Abstract
Since the early 1960s, a compelling body of evidence has accumulated to show that proteinases play critical roles in airspace enlargement in chronic obstructive pulmonary disease (COPD). However, until recently the causative enzymes and their exact roles in pathologic processes in COPD have not been clear. Recent studies of gene-targeted mice in murine models of COPD have confirmed roles for proteinases not only in airspace enlargement, but also in airway pathologies in COPD. These studies have also shed light on the specific proteinases involved in COPD pathogenesis, and the mechanisms by which these proteinases injure the lung. They have also identified important interactions between different classes of proteinases, and between proteinases and other molecules that amplify lung inflammation and injury. This review will discuss the biology of proteinases and the mechanisms by which they contribute to the pathogenesis of COPD. In addition, I will discuss the potential of proteinase inhibitors and anti-inflammatory drugs as new treatment strategies for COPD patients.
Introduction
COPD is currently the fourth most common cause of death in the USA, and its incidence is increasing, especially in women, worldwide and in third-world countries. The National Heart, Lung, and Blood Institute has estimated the annual costs of COPD in the USA in 2005 to be US$38.8 billion. COPD is the least well-funded disease relative to its global heath burden. As a result of the huge healthcare burden associated with COPD, there has been a resurgence of interest in its cellular and molecular mechanisms, and in the development of new treatment strategies to limit the deleterious effects of proteinases in the lungs of COPD patients.
Pathology
In developed countries, the main risk factor for COPD is smoking cigarettes, which accounts for more than 95% of all cases. Other risk factors include inhalation of pollutants, wood smoke, and biomass fuels in enclosed spaces in third-world countries. Genetic factors may also increase individual susceptibility to the adverse effects of cigarette smoke, or alter normal lung repair processes. Inhalation of cigarette smoke and other pollutants leads to a chronic inflammatory process in the small airways and the lung parenchyma including macrophages, polymorphonuclear neutrophils (PMN), T lymphocytes (with CD8+ T cells exceeding the numbers of CD4+ T cells), and B lymphocytes (CitationDi Stefano et al 1996; CitationSaetta 1999; CitationTurato et al 2001; CitationHogg et al 2004). Over time, there is destruction of the alveolar walls leading to airspace enlargement, loss of lung elasticity, closure of small airways, and irreversible airflow obstruction. Pathological changes also develop in the airways, including mucus metaplasia and mucus hypersecretion. Narrowing of the small airways develops as a result of mucus plugging, inflammation in the airway walls and lumen, and subepithelial fibrosis. This small airway obstruction is also an important determinant of the fixed airflow obstruction that occurs in COPD patients (CitationHogg et al 2004). Although COPD is a complex disorder caused by multiple mediators and pathways (including reactive oxygen species [ROS], pro-inflammatory mediators, apoptosis of structural cells, and inadequate repair processes), there is strong evidence that proteinases make critical contributions to all the pathologic processes detected in the lungs of COPD patients.
Historical aspects of proteinases in COPD: The proteinase-antiproteinase hypothesis
Two observations in the 1960s, one clinical and one experimental, led to the proteinase/antiproteinase hypothesis for the pathogenesis of emphysema. The first observation was that genetic deficiency of α1-proteinase inhibitor ([α1-PI], which is the major inhibitor of neutrophil elastase [NE] in the lower respiratory tract) is associated with early-onset, severe panlobular pulmonary emphysema (CitationLaurell and Eriksson 1963). The second observation was that instillation of papain (a metalloproteinase with elastin-degrading activity) into rat lungs results in progressive airspace enlargement (CitationGross et al 1965). Since then, other proteinases that degrade lung elastin, including porcine pancreatic elastase (CitationKarlinsky et al 1983) and subsequently NE and proteinase 3 (PR3), which are more relevant to human COPD compared to porcine pancreatic elastase, were shown to enlarge airspaces when instilled into the lungs of experimental animals (CitationSenior et al 1977; CitationKao et al 1988). Based upon these observations the proteinase-anti-proteinase hypothesis was formulated: Inhalation of cigarette smoke (or other pollutants) leads to the recruitment of inflammatory cells into the lungs. Inflammatory cells release various proteinases that exceed the proteinase inhibitor defense of the lung. Uncontrolled proteinases degrade the extracellular matrix (ECM) protein components of the alveolar walls (especially the elastic fibers) leading to destruction and loss of the alveolar walls and airspace enlargement ().
Figure 1 Mechanisms by which different classes of proteinases contribute to pathologies in COPD. Cigarette smoke stimulates inflammatory cell recruitment, proteinase production, and proteinase release from inflammatory, immune, and structural cells in the lung. Proteinases contribute to airspace enlargement by degrading ECM and promoting death of structural cells of the alveolar walls. Proteinases also amplify lung inflammation and promote mucus hypersecretion and small airway fibrosis.
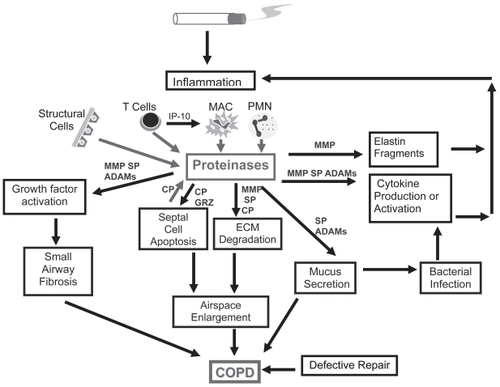
Because of the association between α1-PI deficiency and pulmonary emphysema, early studies focused on the role of NE in airspace enlargement. While unrestrained NE activity in the lung is likely to be important in the panlobular pulmonary emphysema associated with α1-PI deficiency, this is probably an oversimplification of mechanisms underlying the majority of COPD patients, who have normal plasma levels of α1-PI. Studies during the last 3–4 decades have identified roles for other proteinases in airspace enlargement, roles for proteinases in airways pathologies (), and important interactions between different classes of proteinases and between proteinases and other molecules (eg, ROS and inflammatory mediators) that amplify inflammation and ECM destruction in COPD.
Classification and biology of proteinases
Proteinases cleave the internal peptide bonds of polypeptides. They can be classified into 4 groups by the chemical nature of their active site: serine, metallo-, cysteine, and aspartic proteinases (). Proteinase inhibitors are generally targeted against individual classes of proteinases (). Serine proteinases and MMPs are optimally active at neutral pH and have the largest role in extracellular proteolysis. Cysteine and aspartic proteinases are optimally active at acidic pH, and their main role is in intracellular degradation of proteins in lysosomes. However, acid proteinases can degrade extracellular proteins if they retain catalytic activity at neutral pH or are released into an environment having an acidic pH, such as the pericellular environment of activated macrophages (CitationMason et al 1986; CitationShi et al 1992). The proteinases implicated in the pathogenesis of COPD belong to the serine, metallo-, and cysteine proteinase classes.
Table 1 Proteinases involved in the pathogenesis of COPD
Serine proteinases
Serine proteinases implicated in COPD include PMN-derived serine proteinases, urokinase-type plasminogen activator (uPA), granzymes, and plasmin ().
PMN-derived serine proteinases
These include NE, PR3, and cathepsin G (CG). PMN and pro-inflammatory monocytes store preformed serine proteinases in their primary granules, from which the enzymes are released when pro-inflammatory mediators induce PMN degranulation (CitationOwen et al 1994; CitationOwen and Campbell 1999). Together, these serine proteinases have a broad spectrum of activity against ECM proteins (especially elastin) and non-ECM proteins (CitationOwen and Campbell 1999).
Urokinase-type plasminogen activator (uPA)
This enzyme is expressed by PMN, monocytes, and macrophages. Preformed uPA is stored in and released from the specific granules of PMN. However, uPA expression is regulated at the transcriptional level in mononuclear phagocytes by pro-inflammatory mediators (CitationGranelli-Peperno et al 1977; CitationVassalli et al 1991). Following its release from cells, uPA binds to a specific receptor (uPA receptor) on phagocyte surfaces, where it functions as a cell-associated proteinase. The main function of uPA is to convert inactive plasminogen to active plasmin, another serine proteinase. Plasmin degrades fibrin during lysis of blood clots. However, plasmin also cleaves and activates latent growth factors, latent proMMPs, and protease-activated receptor-1 (PAR-1) on macrophages, which drives macrophage MMP-12 production (CitationSaksela and Rifkin 1988; CitationTaipale et al 1992; CitationRaza et al 2000; CitationChurg et al 2007b). Thus by generating plasmin, uPA regulates not only fibrinolysis, but also ECM degradation and fibrotic processes in the lung.
Granzymes (GRZ)
Granzymes are granule-associated enzymes that are predominantly expressed by CD8+ T lymphocytes and are stored in the lytic granules of these cells (CitationSmyth et al 1996). The main GRZ family members in human CD8+ T cells are GRZ A and B. Activation of CD8+ T cells by antigen leads to rapid exocytosis of GRZ and perforin-containing granules. Perforin alters the properties of the cell membrane of the target cells, allowing entry of GRZ into the target cell, and GRZ A and GRZ B then initiate caspase-independent and caspase-dependent apoptosis, respectively.
