Abstract
The challenge posed by resistance among Gram-positive bacteria, epitomized by methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus (VRE) and vancomycin-intermediate and -resistant S. aureus (VISA and VRSA) is being met by a new generation of antimicrobials. This review focuses on the new β-lactams with activity against MRSA (ceftobiprole and ceftaroline) and on the new glycopeptides (oritavancin, dalbavancin, and telavancin). It will also consider the role of vancomycin in an era of existing alternatives such as linezolid, daptomycin and tigecycline. Finally, compounds in early development are described, such as iclaprim, friulimicin, and retapamulin, among others.
Introduction
Bacterial resistance to antibiotics certainly does not represent a novel event. Since the introduction of benzylpenicillins (), it has been observed that given enough time, bacteria will develop resistance to virtually any antibiotic introduced into clinical practice (). Currently, antibiotic resistance largely defines the epidemiology and pharmacologic therapy of infectious diseases. β-lactam resistant Gram-positive bacteria provide the best illustration of this phenomenon. The “persistent pathogen”, Staphylococcus aureus, is even more formidable since it manifested resistance to methicillin and other semi-synthetic penicillins, giving rise to the acronym “MRSA” (methicillin resistant S. aureus) (CitationSabath and Finland 1962). Presently, and perhaps in a more virulent fashion due to its association with Panton-Valentine leucocidin (PVL, an exotoxin active against neutrophils), MRSA has been implicated in a surge of community-acquired infections (CA-MRSA) that is reaching epidemic proportions (CitationVandenesch et al 2003; CitationKing et al 2006; CitationVoyich et al 2006; CitationLabandeira-Rey et al 2007). Unfortunately, the entire class of β-lactam antibiotics currently in use is ineffective for the treatment of methicillin-resistant S. aureus (MRSA) infections. By virtue of its unique mechanism of action on the cell wall, vancomycin (), the preeminent glycopeptide, became the antibiotic of “last resort” against resistant Gram-positive bacteria. Unfortunately, Enterococcus spp. established itself in the 1990s as a “respected pathogen” in the nosocomial setting, more problematic than in the past because of its frequent resistance to vancomycin (vancomycin-resistant Enterococcus; VRE) (CitationMurray 1990). This unwelcome development, together with the threat of the emergence of vancomycin-intermediate and -resistant S. aureus (VISA and VRSA) (CitationSmith et al 1999), conspire to imperil the status of vancomycin as the “workhorse” antibiotic for the treatment of infections caused by Gram-positive bacteria.
Table 1 Timeline of introduction of antibiotics active against Staphylococcus aureus; first report of resistance and description of mechanism of resistance
Figure 1 Chemical structures of select antibiotics active against Gram-positive cocci.
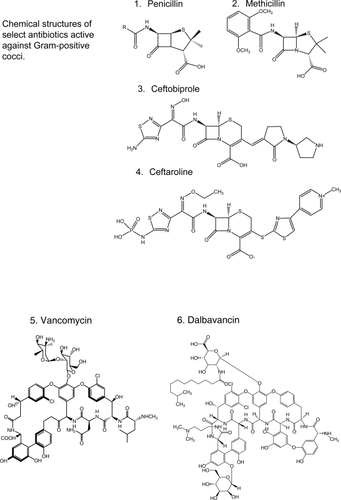
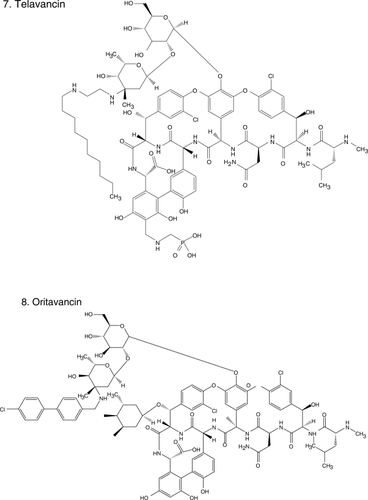
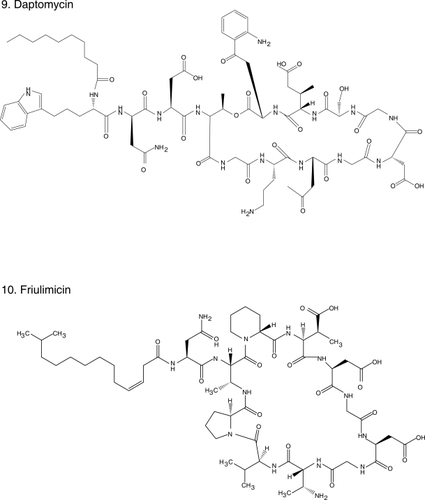
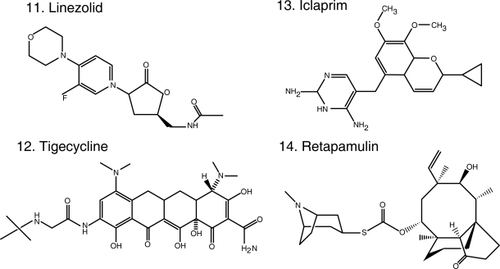
Efforts to find solutions to the “Gram-positive problem” have been fruitful. In the past decade, we witnessed the introduction into contemporary practice of oxazolidinones (eg, linezolid [CitationStevens et al 2002] ), cyclopeptides (eg, daptomycin [CitationFowler et al 2006] ) and glycylcyclines (eg, tigecycline [CitationBabinchak et al 2005; CitationEllis-Grosse et al 2005] ), new classes of antibiotics with in vitro and clinical activity against MRSA and VRE. Furthermore, the rational design of new drugs based on “old molecules”, the discovery of agents with novel mechanisms of action, and the innovative application of the principles of pharmacokinetics (PK) and pharmacodynamics (PD) to improve the use of existent drugs, offer great promise. Thus, we are at an important time to review the new antibiotics and those that are in development which target resistant Gram-positive pathogens. This article will summarize: i) the new generation of antibiotics effective against Gram-positive bacteria based on the modification of the parent β-lactam and glycopeptide structures; ii) the insights gained from the use of vancomycin; iii) the experience accumulated with the introduction of recently developed classes of antibiotics in the clinical arena; and iv) novel compounds in development. We also offer a clinical perspective to these emerging issues as infectious disease physicians.
New β-lactams against MRSA
The construction of the peptidoglycan backbone of the Gram-positive bacterial cell wall requires the polymerization of a polysaccharide chain formed by discaccharide-pentapeptide monomers, with alternating N-acetylglucosamine (NAG) and N-acetylmuramic (NAM) acid moieties, and cross-linked by peptide bridges. Enzymes termed D-alanyl-D-alanine transpeptidases mediate this last key step in the process. β-lactam antibiotics inhibit these enzymes, known as penicillin binding proteins (PBPs), by covalently binding to their serine site (CitationLivermore 2006). Recent studies reveal that the bacterial peptidoglycan takes a helical shape with a defined periodicity (CitationMeroueh et al 2006). These finding forms the basis of novel approaches to inhibiting cell wall synthesis in bacteria.
The identification in 1961 in England of a strain of S. aureus resistant to methicillin marked the prelude of the end of β-lactam therapy against this organism. This strain spread successfully and rapidly in hospitals around the world, carrying the mecA gene that codes for PBP2a, which has very low affinity to any of the existent β-lactams. MRSA has since become the nosocomial pathogen “par excellence”; now, almost five decades later, we are in the midst of an epidemic of CA-MRSA that is changing the landscape of antimicrobial therapy. Translation from the laboratory to the clinic of β-lactams with the capacity to bind to PBP2a has become a therapeutic imperative. The current generation of anti-MRSA β-lactams, once approved for use by regulatory authorities after their validation in clinical trials, will represent a major breakthrough (CitationPage 2006; CitationRossolini 2007).
