Abstract
Patients affected by chronic obstructive pulmonary disease (COPD) have an increased risk of atherothrombotic acute events, independent of smoking and other cardiovascular risk factors. As a consequence, myocardial ischemia is a relevant cause of death in these patients. We reviewed studies concerning the potential mechanisms of atherothrombosis in COPD. Bronchial inflammation spreads to the systemic circulation and is known to play a key role in plaque formation and rupture. In fact, C-reactive protein blood levels increase in COPD and provide independent prognostic information. Systemic inflammation is the first cause of the hypercoagulable state commonly observed in COPD. Furthermore, hypoxia is supposed to activate platelets, thus accounting for the increased urinary excretion of platelet-derived thromboxane in COPD. The potential metabolic risk in COPD is still debated, in that recent studies do not support an association between COPD and diabetes mellitus. Finally, oxidative stress contributes to the pathogenesis of COPD and may promote oxidation of low-density-lipoproteins with foam cells formation. Retrospective observations suggest that inhaled corticosteroids may reduce atherothrombotic mortality by attenuating systemic inflammation, but this benefit needs confirmation in ongoing randomized controlled trials. Physicians approaching COPD patients should always be aware of the systemic vascular implications of this disease.
Introduction
Chronic obstructive pulmonary disease (COPD) is a leading cause of death worldwide (CitationCalverley and Walker 2003). In addition to the healthcare costs, COPD imposes a significant burden in terms of disability and impaired quality of life. Unlike many other leading causes of death and disability, COPD prevalence will raise in the majority of countries as smoking frequency increases and the population ages (CitationHalbert et al 2006). The World Health Organization predicts that by 2020 COPD will be the 5th most prevalent disease worldwide (presently the 12th) and the 3th most common cause of death (presently the 6th). Not respiratory outnumber respiratory causes of death, and most of them are cardiovascular causes (CitationHansell et al 2003). This is the rationale for reviewing the main mechanisms of the COPD-related atherothrombotic risk.
We performed a series of MEDLINE database searches for English language literature published from 1970 to April 2007 by combining the medical subject heading (MeSH) terms chronic obstructive pulmonary disease, chronic bronchitis and pulmonary emphysema with the following MeSH terms atherosclerosis, thrombosis, platelet activation, platelet aggregation, thromboxane, inflammation, inflammation mediators, C-reactive protein, blood coagulation, blood coagulation mediators, blood coagulation factor inhibitors, corticosteroids, catecholamines, sympathetic nervous system, oxidative stress, isoprostanes, F2 isoprostanes. We also supplemented references by cross-checking bibliographies of retrieved articles to identify additional studies.
COPD and atherothrombotic risk: the epidemiological evidence
Reduced forced expiratory volume in 1 second (FEV1) was associated with increased pulse wave velocity, a surrogate measurement for central arterial stiffness, endothelial dysfunction and atherosclerosis (CitationZureik et al 2001).
Although not generally recognized, a low FEV1 has been shown to be as powerful a predictor of cardiac mortality as total serum cholesterol, irrespective of the effect of smoking and other possible confounders (CitationHole et al 1996). In a recent meta-analysis (CitationSin et al 2005b), the relative risk of cardiovascular death in the group with the lowest FEV1 compared with that with the highest FEV1 was 3.36 (1.54–7.34); the corresponding figure for mortality due to myocardial ischemia was 5.65 (2.26–14.13). As a consequence, coronary artery disease is one of the leading causes of death in COPD patients (CitationHansell et al 2003).
FEV1/forced vital capacity (FEV1/FVC) ratio, a more sensitive parameter of obstructive disease than FEV1, was also found to be independently related to vascular events (CitationEngstrom et al 2001).
The overall atherothrombotic impact of COPD is further proved by the increased risk of ischemic stroke in COPD patients. In a 15-year follow up, the relative risk of fatal stroke was 1.1 (1.03–1.2) for every 10% decrease of FEV1 (CitationTruelsen et al 2001).
