverman et al 2002; CitationWilk et al 2003; CitationDeMeo et al 2004). In healthy young adults of the Framingham Heart cohort a genome-wide scan of 330 families with over 1500 members found that loci strongly influencing FEV1 occurred on chromosome 6 [LOD score 2,4] and for FVC on chromosome 21 [LOD 2,6] (CitationJoost et al 2002). In another study including more than 2100 healthy subjects from 391 families who participated in the National Heart, Lung and Blood Institute (NHLBI) Family Heart Study FEV1 and FVC were suggestively linked to regions of chromosome 18 [LOD 2,4]/ [LOD 1.5 / 2,4] (CitationWilk et al 2003). Interestingly, in a genomewide screen for pulmonary function in families of patients with asthma significant evidence for linkage of pre- and postbronchodilator FEV1%VC was obtained for chromosome 2q (2q32, LOD 4,9/6.03) (Postma 2005). The Boston Early-onset (BEO) studies families with subjects suffering from severe early-onset COPD. A genome-wide scan of 585 family members participating in this study showed the highest LOD score for FEV1 was 1.53 on chromosome 12 and 2.05 for FVC on chromosome 1 (CitationSilverman et al 2002). If only smoking subjects were included, evidence for linkage to FEV1/FVC ratio was increased on chromosome 2 (LOD 4.13) and for FEV1 on chromosome 12p (LOD 3,26). Evidence for linkage of chronic bronchitis or airway obstruction was shown for chromosome 22 (LOD 2.08)/12p (LOD 2,13), respectively. summarizes the main results of these linkage studies.
Table 1 Linkage analysis: genomewide scans
Instance areas of interest from linkage studies included SERPINE2 and IL-8 as potential candidate genes (both on chromosome 2), as well as microsomal GST1 and TGFß genes (chromosome 12 and 19) in terms of the severity of pulmonary function in COPD. However, there is no direct evidence for the significance of linkage studies for COPD pathogenesis.
Candidate genes – association studies
Variations, or polymorphism, within genes are classified by means of a change in DNA alone or effect on the protein it codes for. Sigle nucleotide polymorphism consists of a sigle base change are the most common in the human genome. They could either be functional or not. Candidate gene analysis compare the frequency of gene variations or polymorphism in groups with the given disease and control individuals. In diseases with complex and multifactorial etiology, such as COPD, the use of candidate genes may help to identify genetic factors of potential importance. In the candidate gene approach genes are tested directly for their involvement in disease process. Genes that are selected are those that could be postulated to be functional related to the disease. The candidate gene approach is limited on the one hand by the fact that only known genes can be examined, controls and patients are difficult to match and the need of a very exact phenotype definition. On the other hand COPD is a multifactorial disease that is influenced by genetic as well as environmental factors resulting in a complex genotype-environment interaction. However, a reasonable large number of successful association studies had analyzed candidate genes selected on the base of COPD pathophysiology.
COPD is characterized by a progressive decline in lung function due to increasing irreversible airway obstruction. This results from ongoing chronic airway inflammation and loss of lung elastic recoil due to parenchymal destruction. Many inflammatory cells, mediators, enzymes and maybe infectious agents are involved. The genes that are implicated in the pathogenesis of COPD are involved in proteolysis and antiproteolysis (CitationShapiro and Senior 1999; CitationStockley 2001), oxidants and anti-oxidants (CitationMacNee 2000), inflammation (CitationO’Donnell et al 2006), metabolism of toxic substances, airway hyperresponsiveness, mucus homöostasis and host defense mechanisms (CitationO’Donnell et al 2006). show a graphical depiction of cells and mediator systems involved in COPD pathogenesis.
Figure 1 Graphical depiction of cells and mediator systems involved in COPD pathogenesis. Oxidative stress, inflammation and proteolysis and their opponents are the important components influencing disease pathogenesis.
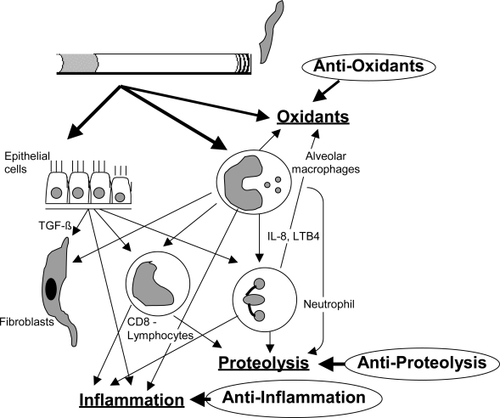
Reactive oxygen species (ROS) are induced as a direct result of cigarette smoke inhalation and/or increased production by activated inflammatory cells and activation of the xantine oxidase pathway. These oxidants may inhibit by anti-oxidative enzymes, such as α1-antitrypsin. Parenchymal damage may be due to several mechanisms, including oxidative stress, inflammation and apoptosis of vascular endothelial cells.