The most advanced among these molecules is ceftobiprole () (CitationBush et al 2007). This “fifth generation” cephalosporin demonstrates potent binding to PBP2a in MRSA (including VISA and VRSA [CitationBogdanovich et al 2005]), as well as to PBP2x in a penicillin-resistant Streptococcus pneumoniae strain and to PBP2 and PBP3 in Escherichia coli and Pseudomonas aeruginosa (CitationDavies et al 2007). Additionally, it has demonstrated activity similar to ampicillin against Enterococcus faecalis, including vancomycin-resistant and β-lactamase-producing isolates (CitationArias et al 2007); it is not active against Enterococcus faecium (CitationJones 2007) (). Although ceftobiprole is resistant to inactivation by a broad range of β-lactamases, it is hydrolyzed by extended-spectrum β-lactamases (ESBLs) found in Escherichia coli and Klebsiella spp. rendering its activity against Gram-negative bacteria comparable with that of cefepime (CitationQueenan et al 2007). The typical minimum inhibitory concentration (MIC) of ceftobiprole against MRSA is 2 μg/ml, contrasted with an MIC of >64 μg/ml for ceftriaxone. PK/PD studies in humans demonstrate that ceftobiprole has an elimination half-life of about 3 hours and predominantly urinary excretion (CitationMurthy and Schmitt-Hoffmann 2008).
Table 2 Activity of different antibiotics against selected Gram-positive bacteria
A phase 3 multi-centered, global, randomized, double-blind trial comparing the efficacy of ceftobiprole with that of vancomycin in patients with complicated skin and skin structure infections (CSSSI) caused by Gram-positive bacteria (MRSA, MSSA, Staphylococcus epidermidis, and β-hemolytic streptococci) showed that both drugs were equivalent. In this trial, 282 patients received ceftobiprole intravenously (IV) 500 mg every 12 h and were compared with 277 patients who received vancomycin 1 gram IV every 12 h. Cure rates for patients with MRSA infections were 91.8% (56/61) with ceftobiprole treatment and 90.0% (54/60) with vancomycin treatment (CitationNoel et al 2008b). A second clinical trial comparing ceftobiprole monotherapy versus treatment with vancomycin plus ceftazidime was also carried out, with important variations (CitationNoel et al 2008a). This design included patients with CSSSIs caused by Gram-negative bacteria (E. coli, P. aeruginosa, Klebsiella pneumoniae, Proteus mirabilis, and Enterobacter cloacae) in addition to Gram-positive bacteria. Thus, patients with diabetic foot infections could be included. A more frequent dosing scheme (ceftobiprole 500 mg IV every 8 hours), and a longer infusion period (120 minutes vs. 60 minutes) were used. These modifications derived from PK studies that demonstrated a higher probability of achieving bactericidal and bacteriostatic targets with this dose; an important caveat derived from this data is that bactericidal concentrations against AmpC producing Enterobacteriaceae were not achievable (CitationLodise et al 2007). Clinical cure rates and microbiological outcomes were similar in both arms across the range of pathogens and CSSSIs. Of note, slightly worse outcomes (but similar in both arms) were observed in patients with diabetic foot infections and MRSA infections; the subgroup of patients infected with MRSA that expressed PVL had better outcomes when treated with ceftobiprole (although statistical significance was not reported). Importantly, adverse events were rare among ceftobiprole-treated patients in both trials; a prolonged infusion time of 120 minutes caused a lower incidence of nausea and vomiting. Clinical trials evaluating the utility of ceftobiprole in the treatment of community- and hospital-acquired pneumonia (CAP and HAP, respectively) are underway.
A second cephalosporin with affinity to PBP2a, ceftaroline (), is also in development. It has demonstrated activity against MSSA, methicillin-resistant S. epidermidis and MRSA. When tested against a collection of CA-MRSA strains, the MIC50 and MIC90 were 0.5 μg/ml (CitationSader et al 2008a). Ceftaroline is also active against strains of S. pneumoniae that are resistant to ceftriaxone, as well as Hemophilus influenzae and Moraxella catharralis. Similar to ceftobiprole, ceftaroline inhibited strains of both vancomycin-susceptible and -resistant E. faecalis, but was inactive against E. faecium. In contrast, its activity against Gram-negative bacteria is inferior to that of extended spectrum cephalosporins. Ceftaroline shows higher ranges of MICs against penicillinase-producing E. coli and Klebsiella spp. and Enterobacter, Citrobacter and Serratia spp. (0.12–1 μg/ml) than ceftriaxone, cefotaxime, ceftazidime, and cefepime. Ceftaroline is inactive against organisms producing ESBLs and AmpC β-lactamases; it is also inactive against anaerobes, Acinetobacter spp., and P. aeruginosa (CitationMushtaq et al 2007).
A phase 2 clinical trial evaluated the safety and efficacy of ceftaroline in the treatment of CSSSI caused by aerobic Gram-positive bacteria, comparing it to “standard therapy” (vancomycin with and without aztreonam). The clinical cure rate was 96.7% for ceftaroline versus 88.9% for standard therapy, but this study was not powered for inferential statistical analysis (CitationTalbot et al 2007).
A great deal of optimism also accompanies the development of anti-MRSA carbapenems. These new compounds feature modifications to the carbapenem structure that have resulted in improved affinity for PBP2a, among other PBPs. In contrast to ceftobiprole and ceftaroline, these molecules are also stable against Class A and C β-lactamases and therefore retain activity against Gram-negative bacteria producing extended-spectrum and AmpC cephalosporinases, as well as activity against anaerobes. These products have not been the subject of clinical trials yet. Some of the strongest candidates for further clinical development include: i) ME1036, which demonstrates very high affinity to miscellaneous PBPs and is active in vitro against a broad collection of Gram-positive and Gram-negative isolates from the United States such as MRSA, penicillin-resistant S. pneumoniae, 3rd generation (extended-spectrum) cephalosporin-resistant Enterobacteriacea (ESBL and AmpC producing) and vancomycin-resistant E. faecalis, but not against vancomycin-resistant E. faecium; ii) PZ-601 (SMP-216601), which has been demonstrated in vitro activity against a similar collection of clinical strains from USA and Japan; iii) FSI-1297, also active in vitro against MRSA and vancomycin resistant E. faecalis (MIC = 1 μg/ml) but less potent against vancomycin resistant E. faecium (MIC = 8 μg/ml); iv) RO4908463 (CS-023), active against MRSA and P. aeruginosa (47th Interscience Conference on Antimicrob Agents Chemother (ICAAC), Abstracts E-280, F1-341, A-29, Chicago, 2007.)
New glycopeptides in clinical development
Vancomycin () belongs to the class of glycopeptides, a family of antibiotics produced by a diverse group of Actinomycetes. Other well-known representatives of this class include teicoplanin, widely employed around the world but not approved for use in the United States, and avoparcin, formerly used in Europe in animal husbandry. The chemical moiety that glycopeptides share in common is a heptapeptide. The heptapeptide backbone forms a carboxylate binding pocket that joins by forming hydrogen bonds to D-alanyl-D-alanine (D-Ala-D-Ala) peptidyl residues of the disaccharide-pentapeptide monomers (composed of N-acetylmuramic acid and N-acetylglucosamine). This prevents the transpeptidation and transglycosilation reactions that the disaccharide-pentapeptide intermediates must undergo in order to polymerize and form the peptidoglycan cell wall. This ultimately leads to the inhibition of bacterial cell wall synthesis.