Serine proteinase inhibitors
Serine proteinase inhibitors in plasma and interstitial fluids include α1-PI, α1-antichymotrypsin, plasminogen activator inhibitors, α2-plasmin inhibitor, and the universal inhibitor, α2-macroglobulin (α2-M), which inhibits all four classes of enzymes (CitationCarrell 1986). Secretory leukocyte proteinase inhibitor (SLPI) and elafin are synthesized locally in the respiratory tract by epithelial cells.
Metalloproteinases
This class of proteinases includes the matrix metalloproteinases (MMPs) and members of the ADAMs family.
MMPs
MMPs have an NH2 terminal pro domain, an active site zinc atom, and a COOH terminal hemopexin domain that regulates the binding of the enzymes to their substrates. MMPs are generally produced as inactive proenzymes (proMMPs). Latency is maintained by an interaction between the active site zinc atom and a conserved cysteine residue in the pro domain. Activation of proMMPs occurs when this interaction is disrupted, which may be achieved by the actions of other proteinases and oxidants in the extracellular space (the cysteine switch mechanism of activation of proMMPs [CitationMurphy et al 1999; CitationFu et al 2001]). Some MMPs are activated in the transgolgi by cleavage of the prodomain by furin, an intracellular serine proteinase (CitationImai et al 1996; CitationCao et al 2005). MMPs are generally synthesized de novo by cells activated by pro-inflammatory mediators or growth factors. However, PMN store preformed MMP-8, MMP-9, and MT6-MMP (MMP-25) in their cytoplasmic granules, from which the enzymes are released when PMN degranulate (CitationOwen and Campbell 1999). Macrophages express MMPs-1, -3, -7, -9, -12, and -14 (CitationShapiro et al 1991; CitationRajavashisth et al 1999), and lung epithelial cells and fibroblasts produce MMPs-2, -9, and -14. MMPs are subdivided into 6 groups based upon a similar domain organization and substrate specificity including: 1) the interstitial collagenases (MMPs-1, -8, and -13); 2) the gelatinases (MMPs-2 and -9); 3) the stromelysins (MMPs-3, -10, and -11); 4) matrilysin (MMP-7); 5) metalloelastase (MMP-12); and 6) membrane-type MMPs (MT-MMPs), which are integral membrane proteinases having either a transmembrane domain or a glycosylphosphatidyl-inositol anchor to the cell membrane (CitationSato et al 1994; CitationTakino et al 1995). The interstitial collagenases degrade interstitial collagens. The other subgroups have broader substrate specificities including denatured collagens (gelatins), basement membrane proteins, and pro-inflammatory mediators. MMPs-7, -9, and -12 also degrade elastin (CitationOwen and Campbell 1999).
ADAM
ADAMs are a family of type I transmembrane proteinases, so called because they contain a disintegrin and a metalloproteinase domain (CitationPrimakoff and Myles 2000). The metalloproteinase domain of ADAMs sheds membrane-anchored cytokines such as pro-tumor necrosis factor (TNF-α), other cytokines, growth factors, apoptosis ligands and receptors for these molecules from cell surfaces to regulate inflammation, apoptosis, and possibly fibrotic processes (CitationBlack et al 1997; CitationPrimakoff and Myles 2000; CitationBlack 2002). The disintegrin domain binds to integrins to regulate integrin-mediated cell adhesion and migration (CitationPrimakoff and Myles 2000).
MMPs are inhibited by α2-M and the four members of the tissue inhibitors of metalloproteinases family (TIMPs1-4), which are synthesized by connective tissue cells and leukocytes and form non-covalent complexes with MMPs (CitationWoessner Jr 1991; CitationMurphy and Docherty 1992). The inhibitors of ADAMs have not been fully elucidated, but ADAM-17 is inhibited by TIMP-3 but not TIMP-1 or -2 (CitationAmour et al 1998; CitationBlack 2004).
Cysteine proteinases
Cathepsins B, H, L, and S have been implicated in COPD (). Cathepsin S and L are potent elastases in vitro (CitationMason et al 1986; CitationShi et al 1992) and contribute to macrophage-mediated ECM degradation. The main inhibitors of cysteine proteinases are the cystatin superfamily, the kininogens, and α2-M (CitationHenskens et al 1996).
Roles of proteinases in COPD
Evidence for roles of proteinases in COPD comes from studies of purified proteinases, studies of clinical samples from COPD patients, and animal models of COPD.
In vitro studies of proteinases
Lung inflammation and airspace enlargement
NE, CG, PR3, and GRZ have the potential to promote lung inflammation in COPD patients, because they stimulate the release of pro-inflammatory mediators from airway epithelial cells and macrophages in vitro (CitationHubbard et al 1991; CitationBedard et al 1993). Proteinases can also proteolytically cleave mediators to alter their biologic activities (). MMPs-8 and -9 cleave and activate various chemokines in vitro (CitationVan Den Steen et al 2000; CitationBalbin et al 2003). ADAM-17 and several MMPs shed and activate membrane-associated pro-TNF-α from macrophage surfaces (CitationPrimakoff and Myles 2000; CitationBlack 2002; CitationChurg et al 2003a). NE, MMP-12, and MMP-9 cleave elastin, and MMPs cleave α1-PI, generating fragments of these two molecules that are chemotactic for inflammatory cells (CitationSenior et al 1980; CitationHunninghake et al 1981). Serine, metallo-, and cysteine proteinases acting together can degrade elastin, interstitial collagens, and basement membrane proteins in vitro (CitationOwen and Campbell 1999). All of these ECM proteins must be degraded when lung airspaces enlarge ().
Airway pathologies
NE, MMP-9, and ADAMs-10 and -17 increase epithelial cell expression of MUC5AC, a major mucin protein, by activating epithelial growth factor receptor (EGFR) through shedding of membrane – bound pro-transforming growth factor (TGF)-α. This releases soluble, active TGF-α, which activates the EGFR (CitationKohri et al 2002; CitationShao et al 2004; CitationDeshmukh et al 2005). NE, CG, and PR3 potently stimulate goblet cell degranulation (CitationSommerhoff et al 1990). Tissue kallikrein is a serine proteinase expressed by inflammatory cells and submucosal glands. It also stimulates mucin synthesis in airway epithelium in vitro by shedding and activating pro-EGF, another EGFR ligand (CitationCasalino-Matsuda et al 2006). NE also damages epithelial cells (CitationAmitani et al 1991) and inhibits ciliary beat frequency of lung epithelial cells (CitationSmallman et al 1984). Increased production and impaired clearance of mucus predispose COPD patients to recurrent bacterial airway infections, which amplify airway inflammation and injury ().
Plasmin, MMP-9, NE, and ADAMs may also induce sub-epithelial fibrosis in COPD airways, because they activate latent growth factors such as TGF-β (CitationTaipale et al 1992; CitationYu and Stamenkovic 2000; CitationChua et al 2007) and insulin-like growth factors in vitro (CitationFowlkes et al 1999; CitationMohan et al 2002) and these growth factors induce fibroblasts to synthesize and secrete interstitial collagens. However, it has not been determined whether these proteinases induce sub-epithelial fibrosis in the small airways of human COPD patients.
Studies of clinical samples from human COPD patients
In addition to the early observation that α1-PI-deficient patients have early-onset emphysema, elegant studies from CitationDamiano and colleagues (1986) further supported a role for NE in pulmonary emphysema. They localized NE bound to lung elastic fibers and showed that the amount of NE bound to lung elastin is strongly correlated with the degree of emphysematous change. Since then, additional studies have confirmed increased levels of NE in lung samples from COPD patients and demonstrated elevated levels of CG, PR3, uPA, and MMPs -1, -2, -8, -9, and -14 in various lung samples from smokers and COPD patients when compared with healthy subjects (CitationDamiano et al 1986; CitationReilly and Chapman Jr 1988; CitationAbboud et al 1998; CitationBetsuyaku et al 1996; CitationFinlay et al 1997; CitationBetsuyaku et al 1999; CitationHill et al 1999; CitationBetsuyaku et al 2000b; CitationCataldo et al 2000; CitationImai et al 2001; CitationBeeh et al 2003; CitationKang et al 2003).
Although most studies have implicated proteinases from inflammatory cells in COPD pathogenesis, proteinases produced by lung structural cells and immune cells also play important roles (). For example, cigarette smoke increases MMP production by lung epithelial cells (CitationImai et al 2001), and fibroblasts (CitationNing et al 2007). T lymphocytes from blood and BAL samples from COPD patients have increased levels of GRZ and perforin compared to samples from asymptomatic smokers and nonsmokers (CitationHodge et al 2006). Elevated levels of GRZ B in BAL samples from COPD patients are correlated with bronchial epithelial cell apoptosis, suggesting that GRZ B promotes epithelial cell death in the lung and contributes to airspace enlargement in COPD patients ().
Animal models of COPD
Animal models of COPD provide the strongest evidence for the roles of proteinases in COPD.