Numerous candidate genes have been studied in COPD case-control association studies and several candidate genes have been reported to be significantly associated with COPD phenotypes (see ). However, the evidence supporting these associations have been inconsistent in most of the cases due to failure in replication by other studies. The inconsistent replication might be due to the study populations (differences in phenotype definition, control subjects or COPD subtypes and/or racial differences), population stratification, multiple testing, random error or small case numbers. However, combination of different study methods, as performed in recent studies, seems to provide more replicatable results. This review will focus on some candidate genes and recent data.
Proteolysis and antiproteolysis
α1-antitrypsin
Severe alpha1-antitrypsin (AAT) deficiency is the one proven genetic risk factor for COPD. AAT is an acute phase protein that provides the major defense against neutrophil elastase. There are four main variants of AAT, classified by their speed of movement in gel electrophoresis: F = fast, M = medium, S = slow and Z = very slow (CitationFagerhol and Laurell 1970). In 1963 Laurell and Eriksson demonstrated that individuals with extremely low levels of AAT had an increased prevalence of emphysema. In the following time it was shown that AAT deficiency was usually associated with the Z isoform of AAT (CitationKueppers et al 1964; CitationEriksson 1965). The AAT variants are inherited in a co-dominant fashion, with the PiM allele being the wild-type and most prevalent. Homozygosity of the Z variant (Glu342Lys) results in severe AAT deficiency due to a phenotype with reduced plasma AAT levels to 10% compared to normal AAT levels (for review see CitationNeedham and Stockley (2004)). However, the risk for COPD among PiMZ heterozygous at the α1-antitrypsin locus remains controversial. A recent meta-analysis compared COPD cases versus control subjects and found an increased risk for COPD among PiMZ subjects, whereas population based studies reported usually similar pulmonary function values (FEV1) in subjects with PiMZ and PiM (CitationHersh et al 2004). There is significant genotype-environment interaction between the Pi Type and pack years of smoking (CitationSandford et al 1999). However, this interaction could not explain the complete phenotype variations (CitationSilverman et al 1992), suggesting gene-gene interaction and/or confounding other factors next to AAT deficiency to be of importance.
Matrix metalloproteinases (MMP’s) and tissue inhibitor of MMP’s (TIMP)
Matrix Metalloproteinases comprise a family of more than 20 related proteolytic enzymes that play an important role in tissue remodeling and repair in development and inflammation. These enzymes degrade collagen, inactivate AAT and activate tumor necrosis factor-α (TNF-α). A functional SNP of the MMP9 gene promoter region had been studied in COPD and emphysema and had been associated in both, Chinese and Japanese populations (CitationMinematsu et al 2001; CitationZhou et al 2004) However, this polymorphim was not associated with COPD in the Lung health Study (CitationJoos et al 2002).
An insertion in the promoter region of MMP1 (G → GG, − 1607) that increases transcription of MMP1 by creating an additional transcription factor binding site, was negatively associated with rapid decline of FEV1 (CitationJoos et al 2002), as well as a haplotype containing the MMP1 together with an MMP12 SNP (Asn357Ser) (CitationJoos et al 2002).
There have been four tissue inhibitors of matrix metalloproteinases (TIMP) described, that inhibit active forms of MMP’s, with different affinities to each MMP. MMP2 is inhibited by TIMP-2 and this system seems to play an important role in emphysema (CitationOhnishi et al 1998). Two SNP’s of the TIMP2 gene have been associated with COPD in a Japanese population (CitationHirano et al 2001). The authors speculate that a linked gene may be associated with genetic susceptibility and probably not TIMP itself.
α1-antichymotrypsin (SERPINA 3)
α1-antichymotrypsin (SERPINA3) is capable of inhibiting cathepsin G and mast cell chymase in a reversible fashion. In the SERPINA3 gene two functional SNPs have been associated with low α1-antichymotrypsin levels and COPD (characterized by airway resistance) in a Swedish population (CitationPoller et al 1990, Citation1993). The association was not replicated in a Japanese Population. However, a third gene variation (non-synonymous mutation with affects to the signal peptide region) was found to be increased in the COPD group. Non of these polymorphims showed differences in an Italian study of patients with airflow obstruction, though next to COPD, the patient group also included subjects with bronchiectasis (CitationBenetazzo et al 1999).
Oxidative stress and antioxidants
An imbalance between oxidative noxes and antioxidants resulting in oxidative stress is postulated to be one of the important mechanisms in the development of COPD. Airway epithelium and lung matrix could be damaged by oxidative stress and enhance lung inflammation with up-regulation of pro-inflammatory cytokines (CitationMacNee 2000). Cigarette smoke is a major source of free radicals, nitric oxide and other oxidants. Moreover, during inflammatory processes radicals are released by leucocytes, that are known to be increased in the lung of patients with COPD. The lung is protected by antioxidative enzymes, including gluthathione-S-transferase, superoxide dismutase and catalase (CitationYoung et al 2006). Therefore, functional genetic variants may be of importance by altering antioxidative capacity of the lung.