The best understood mechanisms of resistance against vancomycin is the elimination of the D-Ala-D-Ala binding target or the replacement of the terminal D-Ala by D-lactate (D-Lac) or D-serine (D-Ser), to which the heptapeptide cannot bind efficiently (CitationCourvalin 2006). Even though this represents the change of a single moiety, the enzymatic machinery necessary to produce it depends on a complex mechanism which involves the acquisition and regulation of several genes (eg, vanR, vanS, van H, vanA, vanX, vanY) through mobile genetic elements or transposons. This results in the expression of different phenotypes of glycopeptide resistance, denominated as VanA, -B, -C, -D, -E, and -G. For example, VanA denotes vancomycin and teicoplanin resistance associated with transposon Tn1546, found mostly among E. faecium and E. faecalis and described in S. aureus. VanB is vancomycin resistant but teicoplanin susceptible, and is associated with transposons Tn1547 and 1549. VanC differs in that it is not associated with a mobile genetic element but is intrinsic to Enterococcus gallinarum and casseliflavus, and depends on a shift of D-Ala to D-Ser (as opposed to D-Lac). An additional mechanism of resistance, the overexpression of D- Ala residues that results in intermediate susceptibility to vancomycin, has been described in S. aureus (see below).
Despite sharing the same basic mechanism of action, the activity of glycopeptides against Gram-positive cocci is not homogeneous, probably explained by differences in their structure outside of the common heptapeptide moiety. These various structure – activity relationships among glycopeptides were exploited to develop a new generation of lipoglycopeptides that are able to overcome resistance to vancomycin, among which dalbavancin (), telavancin (), and oritavancin () are the more advanced representatives. The enhanced antibacterial activities of these lipoglycopeptides may derive from the ability of a hydrophobic side chain to cause both dimerization and binding to bacterial membranes (CitationAllen and Nicas 2003). These cooperative interactions enhance binding at the traditional glycopeptide target. Additionally, the novel glycopeptides take advantage of alternative mechanisms of action; for instance, they may act by direct binding and inhibition of transglycosylation, inhibition of RNA and lipid synthesis or direct disruption of the membrane. The coexistence of multiple mechanisms of action in one drug results in increased activity as demonstrated by lower MICs, when compared against vancomycin (). For example, the MIC of vancomycin against S. pneumoniae ranges from 0.25 to 2 μg/ml, whereas for the new glycopeptides it ranges from 0.002 to 0.12 μg/ml; the MIC for VanA and VanB Enterococcus spp. drops eight-fold (CitationPace and Yang 2006).
Oritavancin (), the first of the novel lipoglycopeptides, illustrates this search for compounds with improved spectrum and PK/PD characteristics based upon the existing structures of vancomycin and teicoplanin. The addition of a clorphenylbenzyl side chain () permits the dimerization of oritavancin molecules and hydrophobic interactions with the bacterial membrane that facilitate binding to peptidoglycan residues, even D-Ala-D-Lac (CitationAllen et al 1997; CitationAllen and Nicas 2003). Oritavancin demonstrates in vitro activity against Enterococcus spp., including VRE (VanA, VanB, and VanC), and Streptococcus (S. pneumoniae, S. pyogenes, S. agalactiae). It is also active against methicillin-susceptible S. aureus (MSSA) and MRSA, with MICs ranging between 1–4 μg/ml, making it less potent than other glycopeptides (). Nevertheless, it is bactericidal against enterococci and staphylococci, unlike vancomycin (CitationLin et al 2005; CitationNoviello et al 2001). Animal models suggest its activity is concentration dependent but a study in humans indicated that percentage of time above MIC was an important parameter of successful therapy in patients with S. aureus bacteremia (CitationBoylan et al 2003; CitationBhavnani et al 2006). Because of its prolonged half-life, once daily dosing is feasible. There are no published clinical data on the use of oritavancin, but it is in development for the treatment of CSSSI. Despite its promise, there has been a several year hiatus in the development of oritavancin.
Dalbavancin (), derived from teicoplanin, is perhaps the most clinically advanced of the novel glycopeptides (CitationBilleter et al 2008). In addition to binding to the D-Ala-D-Ala terminals of the growing peptidoglycan chain, dalbavancin interacts with the bacterial cell membrane, conferring it potent in vitro activity against MSSA, MRSA, S. pneumoniae, and β-hemolytic streptococci (CitationJones et al 2006). Dalbavancin was equally potent against enterococci, including VRE but not VanA-resistant phenotypes (CitationJones et al 2005; CitationStreit et al 2005). It is active against VRSA (CitationBozdogan et al 2003) and miscellaneous Gram-positive anaerobes (CitationGoldstein et al 2006). Its unique characteristic is that it is intensely protein-bound, leading to an extended half-life (mean 181 h), so that it can be dosed once weekly.
Phase 2 and phase 3 trials demonstrate equal effectiveness of dalbavancin in the treatment of CSSSI when compared with 14-day courses of vancomycin and linezolid, respectively (CitationJauregui et al 2005; CitationSeltzer et al 2003). A phase 2 study of patients with staphylococcal (including MRSA) catheter-related bacteremias demonstrated superiority of dalbavancin administered twice daily for 14 days compared with vancomycin. Dalbavancin was administered at 1 gm on the first day and then 500 mg on the eighth day of treatment in all of these trials. This unusual dosing schedule makes it a very appealing compound for outpatient therapy. The concern is the potential occurrence of adverse effects, which logically would also be long lasting given the long-half life of the compound. The above-mentioned studies, however, suggest a very favorable safety profile.
Telavancin () is structurally similar to vancomycin. The modifications on the original glycopeptide, namely the alkylation of the vancosamine substituent with a hydrophobic moiety, result in a multifunctional mechanism of action. The side chain promotes interaction with the cell membrane, improving affinity for D-Ala-D-Ala and altering the bacterial cell membrane potential and permeability (CitationHiggins et al 2005). This results in greater bactericidal activity and potency against staphylococci and enterococci, even VISA, VRSA, and VRE with Van A phenotype (). Similar to the other novel glycopeptides, telavancin has excellent in vitro activity against β-hemolytic streptococci and S. pneumoniae (CitationLeuthner et al 2006; CitationClouse et al 2007; CitationJansen et al 2007). Additionally, when compared with vancomycin, telavancin has a longer half-life (up to 9 h) so that it can be given once a day (CitationShaw et al 2005).
There are two published phase 2 clinical trials comparing telavancin (dosed at 7.5 mg/kg/day and 10 mg/kg/day, respectively) against standard therapy with a β-lactam or vancomycin for the treatment of CSSSI caused by Gram-positive bacteria. Overall, the outcomes were equivalent beween the two arms, except in the group with MRSA infection in which microbiological eradication and clinical cure appeared better in patients treated with telavancin (CitationStryjewski et al 2005, Citation2006). Side effects consisted mostly of nausea and vomiting; these occurred with equal frequency in the comparator arm. Further studies are underway to evaluate the efficacy of telavancin in the treatment of pneumonia, which is supported by animal and preliminary human data (CitationGotfried et al 2008; CitationHegde et al 2008).