Pro-atherothrombotic mechanisms in COPD
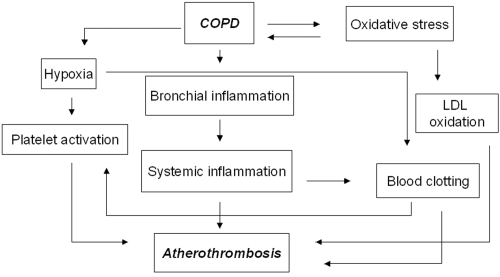
The mechanisms responsible for the association between COPD and atherothrombosis are still largely unknown. However, at least four factors seem to be pathogenetically important: chronic systemic inflammation, hypercoagulable state, platelet activation and oxidative stress ().
Figure 1 The COPD-atherothrombosis network.
Systemic inflammation ()
Table 1 Mechanisms of systemic inflammation in COPD
In the context of the complex and multifactorial pathogenesis of atherothrombosis, low grade systemic inflammation is believed to be a crucial mechanism in plaque formation and rupture (CitationPasceri et al 2000). This concept is strongly supported by experiments showing that some inflammatory markers, such as C-reactive protein (CRP) and fibrinogen, are implicated in plaque formation. CPR upregulates the production of pro-inflammatory cytokines and tissue factor by monocytes, increases the uptake of low-density lipoproteins (LDL) by macrophages with foam cells formation and directly induces expression of adhesion molecules by human endothelial cells (CitationPasceri et al 2000).
Alveolar macrophages, bronchial epithelial cells and lymphocytes, which are implicated in bronchial and alveolar inflammation, produce interleukin (IL)-6 and IL-1β. These cytokines, besides inducing local pro-inflammatory changes, “spill-over” into the systemic circulation and stimulate hepatocytes to synthesize CRP and fibrinogen. Accordingly, systemic blood concentrations of tumor necrosis factor (TNF)-α, IL 6, IL 8, CRP and fibrinogen are higher in COPD than in control subjects (CitationGan et al 2004). Interestingly, at variance from the response observed in healthy subjects and diabetic patients, moderate-intensity exercise abnormally increases plasma TNF-α levels in COPD patients (CitationRabinovich et al 2003; CitationZoppini et al 2006). Circulating TNF-α levels have been found to increase in malnourished COPD patients, likely because systemic hypoxia stimulates TNF-α synthesis (CitationTakabatake et al 2000). TNF-α can cause the expression of tissue factor on monocytes and, possibly, endothelium, thereby initiating the coagulation cascade (CitationEsmon 2000).
CRP serum levels have also been found to increase for increasing severity of bronchial obstruction, ie, in COPD, the level of “systemic” inflammation (CRP) strictly parallels that of “local” bronchial inflammation and obstruction (FEV1) (Sin and Man 2003). In addition, COPD patients with high CRP serum levels have increased risk of either atherothrombotic events (fatal and nonfatal coronary artery disease and stroke) or all-cause mortality, after adjusting for age, sex, smoking and lung function (CitationMan et al 2006). These recent data support the role of the systemic inflammation in the development of atherothrombotic disorders in COPD. In addition, they suggest that CRP measurements should be considered in prognostic models for COPD patients.
Pulmonary hypertension is highly prevalent also in nonhypoxemic COPD and predicts a poor prognosis (CitationWeitzenblum et al 1981). In a recent study, CRP and hypoxia were the only significant correlates of systolic pulmonary arterial pressure, suggesting that systemic inflammation may be involved in the pathogenesis of COPD-related pulmonary hypertension (CitationJoppa et al 2006).