Glutathione-S-transferase (GST)
The family of glutathione-S-transferase comprises enzymes that can detoxify some noxes of tobacco smoke. There are functional polymorphisms, including one SNP in GSTP1 (A313G → Ile105Val) and three alleles of GSTM1 each affecting enzyme activity.
The 105Ile variant of the GSTP1 gene was associated with airflow obstruction in a Japanese population and with rapid decline of FEV1 as well as low baseline lung function in the Lung CitationHealth Study (He et al 2002; He 2004).
The GSTM1 alleles include one null allele, having no detectable GSTM1 activity in case of homozygosity. This genotype had been previously linked to emphysema and COPD (CitationBaranova et al 1997; CitationHarrison et al 1997). However, there are negative studies concerning airflow obstruction and rapid decline in lung function (CitationYim et al 2000; CitationHe et al 2002).
Superoxide dismutase (SOD)
Superoxide dismutase belongs to three superoxides that can scavenger reactive oxygen species (ROS). They are part of the protection system of the lung, particularly during inflammation. A functional SNP (C760G) in the SOD3 gene was found to have a protective effect in Caucasian COPD populations (CitationJuul et al 2006; CitationYoung et al 2006), probably dependent on gene-smoking interaction, as the genetic variant of the SOD3 gene may confer protection from adverse affects of cigarette smoke.
Microsomal epoxide hydrolase
Microsomal epoxide hydrolase (mEH) is expressed in human bronchial epithelial cells and detoxify highly reactive intermediates present in cigarette smoke (CitationBartsch et al 1992). There are two functional polymorphisms that affect enzyme activity that account for a modest change in activity level. For individuals carrying both His variants (exon3: Tyr113His; exon4: His139Arg) a higher risk for development COPD and emphysema were demonstrated (CitationYoshikawa et al 2000). SNPs in the microsomal epoxide hydrolase (mEH) gene have been associated with severe COPD cases, emphysema and COPD phenotypes (CitationSmith and Harrison 1997; CitationKoyama and Geddes 1998; CitationYoshikawa et al 2000; CitationPark et al 2005), as well as with rapid decline in FEV1 in smokers (CitationSandford et al 2001; CitationHe et al 2002). However, the association failed to be replicated in a Corean population (CitationYim et al 2000).
Inflammation and inflammatory mediators
COPD is characterized by chronic airflow obstruction due to chronic inflammation based on an abnormal inflammatory response (CitationCelli and MacNee 2004). Different mediators and cells have been implicated in the pathogenesis of COPD, including TNFα, IL8 and TGF-ß. This extends beyond the lung to systemic manifestations, with circulating mediators and/or cytokines in patients serum (CitationSevenoaks and Stockley 2006). Cigarette smoke activates macrophages to release TNF-α, LTB4, IL-8 and other neutrophil chemotactic factors, as well as antiproteases. TNF-α promotes further IL-8 release from other cells in the respiratory tract by NF-κB mediated effects on gene transcription. This increases local neutrophilic inflammation, and hence the release of proteases. Epithelial cells also stimulate fibroblasts via TGFß, a key-player in the pathogenesis of fibrosis.
Tumor necrosis factor – alpha (TNF-α)
There is one SNP in the promoter region of the TNF-α gene, that is known to be functional by directly affecting gene regulation (CitationWilson et al 1997). Heterogeneity of study results demonstrate a relevance for the SNP – 308 in Asian but not in Caucasian populations, as in Taiwanese and Japanese collectives there is an increased prevalence in COPD compared to controls (CitationHuang et al 1997). Additionally, airflow obstruction without chronic bronchitis, and severity of emphysema had been associated with this polymorphism in Japanese subjects (CitationSakao et al 2001, Citation2002). However, these results could not be confirmed in Caucasian collectives (CitationSandford et al 2001; CitationHersh et al 2005; CitationSeifart et al 2005), maybe due to variation in genotype frequencies between races, or by linkage disequilibrium with HLA alleles, seen previously in the Caucasian population (CitationWilson et al 1993).
Transforming growth factor-beta (TGFß)
TGFß regulates extra-cellular matrix production, cell growth and differentiation, tissue repair and some immune responses (CitationBlobe et al 2000). Animal experiments suggests that disordered activation of TGFß relates to the pathogenesis of COPD.