A novel compound, TD-1792, is described as a heterodimer in which a cephalosporin moiety is covalently linked to vancomycin. TD-1792 therefore inhibits both key cell wall synthesis functions, transpeptidation and transglycosilation. It has demonstrated bactericidal activity against MRSA, with very low MICs (0.03 μg/ml). The administration of its individual components, the cephalosporin and vancomycin, by themselves or together (but not covalently linked) failed to achieve such good parameters of activity (47th ICAAC, Abstract F1-2110, Chicago IL, 2007). Its eventual use in the clinical arena depends on further development and clinical trials that support its efficacy.
Enduring utility of vancomycin against S. aureus
There is an ongoing debate among clinicians regarding the continued utility of vancomycin for the treatment of infections caused by MRSA. Trusting more than 50 years of experience and an improved compound free of major toxicities, physicians rely heavily on vancomycin. Furthermore, vancomycin retains activity against the vast majority of S. aureus isolates. In contrast, many have long considered vancomycin a sub-optimal antibiotic, for instance when compared with β-lactams for the treatment of MSSA. There is fear that vancomycin may be reaching obsolescence given the emergence of resistance. As argued in the recent point-counterpoint arguments published in infectious diseases journals, there are many subtleties involved (CitationDeresinski 2007; CitationMohr and Murray 2007).
In contrast to Enterococcus spp., high-level resistance to vancomycin among S. aureus (MIC ≥ 16 μg/ml) has been reported only sporadically, amounting to seven cases (CitationSievert et al 2008), all from the United States. Analysis of the index case of VRSA from Detroit, Michigan in 2002 identified the transfer of vanA, a vancomycin resistance determinant linked to Tn1546, from E. faecalis to S. aureus, a finding replicated in the subsequent six isolates (CitationChang et al 2003). Because the potential universal dissemination of VRSA has not materialized yet (CitationBush, 2004), the more common and immediate threat may be VISA (also referred to as glycopeptide intermediate S. aureus; GISA). First identified in 1996 in Japan and 1997 in the United States, VISA strains are characterized by thicker extracellular matrixes due to the overproduction of D-Ala D-Ala or decreased turnover in the peptidoglycan wall that serve as a decoy target for vancomycin, resulting in MICs 4–8 μg/ml (CitationSmith et al 1999). Additionally, there is concern about the phenomenon of heteroresistance to vancomycin in S. aureus (hVISA). Subpopulations (MIC > 2 μg/ml) exist within cultures of the same strain of S. aureus that are susceptible to vancomycin (MIC ≤ 1 μg/ml). Growth medium containing vancomycin selects for these subpopulations.
The gradual loss of S. aureus susceptibility to vancomycin observed in a patient with MRSA endocarditis during unsuccessful treatment with vancomycin, is the origin of a remarkable collection of studies investigating VISA in vivo (CitationMcAleese et al 2006; CitationMwangi et al 2007; CitationSieradzki et al 2003; CitationSieradzki et al 1999). Tomasz and his group demonstrated the evolution of vancomycin resistance in S. aureus (from an MIC of 1 and 2 μg/ml to 8 μg/ml) through whole-genome sequencing, identifying the key mutations differentiating isogenic strains with progressive increases in the MIC of vancomycin isolated from a single patient. Thirty-five mutations appeared sequentially during the course of treatment. Furthermore, resistance to rifampin resulted as well, after administration of only one dose of this antibiotic. More intriguing, a 100-fold increase in the MIC of daptomycin (0.01 to 1 μg/ml) was observed, even though that antibiotic was not used as treatment. The mutations associated with these phenotypes were also identified.
Sakoulas and colleagues revealed another important component that furthers our understanding of hVISA: the loss of function of the accessory gene regulator (agr) operon. Changes in this genetic element, identified as the group II polymorphism, may confer higher MICs of vancomycin to S. aureus. This polymorphism is present in isolates with VISA and hVISA phenotypes and may be associated with vancomycin failure (CitationSakoulas et al 2003, Citation2005, Citation2006; CitationMoise-Broder et al 2004b).
The difficult detection of hVISA in the microbiology laboratory hinders the assessment of the epidemiological and clinical impact of this phenotype. The method of reference, population-analysis profile (PAP) testing, is laborious and resource intensive (CitationWalsh et al 2001). Perhaps the best screening test for hVISA is the Etest macromethod, in which a high inoculum of bacteria (McFarland of 2) is used against Etest strips of vancomycin and teicoplanin (CitationVoss et al 2007). The clinical impact of hVISA remains unclear. A study from Australia identified five patients with bacteremia caused by hVISA (defined by PAP); they were more likely to have high infections with high inoculum of bacteria, treatment failure and low vancomycin levels. A second survey by the same group looked at the outcomes of 25 additional patients with infections caused by S. aureus with reduced susceptibility to vancomycin; they had serious infections ranging from endocarditis to osteomyelitis, received prolonged and ineffective courses of vancomycin and had been previously infected with MRSA. It is difficult to ascertain in both cases whether the detection of hVISA in these patients was a cause or an effect of the above-mentioned factors (CitationCharles et al 2004; CitationHowden et al 2004). CitationFalagas and colleagues (2008a) reviewed the studies reporting the clinical outcome of infection with hVISA, and did not find a consistent negative impact but rather conflicting results.
In 2006 the Clinical Laboratory Standards Institute (CLSI) lowered the vancomycin MIC breakpoints for S. aureus (from ≤4 μg/ml to ≤2 μg/ml for “susceptible,” from 8–16 μg/ml to 4–8 μg/ml for “intermediate,” and from ≥32μg/ml to ≥16 μg/ml for “resistant”) (CitationTenover and Moellering 2007). The rationale for this change was, among other considerations, to signal that strains with MICs close to 4 μg/ml (until then considered fully susceptible) are likely to be hVISA and therefore would go unrecognized by the microbiology laboratory and may be associated with vancomycin treatment failure. Moreover, there are reports of poor clinical outcomes associated with strains of S. aureus with MICs of 1–2 μg/ml, which is within the new susceptible range as determined by CLSI (CitationSchwaber et al 2003; CitationSakoulas et al 2004). The application of a logistic regression model to a database of patients from Barcelona with MRSA bacteremia yielded similar results (CitationSoriano et al 2008). Treatment with vancomycin in the instance of bacteremia caused by an isolate with a vancomycin MIC of 2 μg/ml – as defined by Etest – was a factor independently associated with mortality. The database did not permit to assess the effect of serum vancomycin concentrations on outcomes, although the dosing regimen used in these patients targeted trough levels above 10 μg/ml.
The investigation of PK/PD parameters of vancomycin may clarify the association between heteroresistance and response to therapy. Studies have determined that the ratio of the area under the concentration-time curve to the MIC (AUC/MIC) predicts the clinical response to vancomycin (CitationRybak 2006). Furthermore, in pneumonia caused by MRSA, an AUC/MIC ≥400 is associated with a better clinical response (CitationMoise-Broder et al 2004a). In contrast, a retrospective study on the dosing levels of vancomycin on MRSA healthcare-associated pneumonia (HCAP) (CitationJeffres et al 2006), demonstrated that vancomycin trough concentrations were similar in patients who lived and in patients who died (13.6 and 13.9 μg/ml, respectively). The investigators did not explore if the vancomycin MICs of the infecting strain may have had an impact on clinical outcome. However, PK data from this study showed that in order to achieve the above mentioned satisfactory AUC/MIC ratio of >400, both a high vancomycin trough concentration (approximately 20 μg/ml) and a low MIC of 0.5 μg/ml are required. Other investigators have shown that there may be an advantage in achieving vancomycin through concentrations >15 μg/ml within the initial 24 hours of treatment, but only in cases in which the infection is caused by strains with an MIC <2 μg/ml (CitationHidayat et al 2006). The American Thoracic Society guidelines for the treatment of HAP caused by MRSA recommend dosing vancomycin to achieve trough concentrations of 15–20 μg/ml (CitationATS 2005). This suggestion, however, does not derive from clinical studies evaluating the effect of vancomycin serum concentrations with respect to patient outcome and has to be balanced with a higher risk of adverse events in the form of renal dysfunction (47th ICAAC, Session 104, Paper K-1096, Chicago, IL). Recently, an in vitro pharmacodynamic model investigated different doses of vancomycin for the treatment of simulated moderate and high inoculum infections with hVISA. In cases of high inoculum infections, up to 105 cfu/ml of hVISA organisms (with MICs 2- to 4- fold higher than at their baseline) remained after the administration of doses of vancomycin as high as 5 g every 12 hours. This data may explain why treatment of high-inoculum infections caused by hVISA with vancomycin often results in failure (CitationRose et al 2008).