Hypercoagulable state ()
Table 2 Mechanisms explaining the procoagulant status which characterizes COPD (see also )
Table 3 Epidemiological evidence and mechanisms of platelet hyperaggregability in COPD
Some case-control studies have clearly established that, independent of current smoking, plasma levels of fibrinogen and other markers of coagulation are significantly higher in stable COPD patients than in healthy subjects (CitationAlessandri et al 1994; CitationXie and Wang 1998; CitationWedzicha et al 2000; CitationAshitani et al 2002). The increased procoagulant activity in COPD may primarily result from inflammation. In fact, inflammation can trigger coagulation by promoting tissue-factor gene expression in endothelial cells (CitationEsmon 2000; CitationLibby 2001). Hypoxia also could either reduce endothelial thrombomodulin expression or activate factor X (CitationOgawa et al 1990). Coagulation, in turn, amplifies inflammation and both are strongly implicated in the pathogenesis of atherothrombosis (CitationLibby and Simon 2001). That a pro-coagulant status may promote atherothrombosis in COPD is suggested by the direct relationship between serum fibrinogen and the incidence of cardiovascular events in the general population (CitationDanesh et al 1998). Interestingly, central pulmonary lesions, indicative of in situ thrombosis and atherosclerosis, are common in stable COPD patients even free from pulmonary hypertension, and their extent is not strictly related to the severity of bronchial obstruction (CitationRusso et al 1999). This finding testifies to a procoagulant and proatherosclerotic status which can be recognized early in the course of COPD.
Importantly, fibrinogen levels rise further during COPD exacerbation (CitationWedzicha et al 2000). The acute release of this and other prothrombotic factors may thus account for the increased rate of myocardial infarctions immediately following low-respiratory tract infections (CitationMeier et al 1998).
Platelet activation ()
The role of platelet in atherothrombosis is clearly supported by the efficacy of aspirin and other antiplatelet drugs in preventing vascular events in the general population (CitationPatrono et al 2004). Earlier studies had showed increased platelet aggregability in hypoxaemic COPD patients (CitationCordova et al 1985; CitationWedzicha et al 1991). More recently, platelet aggregation has been studied in vivo by measuring 11-dehydro-thromboxane B2 (11-d-TxB2), the urinary metabolites of TxA2 (CitationPatrono et al 1995). This eicosanoid is generated by activated platelets through the enzyme cyclooxygenase-1, which is specifically inhibited by aspirin. Once released, TxA2 amplifies platelet aggregation and stimulates smooth muscle constriction and proliferation (CitationRolin et al 2006). TxA2 is also a strong constrictor of bronchial smooth muscle cells and has been involved in the pathogenesis of asthma (CitationTamaoki et al 2000). The measurement of urinary 11-d-TxA2 directly reflects biosynthesis and is therefore a measure of platelet function (CitationPatrono et al 1995). High excretory values identify patients at increased risk of myocardial infarction and cardiovascular death (CitationEikelboom et al 2002). Importantly, urinary 11-d-TxB2 values are significantly greater in patients with stable COPD than in control subjects, irrespective of smoking status, inversely correlated with arterial oxygen tension and are significantly lowered by short – term oxygen supplementation (CitationDavì et al 1997). These data suggest a link between hypoxia and platelet activation likely because hypoxia induces metabolic changes on the platelet membrane, leading to increased activation of cyclooxygenase-1 with thromboxane formation (CitationPonicke et al 1987). In addition, platelet stimulation may result from clotting activation with thrombin generation, that, in turn, is well known to enhance platelet thromboxane biosynthesis (CitationPatrono 1990).
Oxidative stress ()
Table 4 Potential mechanisms of oxidative stress in COPD
The development of COPD is associated with oxidative stress and reduced antioxidant properties (CitationBoots et al 2003). Hydrogen peroxide (H2O2) in exhaled breath condensate is a marker of oxidative stress in the lungs and have been found to be elevated in COPD patients irrespective of smoking status (CitationDekhuijzen et al 1996; CitationNowak et al 1999), as well as in smokers without the disease (CitationNowak et al 2001). Oxidative stress can promote the peroxidation of polyunsaturated fatty acids. Thiobarbituric acid-reacting substances represent a measure of such a lipid peroxidation and are increased in exhaled breath condensate of patients with COPD (CitationNowak et al 1999).