Acute cigarette smoke exposure models
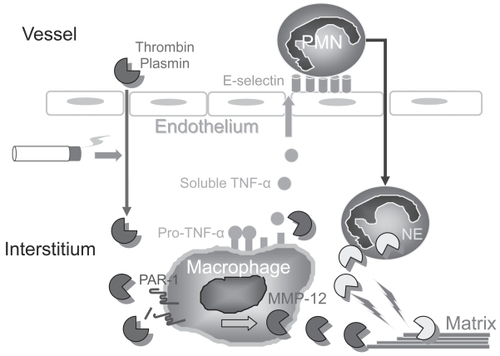
Acute exposure of mice to cigarette smoke for up for 30 days results in increases in lung PMN and macrophages and breakdown of lung collagen and elastin (CitationChurg et al 2002). Studies of mice genetically deficient in proteinases in these acute exposure models have identified critical roles for MMP-12 in regulating PMN influx and for thrombin and plasmin in regulating MMP-12 production (). Cigarette smoke acutely upregulates macrophage MMP-12 levels by injuring lung capillaries (CitationBurns et al 1989; CitationLi et al 1996), leading to leakage of thrombin and plasmin into the alveolar space. Thrombin and plasmin cleave, thereby activating protease-activated receptor -1 (PAR-1) on macrophages. Signaling through PAR-1 increases macrophage MMP-12 synthesis (CitationRaza et al 2000; CitationChurg et al 2007b). Macrophage-derived MMP-12 regulates PMN influx into the lung by shedding pro-TNF-α from activated macrophages, which likely up-regulates E-selectin expression on endothelial cells to promote PMN transendothelial migration (CitationChurg et al 2003a) and lung ECM degradation by PMN-derived serine proteinases (). It is noteworthy that delivering human α1-PI to mice acutely exposed to cigarette smoke prevents PMN influx and ECM destruction. This is probably due to α1-PI inhibiting both PMN serine proteinase-mediated ECM destruction and thrombin- or plasmin-induced increases in macrophage MMP-12 production (CitationChurg et al 2003b, Citation2007b).
Figure 2 Interactions between proteinases regulate inflammation and matrix destruction in mice acutely exposed to cigarette smoke. Cigarette smoke drives macrophage MMP-12 production, at least in part by inducing thrombin- and plasmin-mediated activation of protease-activated receptor-1 (PAR-1) on macrophages. MMP-12 stimulates PMN accumulation in the lung by shedding pro-TNF-α from the macrophage surface, generating soluble, active TNF-α. Active TNF-α stimulates PMN trans-endothelial migration by up-regulating endothelial E selectin expression. PMN proteinases, such as neutrophil elastase (NE), amplify macrophage MMP-12 mediated destruction of the lung ECM.
Chronic smoke exposure models
Exposure of WT mice to cigarette smoke for 3–6 months results in airspace enlargement, inflammation, and subepithelial fibrosis in the small airways, similar to that reported in human cigarette smokers (CitationHautamaki et al 1997; CitationMartin et al 2001). Studies of proteinase-deficient mice in this model have confirmed roles for MMP-12 and NE in regulating chronic lung inflammation and airspace enlargement () and for MMP-9 and/or MMP-12 in inducing subepithelial fibrosis in the small airways of smoke-exposed mice.
Figure 3 Interactions between proteinases regulate inflammation and ECM destruction in mice chronically exposed to cigarette smoke. Neutrophil elastase (NE) promotes inflammation and ECM destruction in mice chronically exposed to cigarette smoke by increasing the influx of PMN and monocytes into the lung (by unknown mechanisms), and by cleaving and inactivating TIMPs to promote MMP-12 mediated ECM degradation. MMP-12 amplifies NE-mediated lung inflammation and destruction by cleaving and inactivating α1-PI, the major inhibitor of NE in the lower respiratory tract. Fragments of elastin generated by MMP-12 (and possibly by NE) amplify MMP-12-mediated lung injury by stimulating the recruitment of blood monocytes into the lung.
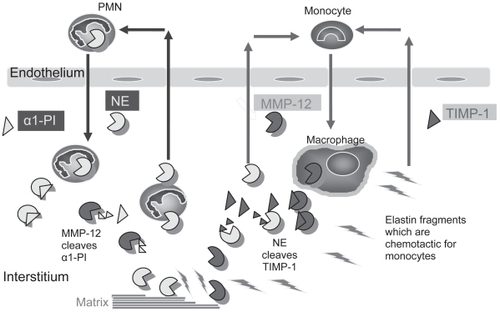
Mice deficient in MMP-12 (MMP-12−/− mice) exposed to cigarette smoke for 6 months are completely protected from developing increased lung macrophage counts and from developing airspace enlargement (CitationHautamaki et al 1997). MMP-12 degrades elastin and other ECM components to cause airspace enlargement (). The decreased macrophage accumulation in MMP-12−/− mice is due to the lack of MMP-12-mediated cleavage of elastin, which generates elastin fragments that are chemotactic for blood monocytes () (CitationHoughton et al 2006b). T lymphocyte products also play a critical role in driving MMP-12-mediated inflammation and airspace enlargement, since CD8+ T-cell-deficient (CD8−/−) mice have a blunted inflammatory response to cigarette smoke and fail to develop emphysema (CitationMaeno et al 2007). This is mediated by a CD8+ T cell product, IFN-γ inducible protein 10 (IP-10), which induces production of MMP-12 and degradation of the lung ECM ( and ). This process may also contribute to COPD pathogenesis in human subjects, since lung tissue from human COPD patients contains increased numbers of Th1 cells associated with increased levels of IP-10 and MMP-12 (CitationGrumelli et al 2004).
NE−/− mice are 60% protected from airspace enlargement and have decreased influx of PMN and monocytes into the lung compared to smoke-exposed WT mice (CitationShapiro et al 2003) (). NE likely contributes to airspace enlargement directly by degrading elastin and other ECM protein components of the alveolar walls (CitationShapiro et al 2003) (), but the mechanisms by which NE promotes lung inflammation are not clear.
When rodent airways are exposed acutely to cigarette smoke, increases in growth factor and collagen production are detectable within 2 h, and before inflammation occurs in the airway walls (CitationChurg et al 2006). This indicates that smoke directly promotes small airway subepithelial fibrosis and that smoke-induced inflammation and proteinase production are unnecessary for this process. However, in guinea pigs chronically exposed to cigarette smoke for up to 6 months, inflammatory cell MMPs amplify this process, since delivering a synthetic dual inhibitor of MMPs-9 and -12 to these animals significantly reduces small airway fibrosis (CitationChurg et al 2007a). Studies of MMP inhibitors in human COPD patients are thus warranted to determine whether these proteinases play important roles in this important pathology in humans as well as mice.
Transgenic murine models
Transgenic mice over-expressing MMP-1 in the lung develop enlarged airspaces (CitationD’Armiento et al 1992), which may either reflect abnormal alveolar development or destruction of mature interstitial collagens by MMP-1. Assessment of transgenic mice inducibly over-expressing cytokines in the adult lung have confirmed a role for immune-mediated inflammation in airspace enlargement. Adult transgenic mice over-expressing a Th1 cytokine (IFN-γ), a Th2 cytokine (IL-13), or a cytokine with Th1 and Th2 activities (IL-18) in airway epithelial cells spontaneously develop striking lung inflammation, increased lung levels of MMPs and cysteine proteinases, and airspace enlargement (CitationWang et al 2000; CitationZheng et al 2000; CitationKang et al 2007). In mice over-expressing IL-13, MMPs -9 and -12 play critical roles in promoting airspace enlargement, and MMP-12 also promotes inflammation and drives the increased expression of other MMPs in the lung (CitationLanone et al 2002). In transgenic mice over-expressing IFN-γ, cathepsin S stimulates lung epithelial apoptosis, lung inflammation, and airspace enlargement (CitationZheng et al 2005).
Alveolar septal cell apoptosis models of airspace enlargement
Apoptosis of alveolar septal cells (CitationAoshiba et al 2001) and leukocytes (CitationAoshiba et al 2001; CitationHodge et al 2005) occurs in the lungs of COPD patients, and apoptosis of the endothelial and epithelial cells that make up the alveolar walls contributes to the development of emphysema. Septal cell apoptosis and airspace enlargement in the absence of overt lung inflammation can be induced rapidly in experimental animals by: 1) pharmacologic blockade of vascular endothelial growth factor receptors in rodents (CitationKasahara et al 2000); and 2) transfection of murine alveolar epithelial cells with caspase-3, a pro-apoptotic cysteine proteinase (CitationAoshiba et al 2003). However, increased elastase activity due to acidic proteinase(s) is detected in BAL samples after transfection of alveolar epithelial cells with caspase-3 (CitationAoshiba et al 2003). Thus, proteinases released from dying structural cells may degrade the lung ECM, thereby acting synergistically with septal cell apoptosis to cause loss of alveolar units and airspace enlargement ().
Interactions between proteinases and other mediators and pathways in COPD
Interactions between different classes of proteinases and between proteinases and other molecules present in COPD lungs either amplify or inhibit proteinase production, lung inflammation, and airspace enlargement in COPD lungs.