How does one reconcile the above observations in order to choose the appropriate therapy against MRSA infections in the clinic? We support the opinion that it is necessary to determine an accurate MIC of vancomycin in the microbiology laboratory, ideally with Etest or a broth-based method, particularly in cases of MRSA infections with high organism loads (eg, pneumonia, endocarditis), in difficult to reach compartments (eye, brain, etc), and in infections requiring prolonged treatment (endovascular infections). It can then be determined whether a satisfactory AUC/MIC ratio is achievable with aggressive vancomycin dosing regimens or if the use of an alternative antibiotic is preferable, a decision that needs to be modulated by the clinical response. Other strategies have been considered plausible improvements upon the limited performance of vancomycin against serious MRSA infections. Combination therapy with vancomycin and other agents has been attempted. A natural candidate has been rifampin, which is extremely bactericidal against S. aureus even in the intracellular compartment. Unfortunately, vancomycin is not able to neutralize rifampin’s notable proclivity to engender resistance (CitationSimon et al 1983; CitationYamaoka 2007). Continuous infusion therapy, although promising for other antimicrobials, did not demonstrate increased effectiveness in the case of vancomycin in a randomized, multi-center study (CitationWysocki et al 2001; CitationKasiakou et al 2005). The prospective evaluation of the above described strategies and other considerations is essential to help define the continued utility of vancomycin, vis-à-vis newer antibiotics.
The experience with recently released antibiotics active against resistant Gram-positive cocci
The approval by the United States Food and Drug Administration (FDA) of linezolid, daptomycin, and tigecycline, each representative of a new class of antibiotics active against MRSA and enterococci offered welcome therapeutic alternatives to vancomycin. Linezolid, approved in 2000, and daptomycin, introduced in 2003, have emerged as the “antibiotics of last resort” for the treatment of infections caused by VRE. A newer addition, tigecycline, combines activity against S. aureus (including MRSA) and Enterococcus spp. with a broad spectrum of activity against Gram-negative and anerobic bacteria. The role of these three agents within the antibiotic armamentarium continues to evolve, shaped by their specific characteristics, the experiences of clinicians, and the performance of these drugs in clinical trials (CitationPaterson 2006; CitationMicek 2007).
Linezolid () was the first oxazolidinone released. By acting on the 50S ribosomal subunit, specifically the peptidyl-transferase center, linezolid blocks the binding of tRNA, interferes with protein synthesis and inhibits bacterial cell growth (CitationLeach et al 2007). Linezolid is bactericidal against streptococci but bacteriostatic against MRSA and VRE in vitro. In contrast, a study carried out in patients with diabetic foot infection caused by MRSA (some of them were additionally VISA or VRSA) demonstrated that serum can potentiate the antimicrobial effect of linezolid resulting in bactericidal activity (CitationStein et al 2007). Its near-complete oral bioavailability makes linezolid a tremendously attractive antibiotic for the treatment of suspected or confirmed MRSA infections. In addition, linezolid offers the opportunity to potentially lower costs that result from hospitalization (CitationMcKinnon et al 2006). These advantages should be balanced with a high cost as an oral agent, compared to generic alternatives widely used in the community. Patients on linezolid suffer from neurological (peripheral neuropathy and optic neuritis) and hematological (anemia, leukopenia and thrombocytopenia) side effects resulting from the inhibition of mitochondrial protein synthesis, as well as drug-drug interactions (serotonin re-uptake inhibitors) (CitationBishop et al 2006). Furthermore, resistance stemming from single nucleotide changes in varying numbers of copies of the genes encoding 23S ribosomal RNA has emerged during the course of treatment (CitationMeka and Gold 2004).
An open label, multi-center, randomized controlled trial demonstrated that linezolid was superior to vancomycin in achieving microbiological cure in the treatment of CSSSIs due to MRSA (CitationWeigelt et al 2005). However, additional trials are needed to confirm this result and ascertain that clinical cure rates with linezolid exceed significantly those of vancomycin (CitationKalil et al 2006). Similarly, there is an ongoing controversy regarding the superiority of linezolid for the treatment of HAP and ventilator-associated pneumonia (VAP) caused by MRSA. Although the initial phase 2 randomized controlled trials showed equivalence between linezolid and vancomycin, further analysis of the same data showed that there was an advantage in survival in patients treated with linezolid; of note, the serum concentrations of vancomycin in patients included in these trials were not reported (CitationRubinstein et al 2001; CitationWunderink et al 2003). A recently published meta-analysis favored linezolid (CitationFalagas et al 2008b), but appropriately designed and powered studies (phase 3 trials) are still needed to settle this question. Although it does not have FDA approval for the treatment of bloodstream infections, a pooled analysis of five randomized studies comparing linezolid versus vancomycin for the treatment of S. aureus bacteremia demonstrated “noninferior” outcomes associated with linezolid (CitationShorr et al 2005). CitationStevens and colleagues (2007) observed that linezolid (as well as clindamycin) suppressed the translation in S. aureus of genes coding for extracellular protein toxins (eg, PVL, alpha-hemolysin, and toxic shock syndrome toxin 1). This supports the notion that linezolid not only inhibits bacterial growth but may also modulate the expression of virulence. Clinicians need to be careful about inferring a clinical advantage from these experimental findings. Despite its utility, there are still many unanswered questions regarding the use of linezolid for its FDA-approved indications (the treatment of CSSSIs and HAP/VAP), as well as its role in the treatment of bacteremia and endocarditis (CitationFalagas et al 2008b).
Daptomycin () is a cyclic lipopeptide antibiotic, unique in its class, with potent in vitro activity against Gram-positive bacteria, including MRSA and VRE. Its mechanism of action is unclear but it seems to involve depolarization of the bacterial membrane and efflux of potassium ions, leading to cell death (CitationSteenbergen et al 2005). It is extremely bactericidal, even in instances of high inoculum/stationary phase infections, as demonstrated in biofilm and endocarditis models (CitationRose et al 2007a; CitationRoveta et al 2008). Even though it penetrates well into vascular structures and the urinary tract, daptomycin does not penetrate into the central nervous system and its use in the treatment of pneumonia is contra-indicated because it is inhibited by pulmonary surfactant (CitationSilverman et al 2005). It has been demonstrated to be safe, with only occasional elevations in creatine kinase observed (CitationKazory et al 2006).