Two recent studies, with new study designs using a combination of different methods, strongly suggests TGFß1 to be of importance for COPD phenotypes. On the basis of the linkage analysis in the Boston Early-Onset COPD study that revealed suggestive linkage between an area on chromosome 19q – containing the TGFß1 gene – and FEV1 (CitationCeledon et al 2004), TGFß was genotyped in subjects of the Boston Early-Onset COPD study. Out of five, three SNPs, one in the TGFß1 promotor region and two SNPs in the 3’ genomic region of TGFß1, had been significantly associated with pre- and post-bronchodilator FEV1. Again, among 304 cases from the National Emphysema Treatment Trail (NETT) and smoking controls, this association was replicated for two of the SNPs (CitationHersh et al 2006), who linked them both to subjective measures of dyspnoea, though not objective exercise capacity. This apparent discordance may be important when defining phenotypes within COPD. The latter two SNPs have an effect on TGFß1 levels, as the one in the promoter region C-509T enhances promoter function, thus increasing levels of TGFß (CitationGrainger et al 1999). While the second is a C -> T change at position 613, that leads to an amino acid substitution (Leu -> Pro) and higher production of TGFß1 (CitationSuthanthiran et al 2000). If both of these SNPs are implicated in COPD subjects, it suggests that TGFß may have a protective role.
Recently, the Pro allele was found to be less common in COPD subjects relative to resistant smokers (OR: 0.59, p = 0.01) and controls (OR: 0.62, p = 0.005) (CitationWood and Stockley 2006).
Vitamin D binding protein
Vitamin D binding protein is a precursor of macrophage activating factor (MAF) and increases the neutrophil chemotactic properties of different peptides. This function could be prevented by neutrophile elastase inhibitors pointing out to a potential relationship between the protease-antiprotease pathway and inflammation.
Two non-synonymous SNPs had been identified in the Vitamin D binding protein (GC2 and GC1S alleles), with the GC2 allele seemed to be protective in studies of Caucasian subjects (CitationHorne et al 1990; CitationSchellenberg et al 1998). The GC1S allele has not been shown to have a significant association with COPD.
The GC1F allele has been associated with an increased risk of developing airflow obstruction, emphysema and rapid decline of FEV1 in Japanese subjects. However, the results for homozygous individuals concerning an increased risk of developing COPD in Caucasian populations are conflicting (CitationHorne et al 1990; CitationSchellenberg et al 1998). Furthermore, the allele failed to be linked to rapid decline in lung function (CitationSandford et al 2001). The study heterogeneity might be due to difference in allele frequency between the racial groups.
Interleukin 13
Mice overexpressing IL-13 develop cathepsine and matrix metalloproteinase dependent emphysema (CitationZheng et al 2000).
A functional polymorphism in the promoter region (C-1055T) of the IL-13 gene leads to increased IL-13 production (Citationvan der Pouw Kraan et al 1999). The T allele had been demonstrated to be more common in COPD patients (Citationvan der Pouw Kraan et al 2002), but this association has not been replicated yet.
Surfactant and SERPINE2
Surfactant
Surfactant proteins are important for normal lung function and play a role in the innate host defense and control of inflammation in the lung. Genetic heterogeneity of the surfactant proteins may explain protein and mRNA content in the lung.
Surfactant protein B is important for the formation of the active surfactant surface film (CitationSchurch et al 1998) and is essential for normal lung function (CitationRobertson et al 1991). SP-B knock out mice show disruption of surfactant film and function and die from respiratory failure (CitationClark et al 1995, Citation1997). Furthermore, those mice are more susceptible to oxidative lung injury (CitationTokieda et al 1999).
A gene variation within intron 4 of the surfactant protein B (SP-B) gene had been associated with respiratory failure in COPD (CitationSeifart et al 2002). One of the known SNPs (B1580 C/T = Thr131Ile) in the SP-B gene was associated with COPD in the Boston Early-Onset study and was confirmed in a following family study (CitationHersh et al 2005). The same polymorphism, had been previously associated as a smoking dependent risk factor in a case-control study in a Mexican population (CitationGuo et al 2001). When gene-environment interaction was taking into account, the same polymorphism was associated with dyspnoea score and exercise capacity in the NETT cohort (CitationHersh et al 2006). Thus, surfactant protein B polymorphisms seemed to be of importance in the genetic background of COPD, probably especially in fact of gene-smoking or gene-environment interaction.
SERPINE 2
Prior linkage analysis in the Boston Early Onset COPD study demonstrated significant linkage of a region on chromosome 2q with COPD. Candidate gene selection in this region were done by integration of the linkage results with results from murine lung development and gene-expression studies of human COPD tissue (CitationDemeo et al 2006). This procedure identified SERPINE2 as a positional candidate susceptibility gene for COPD. Significant association of 18 SNPs in the SERPINE2 gene with COPD phenotypes were demonstrated in a family-based study, while five of these could be confirmed in a case control analysis with cases from the National Emphysema Treatment Trail.
SERPINE2 is known to be an inhibitor of thrombin, urokinase and plasmin. There are no studies investigating a separate different function of SERPINE2 in the lung so far. Nevertheless, SERPINE2 is postulated to be a COPD – susceptibility gene that is likely influenced by gene-by-smoking interaction (CitationDemeo et al 2006).