Studies of the NE−/− and MMP-12−/− mice exposed chronically to cigarette smoke demonstrated interactions between these two classes of proteinases (), with MMP-12 cleaving and inactivating α1-PI to increase NE-mediated lung injury, and NE cleaving and inactivating TIMP-1 to amplify MMP-12-mediated lung destruction (CitationShapiro et al 2003). Proteinases also interact with ROS present in cigarette smoke itself and are generated by phagocytes activated by cigarette smoke. ROS activate proMMPs in vitro and have been thought to exacerbate lung inflammation and injury in COPD patients (CitationOwen 2005). Consistent with this hypothesis, mice transgenically over-expressing the antioxidant enzyme Cu-Zn superoxide dismutase in the lung are protected from developing chronic lung inflammation, increased lung MMP levels, and emphysema in response to intratracheal instillation of porcine pancreatic elastase, or chronic exposure to cigarette smoke (CitationForonjy et al 2006). However, mice deficient in a phagocyte-specific component of the NADPH oxidase, which generates superoxide anions (O2−), develop greater airspace enlargement in response to cigarette smoke than WT mice (CitationKassim et al 2005). This is due to ROS-mediated inactivation of MMPs via oxidative inactivation of residues in the catalytic domain of MMPs (CitationFu et al 2003a). Thus, phagocyte-derived O2− (and ROS derived from O2−) in COPD lungs may constrain rather than promote phagocyte MMP-mediated lung injury (CitationFu et al 2003b; CitationKassim et al 2005). This may be one reason that clinical trials have failed to demonstrate protective effects of antioxidant supplementation in COPD patients (CitationRahman and MacNee 1996).
Molecular mechanisms for proteinase-mediated lung injury in COPD
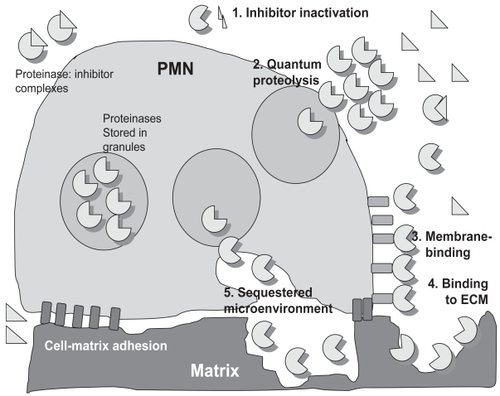
In order to contribute to pathologies in COPD, proteinases must overcome the effects of proteinase inhibitors, which are present at micromolar concentrations in extracellular fluids. Proteinases circumvent the effects of extracellular inhibitors by inactivating, evading, or overwhelming them ().
Figure 4 Mechanisms by which proteinases circumvent proteinase inhibitors in the extracellular space to cause lung injury in COPD. PMN store preformed proteinases within intracellular granules, and proteinases are released into the extracellular space when pro-inflammatory mediators induce PMN degranulation. Proteinases freely released by PMN are inhibited when they form complexes with extracellular inhibitors. However, proteinases can circumvent inhibitors by: 1) cleaving or degrading inhibitors; 2) being released at very high concentrations into the extracellular space, thereby overwhelming inhibitors; 3) binding to cell membranes in inhibitor-resistant forms; 4) binding to matrix substrates in inhibitor-resistant forms; or 5) being released into sequestered microenvironments formed by tight adhesion of PMN to ECM into which diffusion of large inhibitors is impaired.
Inactivation of proteinase inhibitors
Serpins can be cleaved and inactivated by MMPs (CitationDesrochers and Weiss 1988; CitationDesrochers et al 1991, Citation1992; CitationSires et al 1994; CitationGronski Jr et al 1997), NE (Cantin et al 1995), cathepsin B (CitationJohnson and Travis 1977), and bacterial proteinases (CitationSponer et al 1991). Serine proteinases cleave and inactivate TIMPs (CitationOkada et al 1988). Proteolytic inactivation of α1-PI and TIMP-1 by MMP-12 and NE occurs in the cigarette smoke exposure model of emphysema in mice (CitationShapiro et al 2003).
ROS present in cigarette smoke or released by leukocytes activated by smoke inactivate α2-M, and α1-PI, and SLPI in vitro by converting the methionine at the active sites of these inhibitors to methionine sulfoxide, which reduces their capacity to inhibit serine proteinases (CitationCarp and Janoff 1979, Citation1980a, Citation1980b; CitationReddy et al 1994). Whether oxidative inactivation of proteinase inhibitors occurs in COPD patients is controversial, since some studies have detected oxidized α1-PI in lung samples from COPD patients but others have not (CitationGadek et al 1979; CitationStone et al 1983; CitationAbboud et al 1985). Also, ROS can inactivate proteinases as outlined above. Global analysis of the oxidation state of proteinase inhibitors in lung samples from COPD patients may not accurately reflect events in cellular microenvironments. ROS are short-lived molecules and are active only at short distances from the cells generating them before they are inactivated by antioxidants. It is likely that ROS and proteinases released into microenvironments around activated leukocytes act synergistically to locally inactivate inhibitors (or proteinases) and promote (or reduce) extracellular proteolysis.
Evasion of inhibitors
Proteinases can evade inhibitors by being released into sequestered microenvironments, binding tightly to substrates, or binding to cell surfaces ().
Sequestered microenvironments
Integrin-mediated adhesion of inflammatory cells to matrix or to cells results in the formation of a “sealed” micro-environment, which prevents large inhibitors such as α1-PI (CitationCampbell and Campbell 1988) and α2-M (CitationWright and Silverstein 1984) from penetrating into zones of contact between the cells and their substrates ().
Tight binding of proteinases to substrates
NE binds very stably to elastin in an active form, and α1-PI and SLPI have reduced effectiveness against elastin-bound NE compared to soluble NE (CitationBruch and Bieth 1986; CitationMorrison et al 1990, Citation1999). Since NE is bound to interstitial elastin in human emphysematous lungs (CitationDamiano et al 1986), lung elastin-bound NE likely retains catalytic activity and contributes critically to destruction of elastin fibers in pulmonary emphysema (). MMPs-1, -2, and -9 bind to various ECM proteins, which may increase the retention, stability, and bioactivity of proteinases in the lung and facilitate their roles in extracellular proteolysis (CitationMurphy et al 1992; CitationAllan et al 1995).
Membrane binding of proteinases
MT-MMP and ADAMs are integral membrane proteinases, and some members of these families are resistant to inhibition by physiologic inhibitors. For example, ADAM-17 is resistant to inhibition by TIMPs-1 and -2 but not TIMP-3 (CitationAmour et al 1998), and MT1-MMP is resistant to inhibition by TIMP-1 but not TIMP-2 (CitationD’Ortho et al 1998). NE, CG, PR3, MMPs-8 and -9 (which lack transmembrane domains or glycosylphosphatidyl-inositol anchors) are also expressed on the surface of activated PMN (CitationOwen et al 1995a, Citation1995b, Citation2003, Citation2004; CitationOwen and Campbell 1998; CitationCampbell et al 2000) (). These surface-bound proteinases potently degrade lung ECM proteins and proteinase inhibitors and induce goblet cell degranulation (CitationTakeyama et al 1998; CitationOwen et al 1995b, Citation2003b, Citation2004). However, unlike the soluble enzymes, the membrane-bound forms of these proteinases are resistant to inhibition by physiologic inhibitors (CitationOwen et al 1995b, Citation2003, Citation2004; CitationOwen and Campbell 1998; CitationCampbell et al 2000). The inhibitor-resistance of membrane-bound NE is due to positive residues in NE binding to negatively charged sulfate groups in PMN plasma membrane proteoglycans (CitationCampbell and Owen 2007), but the mechanism underlying the resistance of other cell-surface proteinases to inhibition is not known. Whatever the mechanism involved, catalytically active but inhibitor-resistant membrane-bound proteinases are well equipped to play critical roles in pathologies in COPD patients.
Overwhelming of inhibitors
Proteinases may overwhelm inhibitors when massive quantities of enzymes are released from large numbers of inflammatory cells, or when high concentrations of proteinases are released from individual cells (quantum proteolysis).
Brisk influx of inflammatory cells
During acute exacerbations of COPD, there is brisk influx of inflammatory cells into the airways. Active forms of NE, MMP-8, and MMP-9 released from these cells are detectable in lung secretions from COPD patients (CitationBurnett et al 1987; CitationYoshioka et al 1995; CitationBetsuyaku et al 1999; CitationHill et al 1999). Macrophage clearance of PMN recruited into the lung can be impaired in COPD patients by several mechanisms. First, cigarette smoke impairs expression of recognition molecules for apoptotic PMN on the macrophage surface (CitationHodge et al 2007). Second, NE cleaves recognition molecules for apoptotic PMN from the macrophage surface (CitationVandivier et al 2002). Third, when PMN ingest Hemophilus influenzae, which frequently colonizes the respiratory tract of COPD patients, PMN necrosis is rapidly induced (CitationNaylor et al 2007). All of these processes hinder noninflammatory macrophage removal of PMN, instead promoting PMN necrosis and release of proteinases into the lung.
Quantum proteolysis and PiZZ α1-PI deficiency
NE is present at millimolar concentrations in each azurophil granule of PMN, which is more than 100-fold higher than the concentration of α1-PI in plasma (CitationLiou and Campbell 1995). The release of an azurophil granule into the extracellular space is thus accompanied by a transient burst of proteolytic activity (), which persists until the granule contents diffuse from this site, and the proteinase-inhibitor ratio falls below 1:1 (CitationLiou and Campbell 1995). Individuals with severe, inherited deficiency of α1-PI have severe reductions in plasma levels of α1-PI (less than 4 μM in PiZZ α1-PI deficient individuals versus ~30 μM in healthy PiMM individuals) due to loop sheet polymerization of PiZ mutant protein within hepatocytes, leading to reduced hepatocyte secretion of PiZ α1-PI (CitationLomas et al 1992). Quantum bursts of NE-mediated proteolytic activity associated with PMN migrating on ECM proteins are 10-fold larger in area and 4-fold longer in duration when PMN are bathed in serum from PiZZ patients compared to serum from healthy PiMM subjects (CitationCampbell et al 1999), due to defective confinement of PMN-derived NE-mediated ECM degradation. Other mechanisms leading to excessive ECM destruction and lung inflammation in patients with severe, inherited deficiency of α1-PI include the formation of polymers of PiZ α1-PI mutant proteins in the lung, which not only are ineffective inhibitors of NE, but also have chemotactic activity for PMN (CitationMahadeva et al 2005; CitationLomas 2006).