Pulmonary oxidative stress “spreads out” to the circulation and becomes a systemic alteration (CitationBoots et al 2003). F2-isoprostanes are stable products of peroxidation of arachidonic acid (CitationDelanty et al 1996). The assay of F2-isoprostanes in the urine is a reliable measure of in vivo, systemic oxidative stress and, more importantly, it is a marker of LDL oxidation (CitationDevaraj et al 2001), that, in turn, is a key event in the pathogenesis of atherosclerosis (CitationBerliner and Heinecke 1996; CitationPatrono et al 2004). Independently of current smoking, the excretion of F2-isoprostane increases significantly in COPD and peaks during exacerbations (CitationPraticò et al 1998). This should suggest a LDL oxidative susceptibility in COPD, an abnormality potentially contributing to plaque formation.
Lipid status and metabolic risk in COPD ()
Table 5 Lipid status and metabolic risk in COPD
The increased vascular risk in COPD cannot be attributed to an atherogenic lipid pattern. In COPD patients, lipid levels are comparable with those measured in healthy subjects, with values of lipoprotein(a) and of APO B-100 being even significantly lower (CitationBasili et al 1999).
The relation between diabetes mellitus, one of the leading atherothrombotic factors, and COPD is unclear. There are data supporting the notion that the lung is a target organ in diabetes, but abnormalities mostly include alveolar microangiopathy with impaired diffusion capacity (CitationHsia and Raskin 2005) and a restrictive ventilatory dysfunction (CitationLawlor et al 2004). Results of prospective observations in COPD patients are confounding. In a study, COPD was a risk factor for the onset of type 2 diabetes mellitus, but spirometric diagnostic data were not reported (CitationRana et al 2004). In another study, an obstructive pattern was not associated with the development of diabetes, that, conversely, was significantly increased in subjects with a restrictive ventilatory pattern (CitationFord and Mannino 2004). Accordingly, metabolic syndrome and insulin-resistance were very frequent in nondiabetic subjects with restrictive dysfunction, but not in COPD patients (CitationFimognari et al 2007).
Neurohumoral activation
There are convincing data demonstrating a COPD-related neurohumoral activation, including sympathetic overactivity, increased release of catecholamines and decreased vagal tone (CitationAndreas et al 2005). This alteration is evident also after interruption of medications for COPD (CitationScalvini et al 1999) and is supposed to contribute to a generic cardiovascular and systemic risk in COPD (arrhythmias, cachexia, muscle wasting with fatigue) (CitationAndreas et al 2005). The specific impact of neurohumoral activation on the atherothrombotic status of COPD, however, is unclear.
Animal models showed that sympathetic activation can promote systemic inflammation (CitationWoiciechowsky et al 1998; CitationBorovikova et al 2000; CitationLi et al 2004). Theoretically, the neurohumoral activation occurring in COPD may thus favour atherothrombosis by stimulating systemic and vascular inflammation, but this potential causative association needs to be proven in patients with COPD.
Another mechanism by which the sympathetic activation may promote atherothrombosis in COPD is platelet activation. It is well known that the catecholamines released during acute stress may directly activate platelets (CitationHjemdahl et al 1991, Citation1994; CitationLarsson et al 1994), but there are no data to demonstrate that this may also take place during chronic neurohumoral activation, like that complicating COPD (CitationFimognari et al 1996). In conclusion, the notion that the neurohumoral activation contributes to the COPD-related platelet hyperfunction is still highly speculative.