The FDA approved the use of daptomycin for the treatment of CSSSI (at 4 mg/kg/day), as well as S. aureus including MRSA- bacteremia and right sided endocarditis (at 6 mg/kg/day). An important randomized controlled clinical trial showed that daptomycin was not inferior to a β-lactam antibiotic or vancomycin for the treatment of patients with S. aureus bacteremia and right-sided endocarditis (CitationFowler et al 2006). However, in this trial daptomycin was associated with a high rate of microbiologic failure. It was also concerning that reduced susceptibility to daptomycin emerged among S. aureus isolates. This resonates with the clinical finding mentioned above that daptomycin resistant S. aureus populations appear to be linked to reduced vancomycin susceptibility, a phenomenon that needs further elucidation (CitationRose et al 2007b). The mechanism of resistance to daptomycin has not been fully explained, but there are specific mutations associated with nonsusceptible S. aureus strains (CitationBoucher and Sakoulas 2007). Despite this, and because of its convenient once-a-day dosing, its bactericidal action, and its favorable safety profile, the role of daptomycin in the clinic is expanding (eg, into the treatment of bone and joint infections) (CitationLalani et al 2008). Nevertheless, its superiority to vancomycin in the treatment of MRSA infections and its efficacy in the treatment of infections caused by VRE has yet to be prospectively demonstrated. Of note, it is significantly less potent against VRE than against MRSA or than other compounds ().
Tigecycline () is the sole representative of the class of glycylcyclines (CitationRose and Rybak 2006), defined by a glycylamido (tert-butyl) moiety attached to the original tetracycline molecule, making it impervious to the efflux pumps (eg, tetA and tetB) that normally extrude tetracyclines from the bacterial cell. It also avoids the ribosomal protection proteins (eg, tet(O)). Since it is a tetracycline, it also binds to the 30S ribosomal subunit blocking the entry of amino-acyl tRNA and ultimately inhibiting protein synthesis. It has a broad spectrum of activity that includes MRSA and enterococci (including VRE) and also enterobacteria and anaerobes. The adequate performance of tigecycline in clinical trials support its use in the treatment of polymicrobial CSSSI and intra-abdominal infections (CitationBabinchak et al 2005; CitationEllis-Grosse et al 2005). Although it is known to be inactive against Pseudomonas and Proteus spp. (CitationDean et al 2003; CitationVisalli et al 2003), tigecycline held promise as a treatment against multidrug-resistant enterobacteria and Acinetobacter spp. However, the results of the clinical experiences in the treatment of Gram-negatives recently published are disappointing (CitationAnthony et al 2008; CitationNavon-Venezia et al 2007). Tigecycline remains an option against MRSA and enterococci CSSTI (CitationMunoz-Price et al 2006).
Of note, tigecycline has excellent penetration into tissues. Experiments with radiolabeled antibiotic demonstrated the AUC value in spleen, liver, and kidney to be at least eight times higher than in plasma. Tissue exposure in lung was more than four times higher than in plasma, whereas in bone it was several hundred-fold higher. It appears that tigecycline is primarily the subject of biliary excretion. Approximately 30% of the tigecycline is excreted unchanged in the urine (CitationMeagher et al 2005). Despite the above data, trials involving urinary tract infections were abandoned (CitationLivermore 2005). Therefore, more studies are needed before tigecycline can be recommended for that indication (CitationCunha et al 2007; CitationKrueger et al 2008). The profile of adverse effects with tigecycline is remarkable for nausea and vomiting in a large proportion of patients, as well as increases in the blood urea nitrogen (BUN) consistent with the catabolic effects of tetracyclines. It must be noted that the original tetracyclines (ie, minocycline and doxycycline) have re-emerged to play a major role in the therapy against MRSA, mostly in the treatment of uncomplicated skin and soft tissue infections and as adjuvants in chronic suppressive regimens (CitationRuhe et al 2005).
On the horizon
Iclaprim (), a novel compound, represents a departure from a preexisting class of antibiotics, dihydrofolate reductase (DHFR) inhibitors. The best representative of this class is the diaminopyrimidine trimethoprim (TMP), currently used in the clinic in combination with sulfamethoxazole (SMX). TMP/SMX largely retains activity against MRSA and is very often used in the treatment of uncomplicated skin and soft tissue infections. However, resistance to TMP does occur, resulting from a single amino acid change in the active site of S. aureus DHFR that prevents the formation of hydrogen bonds with TMP. Iclaprim, also a diaminopyrimidine, incorporates a cyclopropyl group that allows for the formation of hydrogen bonds even in TMP resistant S. aureus (CitationSchneider et al 2003; CitationHawser et al 2006). Furthermore, iclaprim is as equally effective as TMP in inhibiting DHFR in S. aureus and E. coli, but is an order of magnitude more efficient against DHFR from S. pneumoniae and Pneumocystis jiroveci. This translates into activity against MRSA, VISA, and VRSA, and against multidrug-resistant S. pneumoniae (β-lactam, macrolide, quinolone and TMP resistant). Iclaprim, like TMP, has adequate enteral bioavailability (CitationKohlhoff and Sharma 2007; CitationLaue et al 2007) and its clinical development is followed with interest.
A second class of DHFR inhibitors currently under development is that of dihydrophthalazine antifolates. Three compounds, BAL-30543, BAL-30544, and BAL-30545, have demonstrated activiy against MSSA, MRSA, VISA, and VRSA, and TMP-resistant S. aureus, as well as MDR-resistant S. pneumoniae. Similar to other DHFR inhibitors, they can be administered enterally and parenterally with excellent bioavailability (47th ICAAC, Abstracts F1-934 and 935, Chicago IL, 2007).
RWJ 416457, a novel oxazolidinone with a pyrrolopyrazolyl substitution, has been tested in vitro against a variety of organisms, including MRSA and VRE. It generally had two- to fourfold lower MICs than those of linezolid against most pathogens tested (CitationFoleno et al 2007). Similarly, when tested against linezolid-resistant enterococci and S. aureus it demonstrated lower MICs than linezolid but only by a couple of dilutions, indicating that RWJ 416457 is susceptible to the same mechanism of resistance than linezolid, namely mutations in genes coding for the 23S rRNA (CitationLivermore et al 2007).
Friulimicin (), the natural product of Actinoplanes friulensis, is structurally similar to daptomycin and therefore is classified as a cyclic lipopeptide. Their mechanism of action, however, seems to differ. Friulimicin prevents cell wall synthesis by forming a calcium dependent complex with the bacroprenol-phosphate carrier whereas daptomycin’s putative mechanism of action is the formation of pores that leads to extrusion of potassium and cell death. In general, friulimicin has a similar bactericidal activity as daptomycin against MRSA, S. pneumoniae and VRE, but typically its MICs are one dilution higher. The exception is Streptococcus pyogenes for which friulimicin has a fourfold higher MIC (4–8 μg/ml). Despite sharing a similar structure, friulimicin does not demonstrate cross-resistance with daptomycin. Unlike daptomycin, it is not inhibited by pulmonary surfactant and it appeared effective in a murine S. pneumoniae pneumonia model, as well as in a S. aureus abscess model (47th ICAAC, Abstracts F1-1640, 1642, 1643, 1647, 1651, 1652, Chicago, IL, 2007).
Api-1252 exploits a novel mechanism of action, the inhibition of bacterial enoyl-acyl carrier protein (enoyl-ACP) reductase (FabI). This compound is in development for the treatment of staphylococcal infections. In preliminary trials, it demonstrated potent in vitro activity against clinical isolates of MRSA (MIC90, 0.015 μg/ml), as well as Staphylococcus epidermidis (CitationKarlowsky et al 2007).
CBR-2092 is a novel rifamycin-quinolone hybrid antibiotic in the preclinical stages of development for the treatment of MRSA. It has shown activity in animal models of endocarditis against quinolone resistant MRSA. (47th ICAAC, Abstract F1-2102 Chicago, IL, 2007).