Potential for proteinase inhibition in COPD
Based upon the available evidence, strategies to directly inhibit proteinases or to decrease the lung proteinase burden by decreasing inflammatory cell influx into the lung may be effective in limiting proteinase-induced lung injury in COPD patients.
Direct proteinase inhibition
Supplementation with physiologic proteinase inhibitors
This strategy is effective in murine models of COPD and in human subjects with COPD secondary to α1-PI deficiency. Delivering α1-PI systemically or by the inhaled route to smoke-exposed mice inhibits smoke-induced lung inflammation and airspace enlargement (CitationChurg et al 2003b; CitationPemberton et al 2006). Alpha1-PI augmentation therapy is being used in the USA in α1-PI-deficient patients who have impaired lung function. Observational studies using this strategy confirm that it reduces bronchial inflammation, slows the rate of decline in lung function, increases quality-of-life scores, and decreases exacerbation frequency in α1-PI-deficient patients (CitationStockley et al 2002a; CitationJuvelekian and Stoller 2004).
Synthetic proteinase inhibitors
Synthetic inhibitors have several advantages over physiologic inhibitors, including their resistance to oxidative and proteolytic inactivation and their effectiveness against both soluble and membrane-bound forms of proteinases (CitationOwen et al 1995b, Citation2003, Citation2004). In animals exposed to cigarette smoke, or in transgenic mice over-expressing IL-13, delivering synthetic inhibitors of serine, metallo-, and cysteine proteinases by the systemic, oral, or inhaled routes blocks lung inflammation and airspace enlargement (CitationChurg et al 2002; CitationLanone et al 2002; CitationStockley et al 2002b; CitationWright et al 2002; CitationPemberton et al 2005). Daily oral delivery of synthetic MMP inhibitors not only prevents airspace enlargement in mice chronically exposed to cigarette smoke, but also prevents progression of lung inflammation and airspace enlargement if therapy is initiated after emphysema has been established (CitationMartin et al 2001). Synthetic inhibitors may also have potential in limiting the airflow obstruction produced by small airway fibrosis, since a synthetic compound that inhibits both MMP-9 and MMP-12 effectively blocks small airway fibrosis in cigarette smoke-exposed guinea pigs (CitationChurg et al 2007a).
Anti-inflammatory strategies
Approaches to reducing inflammatory cell recruitment into the lung and activation of inflammatory cells would not only reduce the lung burden of inflammatory cell-derived proteinases but also that of other pathogenetic molecules generated by inflammatory cells in COPD patients such as ROS and pro-inflammatory mediators. Inhibitors of phosphodiesterase E4 (PDE4), the major PDE isoenzyme in inflammatory cells, decrease inflammatory cell migration, activation, and release of proteinases in vitro. Roflumilast (a PDE4 inhibitor) also protects mice from cigarette-smoke induced lung inflammation and airspace enlargement (CitationMartorana et al 2005). Short-term clinical trials of phosphodiesterase E4 inhibitors in COPD patients have indicated that these inhibitors decrease lung inflammation, lung proteinases, and pro-inflammatory mediators, increase post bronchodilator forced expiratory volume in one second (CitationMartina et al 2006; CitationCalverley et al 2007; CitationGrootendorst et al 2007), and reduce the frequency of acute exacerbations (CitationMartina et al 2006; CitationCalverley et al 2007). Statins (hydroxymethylglutaryl CoA reductase inhibitors) have diverse anti-inflammatory effects and also represent a potential new approach to COPD. This is supported by a recent study showing that simvastatin reduces lung inflammation, airspace enlargement, and pulmonary hypertension in cigarette smoke-exposed rats (CitationLee et al 2005). Several recent retrospective analyses have reported reduced morbidity and mortality in COPD patients taking statins for cardiovascular disease (CitationGueders et al 2005; CitationMancini et al 2006).
Other potential approaches to reduce inflammatory cell influx into the lung include anti-oxidant supplementation, inhibiting the transcription factor NF-κB, which drives the production of several pro-inflammatory molecules causing inflammation in COPD lungs (CitationRetamales et al 2001; CitationSzulakowski et al 2006) and inhibitors of chemokine receptors (CitationDonnelly and Barnes 2006). Histone deacetylases, which are enzymes that switch off transcription of pro-inflammatory genes, are inactivated in COPD patients (CitationIto et al 2005) and represent another potential drug target in COPD patients.
Conclusions and future directions
There is now substantial evidence from animal model systems that proteinases make important contributions to pathologies in COPD and that proteinase inhibition and anti-inflammatory strategies effectively limit smoke-induced lung injury in mice. However, there are critical gaps in our knowledge about the roles of proteinases not only in pathogenesis of human COPD, but also in repair processes in the lung in COPD, and in lung biology in general.
It is important to note that murine model systems of COPD have limitations. Mice lack submucosal glands and do not develop mucus hypersecretion or acute exacerbations in the murine cigarette smoke exposure model. Mice also have fewer circulating PMN than humans and do not express MMP-1. The role of MMP-12 in human disease must be clarified. Early studies failed to detect increased expression of MMP-12 in lung samples (CitationFinlay et al 1997; CitationImai et al 2001), but more recent studies using other techniques have demonstrated increased levels of MMP-12 in human COPD (CitationGrumelli et al 2004; CitationMolet et al 2005; CitationWoodruff et al 2005; CitationDemedts et al 2006). Thus, PMN-derived serine proteinases and MMPs in addition to MMP-12 may be important in human COPD. The challenge for the future will be to determine which proteinases play critical roles not only in airspace enlargement but also in airway pathologies in human COPD patients.
The biologic roles of proteinases expressed in the lung have also not been fully elucidated. Evidence is accumulating that some proteinases have both beneficial as well as deleterious roles in the murine lung. NE plays critical roles in bacterial killing in mice (CitationBelaaouaj et al 1998), MMP-8 reduces lung inflammation (CitationOwen et al 2004; CitationGueders et al 2005), and MMP12 has anti-tumor activities (CitationHoughton et al 2006a). If these proteinases have similar beneficial activities in the human lung, this may limit the usefulness of inhibitors of these proteinases in COPD patients, who are at increased risk for developing respiratory tract infections and lung cancer (CitationSkillrud et al 1986). Little is also known about repair processes in the COPD lung in general, or whether proteinases participate in lung repair in COPD. Studies of MMP-9 deficient mice in bleomycin-mediated lung injury suggest that MMP-9 might play roles in epithelial repair processes in the injured lung (CitationBetsuyaku et al 2000a), and it is likely that other proteinases contribute to repair of the injured lung in COPD patients.
There have been no long-term clinical trials of synthetic proteinase inhibitors or anti-inflammatory agents in COPD patients due mainly to the high cost of such trials. In addition, we currently lack knowledge about appropriate biomarkers for studying the effectiveness of new treatment strategies in COPD patients. Nevertheless, based upon the evidence available, randomized clinical trails to test the safety and efficacy of proteinase inhibitors and anti-inflammatory agents are justified in COPD patients.
Abbreviations
| ADAM | = | proteinase with a metalloproteinase and a disintegrin domain |
| α1-PI | = | α1-proteinase inhibitor |
| α1-Ach | = | α1-antichymotrypsin |
| α2-M | = | α2-macroglobulin |
| CG | = | cathepsin G |
| COPD | = | chronic obstructive pulmonary disease |
| ECM | = | extracellular matrix |
| EGFR | = | epithelial growth factor receptor |
| GRZ | = | granzyme |
| IGF | = | insulin like growth factor |
| IGFBP | = | insulin like growth factor binding protein |
| MMP | = | matrix metalloproteinase |
| MT-MMP | = | membrane-type MMP |
| NE | = | neutrophil elastase |
| MNP | = | mononuclear phagocyte |
| PAI | = | plasminogen activator inhibitor |
| PAR | = | proteinase activated receptor |
| PDE | = | phosphodiesterase |
| PMN | = | polymorphonuclear neutrophil |
| PR3 | = | proteinase 3 |
| ROS | = | reactive oxygen species |
| TGF | = | transforming growth factor |
| TIMP | = | tissue inhibitor of metalloproteinases |
| TNF-α | = | tumor necrosis factor-α |
| uPA | = | urokinase type plasminogen activator |
Acknowledgments
Dr Owen is supported by Public Health Services, NHLBI RO1 # HL63137, NHLBI RO1 # HL086814 and by a Clinical Innovator award from the Flight Attendants Medical Research Institute (FAMRI). There are no other conflicts of interest to report.