Potential therapies ()
Table 6 Potential therapies of systemic inflammation in COPD
The role of systemic inflammation in precipitating vascular events in COPD is strongly supported by accumulating data showing the potential benefits of inhaled corticosteroids. These drugs, commonly used to attenuate lung inflammation, have been demonstrated to reduce plasma CRP levels in stable COPD, suggesting a “cooling” effect also on systemic inflammation (CitationSin et al 2004; CitationPinto-Plata et al 2006). In a recent meta-analysis of randomized controlled trials (RCTs), the mortality of COPD patients on inhaled corticosteroids was 27% lower than that reported in the placebo group (adjusted hazard ratio, 0.73; 95% confidence interval, 0.55–0.96) (CitationSin et al 2005a). Of note, this is the same mortality reduction obtained by a 3-year therapy with simvastatin in the Scandinavian Simvastatin Survival Study (CitationScandinavian Simvastatin Survival Study Group 1994). Because the trials included in the meta-analysis were not planned to test the effect of inhaled steroids on survival, the result needs to be confirmed in RCTs specifically designed for this aim. However, the improved survival may be due to a reduction in cardiovascular deaths, in turn determined by the effect of inhaled corticosteroids on vascular plaque inflammation. This intriguing hypothesis is suggested by some important observational studies. CitationHuiart and collegues (2005) found a 18% nonsignificant reduction of myocardial infarctions amongst users of inhaled corticosteroids, but in a subgroup of patients taking 50–200 μg/day of steroids the risk reduction increased to 32% (p < 0.05). A recent study, while confirming improved survival in corticosteroid users (CitationMacie et al 2006), found that this benefit largely derived from a 38% reduction in cardiovascular mortality, while the reduction in respiratory deaths was nonsignificant. Recently, the EUROSCOP (European Respiratory Society Study on Chronic Pulmonary Disease) trial reported a 40% reduction in the rate of coronary artery disease events in patients taking inhaled corticosteroids compared to the placebo group (CitationLofdahl et al 2005).
Very recently, the effect on mortality of inhaled corticosteroids has been questioned by the results of an important RCT, the TORCH (Toward a Revolution in COPD Health) study (CitationCalverley et al 2007). The mortality rate at 3 years in the fluticasone group (16%) was comparable to that observed in the placebo group (15.2%). In patients treated with the combination therapy of fluticasone plus the long-acting β2-agonist salmeterol (mortality rate:12.6%) there was an interesting 17.5% reduction in the risk of death compared with placebo, with a nonsignificant difference (p = 0.052). The impact of these drugs on cardiovascular mortality only was not reported. It is possible that the combination therapy does improve survival in COPD patients and that this study was underpowered to detect this effect. In addition, because β2-agonists activate glucocorticoid receptors in the lung (CitationEickelberg et al 1999), the association with β2-agonists may be crucial to highlight any effect of inhaled corticosteroids on mortality.
The only treatment improving survival in COPD is long-term oxygen therapy in hypoxemic patients (CitationAfessa et al 2002). Oxygen therapy may prevent the hypoxia-induced platelet activation and blood clotting, and part of its clinical benefit may come from a reduction of atherothrombotic fatal events by these effects. Furthermore, oxygen supplementation prevents exercise-induced oxidative stress in normoxemic, muscle-wasted patients with COPD (Citationvan Helvoort et al 2006). However, short-term oxygen supplementation has also been reported to promote oxidative stress and airway inflammation (CitationCarpagnano et al 2004).
Conclusion
COPD is now recognized as a systemic inflammatory disease that may adversely affect the arterial district, predisposing patients to an increased risk of atherosclerotic plaque formation and rupture. Systemic inflammation plays a leading role in this process, but other mechanisms, such as platelet activation, coagulation and oxidative stress, can promote atherosclerosis in COPD. Research is needed to further clarify mechanisms of atherothrombosis, as well as to identify therapeutic and preventive strategies. Current evidence is consistent with an optimal care of bronchial inflammation likely translating in some reduction of atherothrombotic risk. However, the very early development of the pro-atherothrombotic status in the course of COPD suggests that researchers should think of COPD as of a systemic disease from its onset. Accordingly, patients should receive advice about nonrespiratory effects of COPD. This communication strategy might improve the awareness of the disease status and of the need of halting its progression by the most important preventive measure: smoking cessation.
Eventually, the evidence of systemic effects of COPD should induce pneumologists to have a multidisciplinary and multidimensional approach to the disease by involving various specialists. The relatively poor progress in the treatment of COPD in the second half of the last century, when compared with therapy of other chronic diseases, likely reflects the purely respiratory-centered, then scotomic, view of the disease.