Finally, in the field of topical antibiotics, retapamulin () received FDA approval for the treatment of impetigo. A semisynthetic pleuromutilin derivative, it is active against MRSA, as well as Streptococcus spp. It inhibits protein synthesis by binding to the 50S portion of the ribosome in a different fashion than other antibiotics (blocks peptidyl transferase activity, and partially inhibits the binding of the initiator tRNA substrate to the ribosomal P-site), explaining the lack of cross-resistance (CitationYan et al 2006; CitationChampney and Rodgers 2007; CitationJacobs 2007; CitationOranje et al 2007). Another promising class of topical agents is that of diphenyl-ureas, active against MRSA and Streptococcus spp. and effective in murine models studying MRSA skin infection and nasal colonization (47th ICAAC, Abstract F1-2094-2100 Chicago, IL, 2007).
Conclusion
The immediate conclusion after reviewing close to twenty of the antibiotics that are already in use or are in development for the treatment of infections caused by resistant Gram-positive bacteria is that there are indeed many options. The state of the antibiotic pipeline for Gram-positive bacteria appears to be very healthy. This poses a stark contrast with the situation in the Gram-negative realm, where there are serious and widespread misgivings about the capacity and willingness of the pharmaceutical industry to continue to meet the medical needs posed by certain infectious pathogens (CitationTalbot et al 2006). The ever present need for new antimicrobials is made more poignant by the emergence of resistance, which either has already been described for many of the agents discussed () or is predictable and waiting around the corner.
It also seems that, despite or because of the many options available, defining the therapy of “choice” for serious Gram-positive bacterial infections has become more difficult than ever. This apparent paradox underscores the notion that the value of an antibiotic does not lie within its chemical structure but in the body of knowledge supporting its use. As illustrated in the above discussion on the continued utility of vancomycin, multiple considerations and points of view apply and there are no simple answers. We hope that similar discussions about the newer compounds can be made in the future, informed by sound microbiological, clinical, and epidemiological studies.
A single drug is not likely to dominate the future antibiotic landscape; different indications may call for different therapies, as suggested by the example of MRSA pneumonia and linezolid. Individual patient responses to antibiotics and the multiplicity of side effects will also be a consideration; pharmacogenomics will be a powerful tool to discern the best therapy for each patient (CitationDavison and Barrett 2003). The longevity and robustness of our new armamentarium against Gram-positive bacterial infections will be determined by factors specific to each drug, such as effectiveness, convenience, cost, and emergence of resistance, as much as by efforts in the general aspects of immunization, infection control, and antibiotic stewardship.
Acknowledgements
The Veterans Affairs Merit Review Program and the National Institutes of Health (RO1 AI072219) support the work of Dr. Robert A Bonomo. The authors report no conflicts of interest.
References
- [ATS] American Thoracic Society, Infectious Diseases Society of America2005Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumoniaAm J Respir Crit Care Med17138841615699079
- AllenNELeTourneauDLHobbsJNJr1997Molecular interactions of a semisynthetic glycopeptide antibiotic with D-alanyl-D-alanine and D-alanyl-D-lactate residuesAntimicrob Agents Chemotherr416671
- AllenNENicasTI2003Mechanism of action of oritavancin and related glycopeptide antibioticsFEMS Microbiol Rev265113212586393
- AnthonyKBFishmanNOLinkinDR2008Clinical and microbiological outcomes of serious infections with multidrug-resistant gram-negative organisms treated with tigecyclineClin Infect Dis465677018199038
- ArcherGLCoughterJPJohnstonJL1986Plasmid-encoded trimethoprim resistance in staphylococciAntimicrob Agents Chemother29733403729338
- AriasCASinghKVPanessoD2007Time-kill and synergism studies of ceftobiprole against Enterococcus faecalis, including beta-lactamase-producing and vancomycin-resistant isolatesAntimicrob Agents Chemother512043717438057
- BabinchakTEllis-GrosseEDartoisN2005The efficacy and safety of tigecycline for the treatment of complicated intra-abdominal infections: analysis of pooled clinical trial dataClin Infect Dis41Suppl 5S35436716080073
- BarberM1961Methicillin-resistant staphylococciJ Clin Pathol143859313686776
- BhavnaniSMPassarellJAOwenJS2006Pharmacokinetic-pharmacodynamic relationships describing the efficacy of oritavancin in patients with Staphylococcus aureus bacteremiaAntimicrob Agents Chemother50994100016495262
- BilleterMZervosMJChenAY2008Dalbavancin: a novel once-weekly lipoglycopeptide antibioticClin Infect Dis465778318199045
- BishopEMelvaniSHowdenBP2006Good clinical outcomes but high rates of adverse reactions during linezolid therapy for serious infections: a proposed protocol for monitoring therapy in complex patientsAntimicrob Agents Chemother50159960216569895
- BogdanovichTEdnieLMShapiroS2005Antistaphylococcal activity of ceftobiprole, a new broad-spectrum cephalosporinAntimicrob Agents Chemother4942101916189100
- BoucherHWSakoulasG2007Perspectives on Daptomycin resistance, with emphasis on resistance in Staphylococcus aureusClin Infect Dis45601817682996
- BoylanCJCampanaleKIversenPW2003Pharmacodynamics of oritavancin (LY333328) in a neutropenic-mouse thigh model of Staphylococcus aureus infectionAntimicrob Agents Chemother471700612709343
- BozdoganBEselDWhitenerC2003Antibacterial susceptibility of a vancomycin-resistant Staphylococcus aureus strain isolated at the Hershey Medical CenterJ Antimicrob Chemother52864814563898
- BrodieJJamiesonWSommervilleT1955Complications of oxytetracycline and tetracycline therapy related to a defined type of resistant staphylococcusLancet269223513243694
- BushK2004Vancomycin-resistant Staphylococcus aureus in the clinic: not quite ArmageddonClin Infect Dis381056715095206
- BushKHeepMMacielagMJ2007Anti-MRSA beta-lactams in development, with a focus on ceftobiprole: the first anti-MRSA beta-lactam to demonstrate clinical efficacyExpert Opin Investig Drug1641929
- ChampneyWSRodgersWK2007Retapamulin inhibition of translation and 50S ribosomal subunit formation in Staphylococcus aureus cellsAntimicrob Agents Chemother513385717562806
- ChangSSievertDMHagemanJC2003Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance geneN Engl J Med3481342712672861
- CharlesPGWardPBJohnsonPD2004Clinical features associated with bacteremia due to heterogeneous vancomycin-intermediate Staphylococcus aureusClin Infect Dis384485114727222
- ClouseFLHovdeLBRotschaferJC2007In vitro evaluation of the activities of telavancin, cefazolin, and vancomycin against methicillin-susceptible and methicillin-resistant Staphylococcus aureus in peritoneal dialysateAntimicrob Agents Chemother514521417908947
- CourvalinP2006Vancomycin resistance in gram-positive cocciClin Infect Dis42Suppl 1S253416323116