Recent study approach
There are recent studies combining several study methods analysing candidate gene to obtain more reliable data. This include the combination of family-based and case-control studies, emerging data from linkage analysis to identify possible candidate genes and including DNA microarray data (CitationCeledon et al 2004; CitationHersh et al 2005; CitationDemeo et al 2006; CitationHersh et al 2006a, Citation2006b).
CitationHersh et al (2005) genotyped selected candidate genes previously reported to be important in COPD in a family based study, followed by a case-control study. Only one polymorphism in the surfactant protein B gene (SFTPB Thr131Ile) and one in the heme oxygenase gene were replicated across both studies. A similar approach was used in 80 markers in 22 positional or biological plausible candidate genes with exact phenotype definition including COPD related phenotypes. The analysis revealed SNPs in the epoxide hydrolase, latent transforming growth factor ß binding protein-4 (LTBP4), TGF-ß1 and again SP-B to be significant associated with COPD related phenotypes (CitationHersh et al 2006).
Data obtained from previous linkage analysis were used in two recent studies. One integrated additional results from gene expression profiling (CitationDemeo et al 2006), and both were followed by association analysis in a family based samples and a case control study. One identified SERPINE2 to be a COPD-susceptibility gene, likely influenced by gene-smoking interaction, while the other associated TGFß1 gene variants with the pathogenesis of COPD among smokers (Celedon et al 2006).
Gene expression profiling
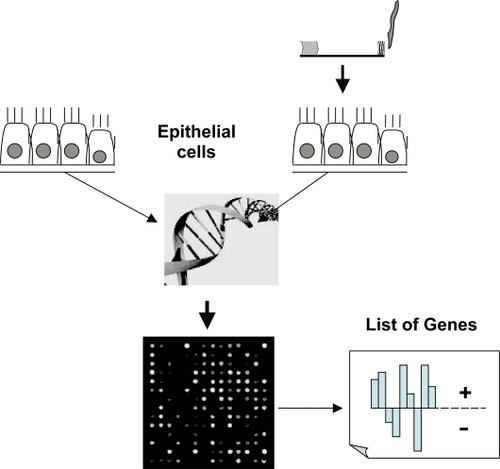
Gene expression profiling using cDNA microarrays may identify the expression of several thousand genes simultaneous. The method point a “photograph” of the current genetic activity in one tissue in comparison to another. shows graphically the principle of the method. Gene expression profiling using cDNA microarrays may provide the possibility to identify candidate genes involved in the pathogenesis of emphysema, though lung tissue represents an end-stage picture of gene expression. Linkage of the obtained gene expression profiles to chromosomal regions that were previously associated with COPD in linkage analyses will focus enclose potential candidate genes. cDNA microarrays are capable of profiling gene expression patterns of thousands of genes and/or gene clusters. It could be used to detect variations in transcriptional programs, including signal transduction and regulatory systems.
Figure 2 Shows graphically the principle of gene expression profiling using cDNA microarrays. The activity of up to 20000 genes could be simultaneous evaluated. Usually two tissues or cell populations are compared and the final result shows the different genetic activity of the sample of interest compared to a reference control.
One study using microarray technology in lung tissue from patients with severe emphysema and controls, with one group of patients that had been previously diagnosed as AAT-deficient, showed surprisingly only modest differences between individuals with normal lung function and patients with end-stage emphysema (CitationGolpon et al 2004). However, 150 genes were significantly different expressed between normal and emphysematous lung tissue samples. Expression of about 30–50 genes differ between emphysema, emphysema due to AAT-deficiency and controls, with a reduction in protein biosynthesis capacity as a clear characteristic of the lung gene expression pattern of patients with AAT deficiency-related emphysema. Significant heterogeneity within the individuals of each group emphasize the multifactorial nature of the disease. In another study, that compares lung tissue of smokers with normal to mild or severe emphysema 102 genes distinguished between the different types of smoking related lung tissue (CitationSpira et al 2004). A total of 76 genes were upregulated in the lungs of smokers with severe emphysema. These include extracellular matrix (ECM)-related genes, whereas many immune and cell signaling-related genes were downregulated in these lungs. Overrepresented gene categories comprises ECM constituents and stress-related genes with oxidoreductase, isomerase, and complement activity. The study revealed distinct molecular subclasses of severe emphysema with the body mass index being the only clinical variable that differed between the groups.
The future
There had been some progress in the last years in genetics of chronic obstructive pulmonary disease. However, there are many areas that require more investigation. Not at least, epigenetic needs have to be considered.
Future genetic studies will include additional linkage analysis with and without combination of gene expression profiling. Focusing on novel phenotypes, defined by molecular or clinical means, will be essential in identifying genetic determinants for COPD. Animal model genetics may help in identifying several pathways of importance and provide more insight in some aspects of pathogenesis.