References
- AbboudRTFeraTRichterA1985Acute effect of smoking on the functional activity of alpha1-protease inhibitor in bronchoalveolar lavage fluidARRD1317985
- AbboudRTOfulueAFSansoresRH1998Relationship of alveolar macrophage plasminogen activator and elastase activities to lung function and CT evidence of emphysemaChest1131257639596303
- AllanJADochertyAJPBarkerPJ1995Binding of gelatinases A and B to type-1 collagen and other matrix componentsBiochem J3092993067619071
- AmitaniRWilsonRRutmenA1991Effects of human neutrophil elastase and Pseudomonas aeruginosa proteinases on human respiratory epitheliumAm J Respir Cell Mol Biol426321898852
- AmourASlocombePMWebsterA1998TNF-alpha converting enzyme (TACE) is inhibited by TIMP-3FEBS Lett43539449755855
- AoshibaKTamaokiJNagaiA2001Acute cigarette smoke exposure induces apoptosis of alveolar macrophagesAm J Physiol Lung Cell Mol Physiol2816L1392L140111704535
- AoshibaKYokohoriNNagaiA2003Alveolar wall apoptosis causes lung destruction and emphysematous changesAm J Respir Cell Mol Biol2855556212707011
- BalbinMFueyoATesterAM2003Loss of collagenase-2 confers increased skin tumor susceptibility to male miceNat Genet353252714517555
- BedardMMcClureCDSchillerNL1993Release of interleukin-8, interleukin-6, and colony-stimulating factors by upper airway epithelial cells: Implications for cystic fibrosisAm J Respir Cell Mol Biol9455627691110
- BeehKMBeierJKornmannO2003Sputum matrix metalloproteinase-9, tissue inhibitor of metalloprotinease-1, and their molar ratio in patients with chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis and healthy subjectsRespir Med976634912814147
- BelaaouajAMcCarthyRBaumannM1998Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsisNat Med461589585238
- BetsuyakuTFukudaYParksWC2000aGelatinase B is required for alveolar bronchiolization after intratracheal bleomycinAm J Pathol1575253510934155
- BetsuyakuTNishimuraMTakeyabuK2000bDecline in FEV(1) in community-based older volunteers with higher levels of neutrophil elastase in bronchoalveolar lavage fluidRespiration6732617
- BetsuyakuTNishimuraMTakeyabuK1999Neutrophil granule proteins in bronchoalveolar lavage fluid from subjects with subclinical emphysemaAm J Respir Crit Care Med15919859110351949
- BetsuyakuTNishimuraMYoshiokaA1996Neutrophil elastase and elastin-derived peptides in BAL fluid and emphysematous changes on CT scansNihon Kyobu Shikkan Gakkai Zasshi34Suppl69749216188
- BlackRA2002Tumor necrosis factor-alpha converting enzymeInt J Biochem Cell Biol3411511733179
- BlackRA2004TIMP3 checks inflammationNat Genet369934515340428
- BlackRARauchCTKozloskyCJ1997A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cellsNature385729339034190
- BruchMBiethJG1986Influence of elastin on the inhibition of leucocyte elastase by alpha1-proteinase inhibitor and bronchial inhibitor. Potent inhibition of elastin-bound elastase by bronchial inhibitorBiochem J238269733492198
- BurnettDAffordSCCampbellEJ1987Characterization of elastase activity in bronchoalveolar lavage samplesARRD135A507
- BurnsARHosfordSPDunnLA1989Respiratory epithelial permeability after cigarette smoke exposure in guinea pigsJ Appl Physiol66521091162473059
- CalverleyPMSanchez-TorilFMcIvorA2007Effect of 1-year treatment with roflumilast in severe chronic obstructive pulmonary diseaseAm J Respir Crit Care Med17621546117463412
- CampbellEJCampbellMA1988Pericellular proteolysis by neutrophils in the presence of proteinase inhibitors: Effects of substrate opsonizationJ Cell Biol106667763279049
- CampbellEJCampbellMABoukedesSS1999Quantum proteolysis by neutrophils: implications for pulmonary emphysema in alpha1-antitrypsin deficiencyJCI10417990
- CampbellEJCampbellMAOwenCA2000Bioactive proteinase 3 on the cell surface of human neutrophils: quantification, catalytic activity, and susceptibility to inhibitionJ Immunol16533667410975855
- CampbellEJOwenCA2007The sulfate groups of chondroitin sulfate- and heparan sulfate-containing proteoglycans in neutrophil plasma membranes are novel binding sites for human leukocyte elastase and cathepsin GJ Biol Chem28219146455417384412
- CantinABilodeauGBeginR1989Granulocyte elastase-mediated proteolysis of alpha1-antitrypsin in cystic fibrosis bronchopulmonary secretionsPediatr Pulmonol71272788857
- CaoJRehemtullaAPavlakiM2005Furin directly cleaves proMMP-2 in the trans-Golgi network resulting in a nonfunctioning proteinaseJ Biol Chem28012109748015637056
- CarpHJanoffA1979In vitro suppression of serum elastase-inhibitory capacity by reactive oxygen species generated by phagocytosing poly-morphonuclear leukocytesJCI637937220283
- CarpHJanoffA1980aInactivation of bronchial mucous proteinase inhibitor by cigarette smoke and phagocyte-derived oxidantsExp Lung Res1225377018895
- CarpHJanoffA1980bPotential mediator of inflammation: Phagocyte-derived oxidants suppress the elastase-inhibitory capacity of alpha1-proteinase inhibitor in vitroJCI66987956253528
- CarrellRW1986Alpha1-antitrypsin: Molecular pathology, leukocytes, and tissue damageJCI781427313537008
- Casalino-MatsudaSMMonzonMEFortezaRM2006Epidermal growth factor receptor activation by epidermal growth factor mediates oxidant-induced goblet cell metaplasia in human airway epitheliumAm J Respir Cell Mol Biol3455819116424381
- CataldoDMunautCNoelA2000MMP-2- and MMP-9-linked gelatinolytic activity in the sputum from patients with asthma and chronic obstructive pulmonary diseaseInt Arch Allergy Immunol12332596711112863
- ChuaFDunsmoreSEClingenPH2007Mice lacking neutrophil elastase are resistant to bleomycin-induced pulmonary fibrosisAm J Pathol1701657417200183
- ChurgATaiHCoulthardT2006Cigarette smoke drives small airway remodeling by induction of growth factors in the airway wallAm J Respir Crit Care Med1741213273417008639
- ChurgAWangRWangX2007aEffect of an MMP-9/MMP-12 inhibitor on smoke-induced emphysema and airway remodelling in guinea pigsThorax62870613
- ChurgAWangRDTaiH2003aMacrophage metalloelastase mediates acute cigarette smoke-induced inflammation via tumor necrosis factor-alpha releaseAm J Respir Crit Care Med167810839
- ChurgAWangRDXieC2003balpha-1-Antitrypsin ameliorates cigarette smoke-induced emphysema in the mouseAm J Respir Crit Care Med1682199207
- ChurgAWangXWangRD2007bAlpha1-antitrypsin suppresses TNF-alpha and MMP-12 production by cigarette smoke-stimulated macrophagesAm J Respir Cell Mol Biol37214451
- ChurgAZayKShayS2002Acute cigarette smoke-induced connective tissue breakdown requires both neutrophils and macrophage metalloelastase in miceAm J Respir Cell Mol Biol2733687412204900
- D’ArmientoJDalalSSOkadaY1992Collagenase expression in the lungs of transgenic mice causes pulmonary emphysemaCell71955611458541
- D’OrthoMPStantonHButlerM1998MT1-MMP on the cell surface causes focal degradation of gelatin filmsFEBS Lett421215964
- DamianoVVTsangAKucichU1986Immunolocalization of elastase in human emphysematous lungsJCI78482933525610
- DemedtsIKMorel-MonteroALebecqueS2006Elevated MMP-12 protein levels in induced sputum from patients with COPDThorax61319620116308335
- DeshmukhHSCaseLMWesselkamperSC2005Metalloproteinases mediate mucin 5AC expression by epidermal growth factor receptor activationAm J Respir Crit Care Med17143051415531749
- DesrochersPEJeffreyJJWeissSJ1991Interstitial collagenase (matrix metalloproteinase-1) expresses serpinase activityJCI872258651645757
- DesrochersPEMookhtiarKVan WartHE1992Proteolytic inactivation of alpha 1-proteinase inhibitor and alpha 1-antichymo-trypsin by oxidatively activated human neutrophil metalloproteinasesJ Biol Chem2675005121311327
- DesrochersPEWeissSJ1988Proteolytic inactivation of alpha-1-proteinase inhibitor by a neutrophil metalloproteinaseJCI811646503259248
- Di StefanoATuratoGMaestrelliP1996Airflow limitation in chronic bronchitis is associated with T-lymphocyte and macrophage infiltration of the bronchial mucosaAm J Respir Crit Care Med1532629328564109
- DonnellyLEBarnesPJ2006Chemokine receptors as therapeutic targets in chronic obstructive pulmonary diseaseTrends Pharmacol Sci27105465316911834
- FinlayGAO’DriscollLRussellKJ1997Matrix metalloproteinase expression and production by alveolar macrophages in emphysema.Am J Respir Crit Care Med15624079230755
- ForonjyRFMirochnitchenkoOPropokenkoO2006Superoxide dismutase expression attenuates cigarette smoke- or elastase-generated emphysema in miceAm J Respir Crit Care Med17366233116387805
- FowlkesJLSerraDMNagaseH1999MMPs are IGFBP-degrading proteinases: implications for cell proliferation and tissue growthAnn N Y Acad Sci878696910415811