References
- AfessaBMoralesIJScanlonPDPrognostic factors, clinical course, and hospital outcome of patients with chronic obstructive pulmonary disease admitted to an intensive care unit for acute respiratory failureCrit Care Med20023016101512130987
- AlessandriCBasiliSVioliFHypercoagulability state in patients with chronic obstructive pulmonary disease Chronic Obstructive Bronchitis and Haemostasis GroupThromb Haemost19947234367855781
- AndreasSAnkerSDScanlonPDNeurohumoral activation as a link to systemic manifestations of chronic lung diseaseChest200512836182416304321
- AshitaniJMukaeHArimuraYElevated plasma procoagulant and fibrinolytic markers in patients with chronic obstructive pulmonary diseaseIntern Med200241181511929177
- BasiliSFerroniPVieriMLipoprotein(a) serum levels in patients affected by chronic obstructive pulmonary diseaseAtherosclerosis19991472495210559510
- BerlinerJAHeineckeJWThe role of oxidized lipoproteins in atherogenesisFree Radic Biol Med199620707278721615
- BootsAWHaenenGRBastAOxidant metabolism in chronic obstructive pulmonary diseaseEur Respir J Suppl20034614s27s14621103
- BorovikovaLVIvanovaSZhangMVagus nerve stimulation attenuates the systemic inflammatory response to endotoxinNature20004054586210839541
- CalverleyPMWalkerPChronic obstructive pulmonary diseaseLancet200336210536114522537
- CalverleyPMAndersonJACelliBTORCH investigatorsSalmeterol and fluticasone propionate and survival in chronic obstructive pulmonary diseaseN Engl J Med20073567758917314337
- CarpagnanoGEKharitonovSAFoschino-BarbaroMPSupplementary oxygen in healthy subjects and those with COPD increases oxidative stress and airway inflammationThorax20045910161915563698
- CordovaCMuscaAVioliFPlatelet hyperfunction in patients with chronic airways obstructionEur J Respir Dis1985669122579841
- DaneshJCollinsRApplebyPAssociation of fibrinogen, C-reactive protein, albumin, or leukocyte count with coronary heart disease: meta-analyses of prospective studiesJAMA19982791477829600484
- DavìGBasiliSVieriMEnhanced thromboxane biosynthesis in patients with chronic obstructive pulmonary disease. The Chronic Obstructive Bronchitis and Haemostasis Study GroupAm J Respir Crit Care Med1997156179499412557
- DekhuijzenPNAbenKKDekkerIIncreased exhalation of hydrogen peroxide in patients with stable and unstable chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1996154813168810624
- DelantyNReillyMPraticoD8-Epi PGF2 alpha: specific analysis of an isoeicosanoid as an index of oxidant stress in vivoBr J Clin Pharmacol19964215198807139
- DevarajSHiranySVBurkRFDivergence between LDL oxidative susceptibility and urinary F(2)-isoprostanes as measures of oxidative stress in type 2 diabetesClin Chem2001471974911673365
- EickelbergORothMLorxRLigand-independent activation of the glucocorticoid receptor by beta2-adrenergic receptor agonists in primary human lung fibroblasts and vascular smooth muscle cellsJ Biol Chem19992741005109873044
- EikelboomJWHirshJWeitzJIAspirin-resistant thromboxane biosynthesis and the risk of myocardial infarction, stroke, or cardiovascular death in patients at high risk for cardiovascular eventsCirculation20021051650511940542
- EngstromGWollmerPHedbladBOccurrence and prognostic significance of ventricular arrhythmia is related to pulmonary function: a study from “men born in 1914,” Malmo, SwedenCirculation200110330869111425773
- EsmonCTDoes inflammation contribute to thrombotic events?Haemostasis200030Suppl 2344011251339
- FimognariFLPasqualettiPMoroLThe association between metabolic syndrome and restrictive ventilatory dysfunction in older personsJ Gerontol A Biol Sci Med2007627605
- FimognariFLPiccirilloGLamaJAssociated daily biosynthesis of cortisol and thromboxane A2: a preliminary reportJ Lab Clin Med1996128115218759943