Recently, analyses using the genomewide association analysis approach (CitationHirschhorn 2005; CitationHirschhorn and Daly 2005) – an aproach that involves genotyping of 300,000–5,000,000 SNPs across the whole genome – is expected to provide new insights into COPD pathogenesis, though it still has limitations concerning statistical calculation, number of investigated SNPs and costs of the studies.
References
- AstemborskiJABeatyTH1985Variance components analysis of forced expiration in familiesAm J Med Genet21741534025399
- BaranovaHPerriotJ1997Peculiarities of the GSTM1 0/0 genotype in French heavy smokers with various types of chronic bronchitisHum Genet9982269187680
- BartschHCastegnaroM1992Expression of pulmonary cytochrome P4501A1 and carcinogen DNA adduct formation in high risk subjects for tobacco-related lung cancerToxicol Lett6465 Spec No:477–83.
- BeckGJDoyleCA1981Smoking and lung functionAm Rev Respir Dis123149557235353
- BenetazzoMGGileLS1999alpha 1-antitrypsin TAQ I polymorphism and alpha 1-antichymotrypsin mutations in patients with obstructive pulmonary diseaseRespir Med936485410542979
- BlobeGCSchiemannWP2000Role of transforming growth factor beta in human diseaseN Engl J Med3421350810793168
- CeledonJCLangeC2004The transforming growth factor-beta1 (TGFB1) gene is associated with chronic obstructive pulmonary disease (COPD)Hum Mol Genet1316495615175276
- CelliBRMacNeeW2004Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paperEur Resp J2393246
- ChenYHorneSL1996Segregation analysis of two lung function indices in a random sample of young families: the Humboldt Family StudyGenet Epidemiol1335478647377
- ClarkJCWeaverTE1997Decreased lung compliance and air trapping in heterozygous SP-B-deficient miceAm J Respir Cell Mol Biol1646528998078
- ClarkJCWertSE1995Targeted disruption of the surfactant protein B gene disrupts surfactant homeostasis, causing respiratory failure in newborn miceProc Natl Acad Sci USA92779487644495
- DeMeoDLCeledonJC2004Genome-wide linkage of forced midexpiratory flow in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med170129430115347563
- DeMeoDLMarianiTJ2006The SERPINE2 gene is associated with chronic obstructive pulmonary diseaseAm J Hum Genet782536416358219
- ErikssonS1965Studies in alpha 1-antitrypsin deficiencyActa Med Scand Suppl4321854160491
- FagerholMKLaurellCB1970The Pi system-inherited variants of serum alpha 1-antitrypsinProg Med Genet7961114911922
- FletcherCM1976Letter: Natural history of chronic bronchitisBr Med J1159231276784
- GivelberRJCouropmitreeNN1998Segregation analysis of pulmonary function among families in the Framingham StudyAm J Respir Crit Care Med1571445519603122
- GivelberRJCouropmitreeNNGottliebDJ1998Segregation analysis of pulmonary function among families in the Framingham StudyAmerican Journal of Respiratory and Critical Care Medicine1571445519603122
- Global Initiative for Chronic Obstuctive Lung Disease2001 NHLBI/WHO Workshop report. National Institutes of Health. Publication number 2701.
- GolponHAColdrenCD2004Emphysema lung tissue gene expression profilingAm J Respir Cell Mol Biol3159560015284076
- GraingerDJHeathcoteK1999Genetic control of the circulating concentration of transforming growth factor type beta1Hum Mol Genet89379887336
- GuoXLinHM2001Surfactant protein gene A, B, and D marker alleles in chronic obstructive pulmonary disease of a Mexican populationEur Respir J184829011589345
- HarrisonDJCantlayAM1997Frequency of glutathione S-transferase M1 deletion in smokers with emphysema and lung cancerHum Exp Toxicol16356609257159
- HeJQRuanJ2002Antioxidant gene polymorphisms and susceptibility to a rapid decline in lung function in smokersAm J Respir Crit Care Med166323812153964
- HershCPDahlM2004Chronic obstructive pulmonary disease in alpha1-antitrypsin PI MZ heterozygotes: a meta-analysisThorax59843915454649
- HershCPDemeoDL2005Attempted replication of reported chronic obstructive pulmonary disease candidate gene associationsAm J Respir Cell Mol Biol3371815817713
- HershCPDemeoDL2006aGenetic association analysis of functional impairment in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med17397784
- HershCPDemeoDL2006bGenetic determinants of functional impairment in chronic obstructive pulmonary diseaseProc Am Thorac Soc3476
- HigginsMKellerJ1975Familial occurrence of chronic respiratory disease and familial resemblance in ventilatory capacityJ Chronic Dis28239511127070
- HiranoKSakamotoT2001Tissue inhibitor of metalloproteinases-2 gene polymorphisms in chronic obstructive pulmonary diseaseEur Respir J187485211757622