- FuXKassimSYParksWC2001Hypochlorous acid oxygenates the cysteine switch domain of promatrilysin (MMP-7). A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidaseJ Biol Chem27644412798711533038
- FuXKassimSYParksWC2003bHypochlorous acid generated by myeloperoxidase modifies adjacent tryptophan and glycine residues in the catalytic domain of matrix metalloproteinase-7 (matrilysin): an oxidative mechanism for restraining proteolytic activity during inflammationJ Biol Chem27831284039
- FuXKassimSYParksWC2003aHypochlorous acid generated by myeloperoxidase modifies adjacent tryptophan and glycine residues in the catalytic domain of matrix metalloproteinase-7 (matrilysin): an oxidative mechanism for restraining proteolytic activity during inflammationJ Biol Chem27831284039
- GadekJEFellsGACrystalRG1979Cigarette smoking induces functional antiprotease deficiency in the lower respiratory tract of humansScience20613156316188
- Granelli-PepernoAVassalliJ-DReichE1977Secretion of plasminogen activator by human polymorphonuclear leukocytesJ Exp Med1461693706200699
- GronskiTJJrMartinRLKobayashiDK1997Hydrolysis of a broad spectrum of extracellular matrix proteins by human macrophage elastaseJ Biol Chem27212189949115292
- GrootendorstDCGauwSAVerhooselRM2007The PDE4 inhibitor roflumilast reduces sputum neutrophil and eosinophil numbers in patients with COPDThorax621081717573446
- GrossPPfitzerEATolkerE1965Experimental emphysema. Its production with papain in normal and silicotic ratsArch Environ Health1150814312390
- GrumelliSCorryDBSongLZ2004An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysemaPLoS Med11e815526056
- GuedersMMBalbinMRocksN2005Matrix metalloproteinase-8 deficiency promotes granulocytic allergen-induced airway inflammationJ Immunol175425899716081833
- HautamakiRDKobayashiDKSeniorRM1997Requirement for macrophage elastase for cigarette smoke-induced emphysema in miceScience277200249302297
- HenskensYMCVeermanECINieuw AmerongenAVN1996Cystatins in health and diseaseBiol Chem Hoppe-Seyler37771868868064
- HillATBayleyDStockleyRA1999The interrelationship of sputum inflammatory markers in patients with chronic bronchitisAm J Respir Crit Care Med1603893810471615
- HodgeSHodgeGAhernJ2007Smoking alters alveolar macrophage recognition and phagocytic ability: Implications in COPDAm J Respir Cell Mol Biol377485517630319
- HodgeSHodgeGHolmesM2005Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessationEur Respir J2534475415738287
- HodgeSHodgeGNairnJ2006Increased airway granzyme b and perforin in current and ex-smoking COPD subjectsCOPD341798717361498
- HoggJCChuFUtokaparchS2004The nature of small-airway obstruction in chronic obstructive pulmonary diseaseN Engl J Med3502626455315215480
- HoughtonAMGrisolanoJLBaumannML2006aMacrophage elastase (matrix metalloproteinase-12) suppresses growth of lung metastasesCancer Res6612614955
- HoughtonAMQuinteroPAPerkinsDL2006bElastin fragments drive disease progression in a murine model of emphysemaJ Clin Invest11637539
- HubbardRCFellsGGadekJ1991Neutrophil accumulation in the lung in α1-antitrypsin deficiency. Spontaneous release of leukotriene B4 by alveolar macrophagesJCI8889171653278
- HunninghakeGWDavidsonJMRennardS1981Elastin fragments attract macrophage precursors to diseased sites in pulmonary emphysemaScience21292577233186
- ImaiKDalalSSChenES2001Human collagenase (matrix metal-loproteinase-1) expression in the lungs of patients with emphysemaAm J Respir Crit Care Med1633Pt 17869111254539
- ImaiKOhuchiEAokiT1996Membrane-type matrix metalloproteinase 1 is a gelatinolytic enzyme and is secreted in a complex with tissue inhibitor of metalloproteinases 2Cancer Res56122707108665498
- ItoKItoMElliottWM2005Decreased histone deacetylase activity in chronic obstructive pulmonary diseaseN Engl J Med3521919677615888697
- JohnsonDTravisJ1977Inactivation of human α1 proteinase inhibitor by thiol proteinasesBiochem J16363941301739
- JuvelekianGSStollerJK2004Augmentation therapy for alpha(1)-antitrypsin deficiencyDrugs641617435615301559
- KangMJHomerRJGalloA2007IL-18 is induced and IL-18 receptor alpha plays a critical role in the pathogenesis of cigarette smoke-induced pulmonary emphysema and inflammationJ Immunol178319485917237446
- KangMJOhYMLeeJC2003Lung matrix metalloproteinase-9 correlates with cigarette smoking and obstruction of airflowJ Korean Med Sci186821714676438
- KaoRCWehnerNGSkubitzKM1988Proteinase 3. A distinct human polymorphonuclear leukocyte proteinase that produces emphysema in hamstersJCI821963733198760
- KarlinskyJFredetteJDavidovitsG1983The balance of lung connective tissue elements in elastase-induced emphysemaJ Lab Clin Med1022151626553073
- KasaharaYTuderRMCoolCD2001Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysemaAm J Respir Crit Care Med1633Pt 17374411254533
- KasaharaYTuderRMTaraseviciene-StewartL2000Inhibition of VEGF receptors causes lung cell apoptosis and emphysemaJ Clin Invest106111311911104784
- KassimSYFuXLilesWC2005NADPH oxidase restrains the matrix metalloproteinase activity of macrophagesJ Biol Chem2803430201515983040
- KohriKUekiIFNadelJA2002Neutrophil elastase induces mucin production by ligand-dependent epidermal growth factor receptor activationAm J Physiol Lung Cell Mol Physiol2833L531L4012169572
- LanoneSZhengTZhuZ2002Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and -12 in IL-13-induced inflammation and remodelingJ Clin Invest11044637412189240
- LaurellC-BErikssonS1963The electrophoretic alpha-1-globulin pattern of serum in alpha-1-antitrypsin deficiencyScand J Clin Lab Invest1513240
- LeeJHLeeDSKimEK2005Simvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungsAm J Respir Crit Care Med17289879316002570
- LiXYRahmanIDonaldsonK1996Mechanisms of cigarette smoke induced increased airspace permeabilityThorax515465718711672
- LiouTGCampbellEJ1995Non-isotropic enzyme-inhibitor interactions: A novel non-oxidative mechanism for quantum proteolysis by human neutrophilsBiochemistry341617178519774
- LomasDA2006Parker B. Francis lectureship. Antitrypsin deficiency, the serpinopathies, and chronic obstructive pulmonary diseaseProc Am Thorac Soc3649950116921127
- LomasDAEvansDLFinchJT1992The mechanism of Z alpha 1-antitrypsin accumulation in the liverNature357637960571608473
- MaenoTHoughtonAMQuinteroPA2007CD8+ T Cells are required for inflammation and destruction in cigarette smoke-induced emphysema in miceJ Immunol178128090617548647
- MahadevaRAtkinsonCLiZ2005Polymers of Z alpha1-antitrypsin co-localize with neutrophils in emphysematous alveoli and are chemotactic in vivoAm J Pathol16623778615681822
- ManciniGBEtminanMZhangB2006Reduction of morbidity and mortality by statins, angiotensin-converting enzyme inhibitors, and angiotensin receptor blockers in patients with chronic obstructive pulmonary diseaseJ Am Coll Cardiol471225546016781387
- MartinRLShapiroSDTongSEHanselTBP2001Macrophage Metalloelastase InhibitorsNew Drugs for Asthma, Allergy and COPDBasel KargerProg Respir Res17780
- MartinaSDIsmailMSVestaKS2006Cilomilast: orally active selective phosphodiesterase-4 inhibitor for treatment of chronic obstructive pulmonary diseaseAnn Pharmacother40101822816985092
- MartoranaPABeumeRLucattelliM2005Roflumilast fully prevents emphysema in mice chronically exposed to cigarette smokeAm J Respir Crit Care Med17278485315961691
- MasonRWJohnsonDABarrettAJ1986Elastinolytic activity of human cathepsin LBiochem J23392573518704
- MohanSThompsonGRAmaarYG2002ADAM-9 is an insulin-like growth factor binding protein-5 protease produced and secreted by human osteoblastsBiochemistry41511539440312484779
- MoletSBelleguicCLenaH2005Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary diseaseInflamm Res54131615723202
- MorrisonHMWelgusHGOwenCA1999Interaction between leukocyte elastase and elastin: quantitative and catalytic analysesBiochim Biophys Acta14301799010082946
- MorrisonHMWelgusHGStockleyRA1990Inhibition of human leukocyte elastase bound to elastin: Relative ineffectiveness and two mechanisms of inhibitory activityAm J Respir Cell Mol Biol226392310584
- MurphyGAllanJAWillenbrockF1992The role of the C-terminal domain in collagenase and stromelysin specificityJ Biol Chem267961281315762
- MurphyGDochertyAJP1992The matrix metalloproteinases and their inhibitorsAm J Respir Cell Mol Biol712051497900
- MurphyGStantonHCowellS1999Mechanisms for pro matrix metalloproteinase activationAPMIS1071384410190278
- NaylorEJBakstadDBiffenM2007Haemophilus influenzae induces neutrophil necrosis: a role in chronic obstructive pulmonary disease?Am J Respir Cell Mol Biol3721354317363778