- FordESManninoDMProspective association between lung function and the incidence of diabetes: findings from the National Health and Nutrition Examination Survey Epidemiologic Follow-up StudyDiabetes Care20042729667015562215
- GanWQManSFSenthilselvanAAssociation between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysisThorax2004595748015223864
- HalbertRJNatoliJLGanoAGlobal burden of COPD: systematic review and meta-analysisEur Respir J2006285233216611654
- HansellALWalkJASorianoJBWhat do chronic obstructive pulmonary disease patients die from? A multiple cause coding analysisEur Respir J2003228091414621089
- HjemdahlPLarssonPTWallenNHEffects of stress and beta-blockade on platelet functionCirculation199184VI44611683610
- HjemdahlPChronosNAWilsonDJEpinephrine sensitizes human platelets in vivo and in vitro as studied by fibrinogen binding and P-selectin expressionArterioscler Thromb19941477847506054
- HoleDJWattGCDavey-SmithGImpaired lung function and mortality risk in men and women: findings from the Renfrew and Paisley prospective population studyBMJ1996313711158819439
- HsiaCCRaskinPThe diabetic lung: relevance of alveolar microangiopathy for the use of inhaled insulinAm J Med20051182051115745714
- HuiartLErnstPRanouilXLow-dose inhaled corticosteroids and the risk of acute myocardial infarction in COPDEur Respir J200525634915802336
- JoppaPPetrasovaDStancakBSystemic inflammation in patients with COPD and pulmonary hypertensionChest20061303263316899829
- LarssonPTWallenNHHjemdahlPNorepinephrine-induced human platelet activation in vivo is only partly counteracted by aspirinCirculation199489195178181117
- LawlorDAEbrahimSSmithGDAssociations of measures of lung function with insulin resistance and Type 2 diabetes: findings from the British Women’s Heart and Health StudyDiabetologia20044719520314704837
- LiMZhengCSatoTVagal nerve stimulation markedly improves long-term survival after chronic heart failure in ratsCirculation2004109120414662714
- LibbyPSimonDIInflammation and thrombosis: the clot thickensCirculation200110317182011282900
- LofdahlCGPostmaDPrideNDoes inhaled budesonide protect against cardio-ischemic events in mild-moderate COPD: a post-hoc evaluation of the EUROSCOP Study [Abstract]European Respiratory Society Conference, Copenhagen 20052005
- MacieCWooldrageKManfredaJInhaled corticosteroids and mortality in COPDChest2006130640616963657
- ManSFConnettJEAnthonisenNRC-reactive protein and mortality in mild to moderate chronic obstructive pulmonary diseaseThorax2006618495316738034
- MeierCRJickSSDerbyLEAcute respiratory-tract infections and risk of first-time acute myocardial infarctionLancet19983511467719605802
- NowakDKasielskiMAntczakAIncreased content of thiobarbituric acid-reactive substances and hydrogen peroxide in the expired breath condensate of patients with stable chronic obstructive pulmonary disease: no significant effect of cigarette smokingRespir Med1999933899610464820
- NowakDKaluckaSBialasiewiczPExhalation of H2O2 and thiobarbituric acid reactive substances (TBARs) by healthy subjectsFree Radic Biol Med2001301788611163535
- OgawaSShreeniwasRBrettJThe effect of hypoxia on capillary endothelial cell function: modulation of barrier and coagulant functionBr J Haematol199075517242169857
- PasceriVWillersonJTYehETDirect proinflammatory effect of C-reactive protein on human endothelial cellsCirculation20001022165811056086
- PatronoCThromboxane synthesis inhibitors and receptor antagonistsThromb Res Suppl19901115232148990
- PatronoCCollerBFitzGeraldGAPlatelet-active drugs: the relationships among dose, effectiveness, and side effects: the Seventh ACCP Conference on Antithrombotic and Thrombolytic TherapyChest2004126234S264S15383474
- PatronoCPatrignaniPRoccaBCharacterization of biochemical and functional effects of antiplatelet drugs as a key to their clinical developmentThromb Haemost1995743964008578493