- HirschhornJN2005Genetic approaches to studying common diseases and complex traitsPediatr Res5774R77R
- HirschhornJNDalyMJ2005Genome-wide association studies for common diseases and complex traitsNat Rev Genet69510815716906
- HorneSLCockcroftDW1990Possible protective effect against chronic obstructive airways disease by the GC2 alleleHum Hered4017362365378
- HuangSLSuCH1997Tumor necrosis factor-alpha gene polymorphism in chronic bronchitisAm J Respir Crit Care Med156143699372657
- HubertHBFabsitzRR1982Genetic and environmental influences on pulmonary function in adult twinsAm Rev Respir Dis125409157200340
- HurdS2005Global impact of COPDExp Lung Res31Suppl 1576216395860
- JoosLHeJQ2002The role of matrix metalloproteinase polymorphisms in the rate of decline in lung functionHum Mol Genet115697611875051
- JoostOWilkJB2002Genetic loci influencing lung function: a genome-wide scan in the Framingham StudyAm J Respir Crit Care Med165795911897646
- JuulKTybjaerg-HansenA2006Genetically increased antioxidative protection and decreased chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1738586416399992
- KauffmannFTagerIB1989Familial factors related to lung function in children aged 6–10 years. Results from the PAARC epidemiologic studyAm J Epidemiol1291289992729263
- KoyamaHGeddesDM1998Genes, oxidative stress, and the risk of chronic obstructive pulmonary diseaseThorax53Suppl 2S101410193341
- KueppersFBriscoeWA1964Hereditary deficiency of serum alpha-L-antitrypsinScience1461678914224514
- LarsonRKBarmanML1970Genetic and environmental determinants of chronic obstructive pulmonary diseaseAnn Intern Med72627325448092
- LebowitzMDKnudsonRJ1984Family aggregation of pulmonary function measurementsAm Rev Respir Dis1298116703487
- MacNeeW2000Oxidants/antioxidants and COPDChest1175 Suppl 1303S17S10843965
- ManninoDM2002COPD: epidemiology, prevalence, morbidity and mortality, and disease heterogeneityChest1215 Suppl121S126S12010839
- ManninoDM2003Chronic obstructive pulmonary disease: definition and epidemiologyRespir Care48118591 discussion 1191–3.14651759
- ManninoDMBuistAS2003Lung function and mortality in the United States: data from the First National Health and Nutrition Examination Survey follow up studyThorax583889312728157
- MinematsuNNakamuraH2001Genetic polymorphism in matrix metalloproteinase-9 and pulmonary emphysemaBiochem Biophys Res Commun2891161911708786
- NeedhamMStockleyRA2004Alpha 1-antitrypsin deficiency. 3: Clinical manifestations and natural historyThorax59441515115878
- O’DonnellRBreenD2006Inflammatory cells in the airways in COPDThorax614485416648353
- OhnishiKTakagiM1998Matrix metalloproteinase-mediated extra-cellular matrix protein degradation in human pulmonary emphysemaLab Invest781077879759652
- ParkJYChenL2005Polymorphisms for microsomal epoxide hydrolase and genetic susceptibility to COPDInt J Mol Med15443815702235
- PauwelsRABuistAS2001Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summaryAm J Respir Crit Care Med16312567611316667
- PollerWFaberJP1993A leucine-to-proline substitution causes a defective alpha 1-antichymotrypsin allele associated with familial obstructive lung diseaseGenomics1774038244391
- PollerWMeisenC1990DNA polymorphisms of the alpha 1-antitrypsin gene region in patients with chronic obstructive pulmonary diseaseEur J Clin Invest2017
- RedlineS1990Genetic and perinatal risk factors for the development of chronic obstructive pulmonary diseaseNew YorkMarcel Dekker
- RedlineSTishlerPV1987Assessment of genetic and nongenetic influences on pulmonary function. A twin studyAm Rev Respir Dis135217223800147
- RedlineSTishlerPVLewitterFI1987Assessment of genetic and nongenetic influences on pulmonary function: atwin studyAmerican Review of Respiratory Diseases135217223800147
- RobertsonBKobayashiT1991Experimental neonatal respiratory failure induced by a monoclonal antibody to the hydrophobic surfactant-associated protein SP-BPediatr Res30239431945561
- RybickiBABeatyTH1990Major genetic mechanisms in pulmonary functionJ Clin Epidemiol43667752370574
- SakaoSTatsumiK2001Association of tumor necrosis factor alpha gene promoter polymorphism with the presence of chronic obstructive pulmonary diseaseAm J Respir Crit Care Med163420211179116
- SakaoSTatsumiK2002Association of tumor necrosis factor-alpha gene promoter polymorphism with low attenuation areas on high-resolution CT in patients with COPDChest1224162012171811
- SandfordAJChaganiT2001Susceptibility genes for rapid decline of lung function in the lung health studyAm J Respir Crit Care Med1634697311179124