- NingWDongYSunJ2007Cigarette smoke stimulates matrix metalloproteinase-2 activity via EGR-1 in human lung fibroblastsAm J Respir Cell Mol Biol3644809017099140
- OkadaYWatanabeSNakanishiI1988Inactivation of tissue inhibitor of metalloproteinases by neutrophil elastase and other serine proteinasesFEBS-Lett229157603162216
- OwenCA2005Proteinases and oxidants as targets in the treatment of chronic obstructive pulmonary diseaseProc Am Thorac Soc243738516267366
- OwenCACampbellEJ1998Angiotensin II generation at the cell surface of activated neutrophils: Novel cathepsin G-mediated catalytic activity that is resistant to inhibitionJ Immunol1601436439570564
- OwenCACampbellEJ1999The cell biology of leukocyte-mediated proteolysisJ Leukoc Biol651375010088596
- OwenCACampbellMABoukedesSS1995aInducible binding of cathepsin G to the cell surface of neutrophils: A mechanism for mediating extracellular proteolytic activity of cathepsin GJ Immunol1555803107499869
- OwenCACampbellMABoukedesSS1994A discrete subpopulation of human monocytes expresses a neutrophil-like pro-inflammatory (P) phenotypeAm J Physiol (Lung Cell Mol Physiol 11)267L775L785
- OwenCACampbellMASannesPL1995bCell-surface-bound elastase and cathepsin G on human neutrophils. A novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinasesJ Cell Biol131775897593196
- OwenCAHuZBarrickB2003Inducible expression of tissue inhibitor of metalloproteinases-resistant matrix metalloproteinase-9 on the cell surface of neutrophilsAm J Resp Cell Mol Biol2928394
- OwenCAHuZLopez-OtinC2004Membrane-bound matrix metalloproteinase-8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase-resistant collagenase and serpinaseJ Immunol17212779180315187163
- PembertonPACantwellJSKimKM2005An inhaled matrix metalloprotease inhibitor prevents cigarette smoke-induced emphysema in the mouseCOPD233031017146995
- PembertonPAKobayashiDWilkBJ2006Inhaled recombinant alpha 1-antitrypsin ameliorates cigarette smoke-induced emphysema in the mouseCOPD32101817175673
- PrimakoffPMylesDG2000The ADAM gene family: surface proteins with adhesion and protease activityTrends Genet16283710652535
- RahmanIMacNeeW1996Role of oxidants/antioxidants in smoking-induced lung diseasesFree Radic Biol Med215669818891669
- RajavashisthTBXuXPJovingeS1999Membrane type 1 matrix metalloproteinase expression in human atherosclerotic plaques: evidence for activation by proinflammatory mediatorsCirculation99243103910377072
- RazaSLNehringLCShapiroSD2000Proteinase activated receptor-1 regulation of macrophage elastase secretion by serine proteinasesJ Biol Chem52412435010993890
- ReddyVYDesrochersPEPizzoSV1994Oxidative dissociation of human alpha 2-macroglobulin tetramers into dysfunctional dimersJ Biol Chem2694683917508448
- ReillyJJChapmanHAJr1988Association between alveolar macrophage plasminogen activator activity and indices of lung function in young cigarette smokersARRD138614228
- RetamalesIElliottWMMeshiB2001Ampification of inflammation in emphysema and its association with latent adenoviral infectionAm J Respir Crit Care Med1644697311500352
- SaettaM1999Airway inflammation in chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1605Pt 2S17S2010556163
- SakselaORifkinDB1988Cell-associated plasminogen activation: Regulation and physiological functionsAnn Rev Cell Biol4931263143380
- SatoHTakinoTOkadaY1994A matrix metalloproteinase expressed on the surface of invasive tumour cellsNature3706158015608
- SeniorRMGriffinGLMechamRP1980Chemotactic activity of elastin-derived peptidesJ Clin Invest664859626903189
- SeniorRMTegnerHKuhnCIII1977The induction of pulmonary emphysema with human leukocyte elastaseARRD11646975
- ShaoMXNakanagaTNadelJA2004Cigarette smoke induces MUC5AC mucin overproduction via tumor necrosis factor-alpha-converting enzyme in human airway epithelial (NCI-H292) cellsAm J Physiol Lung Cell Mol Physiol2872L420L2715121636
- ShapiroSDCampbellEJSeniorRM1991Proteinases secreted by human mononuclear phagocytesJ Rheumatol27958
- ShapiroSDGoldsteinNMHoughtonAM2003Neutrophil elastase contributes to cigarette smoke-induced emphysema in miceAm J Pathol163623293514633606
- ShiG-PMungerJSMearaJP1992Molecular cloning and expression of human alveolar macrophage cathepsin S, an elastinolytic cysteine proteaseJ Biol Chem2677258621373132
- SiresUIMurphyGWelgusHG1994Matrilysin is much more efficient than other metalloproteinases in the proteolytic inactivation of alpha 1-antitrypsinBiochem Biophys Res Commun204613207980522
- SkillrudDMOffordKPMillerRD1986Higher risk of lung cancer in chronic obstructive pulmonary disease. A prospective, matched, controlled studyAnn Intern Med105450373752756
- SmallmanLAHillSLStockleyRA1984Reduction of ciliary beat frequency in vitro by sputum from patients with bronchiectasis: A serine proteinase effectThorax3966376382675
- SmythMJO’ConnorMDTrapaniJA1996Granzymes: a variety of serine protease specificities encoded by genetically distinct subfamiliesJ Leukoc Biol605555628929545
- SommerhoffCPNadelJABasbaumCB1990Neutrophil elastase and cathespin G stimulate secretion from cultured bovine airway gland serous cellsJCI858629
- SponerMNickH-PSchnebliH-P1991Different susceptibility of elastase inhibitors to inactivation by proteinases from Staphylococcus aureus and Pseudomonas aeruginosaBiol Chem Hoppe-Seyler372963701686554
- StockleyRABayleyDLUnsalI2002aThe effect of augmentation therapy on bronchial inflammation in alpha1-antitrypsin deficiencyAm J Respir Crit Care Med1651114948
- StockleyRABayleyDLUnsalI2002bThe effect of augmentation therapy on bronchial inflammation in alpha1-antitrypsin deficiencyAm J Respir Crit Care Med1651114948
- StonePJCaloreJDMcGowanSE1983Functional alpha-1-protease inhibitor in the lower respiratory tract of cigarette smokers is not decreasedScience221118796612333
- SzulakowskiPCrowtherAJJimenezLA2006The effect of smoking on the transcriptional regulation of lung inflammation in patients with chronic obstructive pulmonary diseaseAm J Respir Crit Care Med1741415016574938
- TaipaleJKoliKKeski-OjaJ1992Release of transforming growth factor-beta 1 from the pericellular matrix of cultured fibroblasts and fibrosarcoma cells by plasmin and thrombinJ Biol Chem2673525378841281156
- TakeyamaKAgustiCUekiI1998Neutrophil-dependent goblet cell degranulation: role of membrane-bound elastase and adhesion moleculesAm J Physiol275L294L3029700090
- TakinoTHSatoHShinagawaA1995Identification of the second membrane-type matrix metalloproteinase (MT-MMP-2) gene from a human placenta cDNA library. MT-MMPs form a unique membrane-type subclass in the MMP family.J Biol Chem27023013207559440
- TuratoGZuinRSaettaM2001Pathogenesis and pathology of COPDRespiration6821172811287822
- Van Den SteenPEProostPWuytsA2000Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-α and leaves RANTES and MCP-2 intactBlood9626738111023497
- VandivierRWFadokVAHoffmannPR2002Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasisJ Clin Invest10956617011877474
- VassalliJ-DSappinoAPBelinD1991The plasminogen activator/plasmin systemJCI881067721833420
- WangZZhengTZhuZ2000Interferon gamma induction of pulmonary emphysema in the adult murine lungJ Exp Med19211158760011104801
- WoessnerJFJr1991Matrix metalloproteinases and their inhibitors in connective tissue remodelingFASEB J52145541850705
- WoodruffPGKothLLYangYH2005A distinctive alveolar macrophage activation state induced by cigarette smokingAm J Respir Crit Care Med1721113839216166618
- WrightJLFarmerSGChurgA2002Synthetic serine elastase inhibitor reduces cigarette smoke-induced emphysema in guinea pigsAm J Respir Crit Care Med16679546012359653
- WrightSDSilversteinSC1984Phagocytosing macrophages exclude proteins from zones of contact with targetsNature309359616374464
- WuKUranoTIharaH1995The cleavage and inactivation of plasminogen activator inhibitor type 1 by neutrophil elastase: the evaluation of its physiologic relevance in fibrinolysisBlood861056617620159
- YoshiokaABetsuyakuTNishimuraM1995Excessive neutrophil elastase in bronchoalveolar lavage fluid in subclinical emphysemaAm J Respir Crit Care Med1526Pt 12127328520785
- YuQStamenkovicI2000Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesisGenes Dev1421637610652271
- ZhengTKangMJCrothersK2005Role of cathepsin S-dependent epithelial cell apoptosis in IFN-gamma-induced alveolar remodeling and pulmonary emphysemaJ Immunol1741281061515944319
- ZhengTZhuZWangZ2000Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysemaJ Clin Invest106910819311067861