- Pinto-PlataVMMullerovaHTosoJFC-reactive protein in patients with COPD, control smokers and non-smokersThorax20066123816143583
- PonickeKSternitzkyRMestHJStimulation of aggregation and thromboxane A2 formation of human platelets by hypoxiaProstaglandins Leukot Med19872949593478738
- PraticòDBasiliSVieriMChronic obstructive pulmonary disease is associated with an increase in urinary levels of isoprostane F2alpha-III, an index of oxidant stressAm J Respir Crit Care Med19981581709149847257
- RabinovichRAFiguerasMArditeEIncreased tumour necrosis factor-alpha plasma levels during moderate-intensity exercise in COPD patientsEur Respir J2003217899412765422
- RanaJSMittlemanMASheikhJChronic obstructive pulmonary disease, asthma, and risk of type 2 diabetes in womenDiabetes Care20042724788415451919
- RolinSMasereelBDogneJMProstanoids as pharmacological targets in COPD and asthmaEur J Pharmacol20065338910016458293
- RussoADe LucaMVignaCCentral pulmonary artery lesions in chronic obstructive pulmonary disease: A transesophageal echocardiography studyCirculation199910018081510534469
- Scandinavian Simvastatin Survival Study GroupRandomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S)Lancet1994344138397968073
- ScalviniSPortaRZanelliEEffects of oxygen on autonomic nervous system dysfunction in patients with chronic obstructive pulmonary diseaseEur Respir J1999131192410836335
- SinDDLacyPYorkEEffects of fluticasone on systemic markers of inflammation in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med2004170760515229100
- SinDDWuLAndersonJAInhaled corticosteroids and mortality in chronic obstructive pulmonary diseaseThorax2005a60992716227327
- SinDDWuLManSFThe relationship between reduced lung function and cardiovascular mortality: a population-based study and a systematic review of the literatureChest2005b1271952915947307
- TakabatakeNNakamuraHAbeSThe relationship between chronic hypoxemia and activation of the tumor necrosis factor-alpha system in patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med200016111798410764309
- TamaokiJKondoMNakataJEffect of a thromboxane A(2) antagonist on sputum production and its physicochemical properties in patients with mild to moderate asthmaChest200011873910893362
- TruelsenTPrescottELangePLung function and risk of fatal and non-fatal stroke. The Copenhagen City Heart StudyInt J Epidemiol2001301455111171876
- van HelvoortHAHeijdraYFHeunksLMSupplemental oxygen prevents exercise-induced oxidative stress in muscle-wasted patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med20061731122916514109
- VestboJThe TORCH (towards a revolution in COPD health) survival study protocolEur Respir J2004242061015332386
- WedzichaJASeemungalTAMacCallumPKAcute exacerbations of chronic obstructive pulmonary disease are accompanied by elevations of plasma fibrinogen and serum IL-6 levelsThromb Haemost2000842101510959691
- WedzichaJASyndercombe-CourtTanKCIncreased platelet aggregate formation in patients with chronic airflow obstruction and hypoxaemiaThorax19914650471877038
- WeitzenblumEHirthCDucoloneAPrognostic value of pulmonary artery pressure in chronic obstructive pulmonary diseaseThorax19813675287330793
- WoiciechowskyCAsadullahKNestlerDSympathetic activation triggers systemic interleukin-10 release in immunodepression induced by brain injuryNat Med19984808139662372
- XieMWangZPrethrombotic state in patients with chronic obstructive pulmonary disease and treatment with heparinHua Xi Yi Ke Da Xue Xue Bao1998294111410743240
- ZoppiniGTargherGZamboniCEffects of moderate-intensity exercise training on plasma biomarkers of inflammation and endothelial dysfunction in older patients with type 2 diabetesNutr Metab Cardiovasc Dis200616543917126770
- ZureikMBenetosANeukirchCReduced pulmonary function is associated with central arterial stiffness in menAm J Respir Crit Care Med20011642181511751184