- SandfordAJWeirTD1999Z and S mutations of the alpha 1-antitrypsin gene and the risk of chronic obstructive pulmonary diseaseAm J Respir Cell Mol Biol20287919922220
- SchellenbergDParePD1998Vitamin D binding protein variants and the risk of COPDAm J Respir Crit Care Med157957619517617
- SchurchSGreenFH1998Formation and structure of surface films: captive bubble surfactometryBiochim Biophys Acta14081802029813315
- SeifartCDempfleA2005TNF-alpha-, TNF-beta-, IL-6-, and IL-10-promoter polymorphisms in patients with chronic obstructive pulmonary diseaseTissue Antigens659310015663746
- SeifartCPlagensA2002Surfactant protein B intron 4 variation in German patients with COPD and acute respiratory failureDis Markers181293612515908
- SevenoaksMJStockleyRA2006Chronic obstructive pulmonary disease, inflammation and co-morbidity – a common inflammatory phenotype?Respir Res77016669999
- ShapiroSDSeniorRM1999Matrix metalloproteinases. Matrix degradation and moreAm J Respir Cell Mol Biol201100210340927
- SherrillDLebowitzMBurrowsB1990Epidemiology of cigarette smoking and ist impact on chronic obstructive pulmonary diseaseClin Chest Med11375872205437
- SilvermanEKMosleyJD2002Genome-wide linkage analysis of severe, early-onset chronic obstructive pulmonary disease: airflow obstruction and chronic bronchitis phenotypesHum Mol Genet116233211912177
- SilvermanEKPalmerLJ2002Genomewide linkage analysis of quantitative spirometric phenotypes in severe early-onset chronic obstructive pulmonary diseaseAm J Hum Genet7012293911914989
- SilvermanEKProvinceMA1992Family study of alpha 1-antitrypsin deficiency: effects of cigarette smoking, measured genotype, and their interaction on pulmonary function and biochemical traitsGenet Epidemiol9317311427021
- SmithCAHarrisonDJ1997Association between polymorphism in gene for microsomal epoxide hydrolase and susceptibility to emphysemaLancet35063039288046
- SpiraABeaneJ2004Gene expression profiling of human lung tissue from smokers with severe emphysemaAm J Respir Cell Mol Biol316011015374838
- SteerSAbkevichV2007Genomic DNA pooling for whole genome accosiation scans in complex diseases: epirical demonstration of efficacy in rheumatoid arthritisGenes Immun8576817159887
- StockleyRA2001Proteases and antiproteasesNovartis Found Symp23418999 discussion 199–204.11199096
- SuthanthiranMLiB2000Transforming growth factor-beta 1 hyperexpression in African-American hypertensives: A novel mediator of hypertension and/or target organ damageProc Natl Acad Sci USA9734798410725360
- TagerIBRosnerB1976Household aggregation of pulmonary function and chronic bronchitisAm Rev Respir Dis11448592970729
- TokiedaKIkegamiM1999Surfactant protein B corrects oxygen-induced pulmonary dysfunction in heterozygous surfactant protein B-deficient micePediatr Res467081410590028
- van der Pouw KraanTCKucukaycanM2002Chronic obstructive pulmonary disease is associated with the −1055 IL-13 promoter polymorphismGenes Immun3436912424628
- van der Pouw KraanTCvan VeenA1999An IL-13 promoter polymorphism associated with increased risk of allergic asthmaGenes Immun161511197307
- WangWYBarrattBJ2005Genome-wide association studies: theoretical and practical concernsNat Rev Genet61091815716907
- WilkJBDeStefanoAL2003A genome-wide scan of pulmonary function measures in the National Heart, Lung, and Blood Institute Family Heart StudyAm J Respir Crit Care Med16715283312637344
- WilsonAGde VriesNPociotF1993An allelic polymorphism within the human tumor necrosis factor alpha promoter region is strongly associated with HLA A1, B8, and DR3 allelesJ Exp Med177557608426126
- WilsonAGSymonsJA1997Effects of a polymorphism in the human tumor necrosis factor alpha promoter on transcriptional activationProc Natl Acad Sci USA94319599096369
- WoodAMStockleyRA2006The genetics of chronic obstructive pulmonary diseaseRespir Res713017054776
- YimJJParkGY2000Genetic susceptibility to chronic obstructive pulmonary disease in Koreans: combined analysis of polymorphic genotypes for microsomal epoxide hydrolase and glutathione S-transferase M1 and T1Thorax55121510639528
- YoshikawaMHiyamaK2000Microsomal epoxide hydrolase genotypes and chronic obstructive pulmonary disease in JapaneseInt J Mol Med5495310601573
- YoungRPHopkinsR2006Functional variants of antioxidant genes in smokers with COPD and in those with normal lung functionThorax61394916467073
- ZhengTZhuZ2000Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysemaJ Clin Invest10610819311067861
- ZhouMHuangSG2004Genetic polymorphism in matrix metalloproteinase-9 and the susceptibility to chronic obstructive pulmonary disease in Han population of south ChinaChin Med J (Engl)1171481415498369