- CunhaBAMcDermottBNausheenS2007Single daily high-dose tigecycline therapy of a multidrug-resistant (MDR) Klebsiella pneumoniae and Enterobacter aerogenes nosocomial urinary tract infectionJ Chemother (Florence, Italy)197534
- DaviesTAPageMGShangW2007Binding of ceftobiprole and comparators to the penicillin-binding proteins of Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, and Streptococcus pneumoniaeAntimicrob Agents Chemother512621417470659
- DavisonDBBarrettJF2003Antibiotics and pharmacogenomicsPharmacogenomics46576512943472
- DeanCRVisalliMAProjanSJ2003Efflux-mediated resistance to tigecycline (GAR-936) in Pseudomonas aeruginosa PAO1Antimicrob Agents Chemother47972812604529
- DemerecM1945Production of Staphylococcus strains resistant to various concentrations of penicillinProc Natl Acad Sci USA31162416588677
- DeresinskiS2007Counterpoint: Vancomycin and Staphylococcus aureus – an antibiotic enters obsolescenceClin Infect Dis441543817516396
- DeshpandeLMFritscheTRMoetGJ2007Antimicrobial resistance and molecular epidemiology of vancomycin-resistant enterococci from North America and Europe: a report from the SENTRY antimicrobial surveillance programDiagn Microbiol Infect Dis581637017368801
- Ellis-GrosseEJBabinchakTDartoisN2005The efficacy and safety of tigecycline in the treatment of skin and skin-structure infections: results of 2 double-blind phase 3 comparison studies with vancomycin-aztreonamClin Infect Dis41Suppl 5S3415316080072
- FalagasMEMakrisGCDimopoulosG2008aHeteroresistance: a concern of increasing clinical significanceClin Microbiol Infect14101418093235
- FalagasMESiemposIIVardakasKZ2008bLinezolid versus glycopeptide or beta-lactam for treatment of Gram-positive bacterial infections: meta-analysis of randomised controlled trialsLancet Infect Dis8536618156089
- FolenoBDAbbanatDGoldschmidtRM2007In vitro antibacterial activity of the pyrrolopyrazolyl-substituted oxazolidinone RWJ-416457Antimicrob Agents Chemother51361517101672
- FowlerVGJrBoucherHWCoreyGR2006Daptomycin versus standard therapy for bacteremia and endocarditis caused by Staphylococcus aureusN Engl J Med3556536516914701
- GalesACSaderHSFritscheTR2008Tigecycline activity tested against 11808 bacterial pathogens recently collected from US medical centersDiagn Microbiol Infect Dis60421718068934
- GoldsteinEJCitronDWarrenYA2006In vitro activities of dalbavancin and 12 other agents against 329 aerobic and anaerobic Gram-positive isolates recovered from diabetic foot infectionsAntimicrob Agents Chemother502875916870792
- GotfriedMHShawJPBentonBM2008Intrapulmonary distribution of intravenous telavancin in healthy subjects and effect of pulmonary surfactant on in vitro activities of telavancin and other antibioticsAntimicrob Agents Chemother5292717923490
- HawserSLociuroSIslamK2006Dihydrofolate reductase inhibitors as antibacterial agentsBiochem Pharmacol71941816359642
- HaydenMKRezaiKHayesRA2005Development of Daptomycin resistance in vivo in methicillin-resistant Staphylococcus aureusJ Clin Microbiol435285716207998
- HegdeSSReyesNSkinnerR2008Efficacy of telavancin in a murine model of pneumonia induced by methicillin-susceptible Staphylococcus aureusJ Antimicrob Chemother611697217993505
- HidayatLKHsuDIQuistR2006High-dose vancomycin therapy for methicillin-resistant Staphylococcus aureus infections: efficacy and toxicityArch Intern Med16621384417060545
- HigginsDLChangRDebabovDV2005Telavancin, a multi-functional lipoglycopeptide, disrupts both cell wall synthesis and cell membrane integrity in methicillin-resistant Staphylococcus aureusAntimicrob Agents Chemother4911273415728913
- HiramatsuKAritakaNHanakiH1997Dissemination in Japanese hospitals of strains of Staphylococcus aureus heterogeneously resistant to vancomycinLancet350167039400512
- HobanDJBouchillonSKJohnsonBM2005In vitro activity of tigecycline against 6792 Gram-negative and Gram-positive clinical isolates from the global Tigecycline Evaluation and Surveillance Trial (TEST Program, 2004)Diagn Microbiol Infect Dis522152716105567
- HowdenBPWardPBCharlesPG2004Treatment outcomes for serious infections caused by methicillin-resistant Staphylococcus aureus with reduced vancomycin susceptibilityClin Infect Dis3852152814765345
- JacobsMR2007Retapamulin: a semisynthetic pleuromutilin compound for topical treatment of skin infections in adults and childrenFuture Microbiol259160018041900
- JansenWTVerelAVerhoefJ2007In vitro activity of telavancin against gram-positive clinical isolates recently obtained in EuropeAntimicrob Agents Chemother513420417606689
- JaureguiLEBabazadehSSeltzerE2005Randomized, double-blind comparison of once-weekly dalbavancin versus twice-daily linezolid therapy for the treatment of complicated skin and skin structure infectionsClin Infect Dis4114071516231250
- JeffresMNIsakowWDohertyJA2006Predictors of mortality for methicillin-resistant Staphylococcus aureus health-care-associated pneumonia: specific evaluation of vancomycin pharmacokinetic indicesChest1309475517035423
- JonesME2007In-vitro profile of a new beta-lactam, ceftobiprole, with activity against methicillin-resistant Staphylococcus aureusClin Microbiol Infect13Suppl 2172417488372
- JonesRNFritscheTRSaderHS2005Antimicrobial spectrum and potency of dalbavancin tested against clinical isolates from Europe and North America (2003): initial results from an international surveillance protocolJ Chemother (Florence, Italy)17593600
- JonesRNStilwellMGSaderHS2006Spectrum and potency of dalbavancin tested against 3322 Gram-positive cocci isolated in the United States Surveillance Program (2004)Diagn Microbiol Infect Dis541495316426793
- KalilACPuumalaSStonerJ2006Is linezolid superior to vancomycin for complicated skin and soft tissue infections due to methicillin-resistant Staphylococcus aureus?Antimicrob Agents Chemother501910 author reply,19101116641477
- KarlowskyJALaingNMBaudryT2007In vitro activity of API-1252, a novel FabI inhibitor, against clinical isolates of Staphylococcus aureus and Staphylococcus epidermidisAntimicrob Agents Chemother511580117220418
- KasiakouSKSermaidesGJMichalopoulosA2005Continuous versus intermittent intravenous administration of antibiotics: a meta-analysis of randomised controlled trialsLancet infect Dis5581916122681
- KazoryADibadjKWeinerID2006Rhabdomyolysis and acute renal failure in a patient treated with daptomycinJ Antimicrob Chemother57578916410267
- KingAPhillipsIKanigaK2004Comparative in vitro activity of telavancin (TD-6424), a rapidly bactericidal, concentration-dependent anti-infective with multiple mechanisms of action against Gram-positive bacteriaJ Antimicrob Chemother5379780315028667
- KingMDHumphreyBJWangYF2006Emergence of community-acquired methicillin-resistant Staphylococcus aureus USA 300 clone as the predominant cause of skin and soft-tissue infectionsAnn Intern Med1443091716520471
- KohlhoffSASharmaR2007IclaprimExpert Opin Investig Drug1614418
- KruegerWAKempfVAPeifferM2008Treatment with tigecycline of recurrent urosepsis caused by extended-spectrum-beta-lactamase-producing Escherichia coliJ Clin Microbiol468172018077630
- Labandeira-ReyMCouzonFBoissetS2007Staphylococcus aureus Panton-Valentine leukocidin causes necrotizing pneumoniaScience (New York, N.Y)31511303
- LalaniTBoucherHWCosgroveSE2008Outcomes with daptomycin versus standard therapy for osteoarticular infections associated with Staphylococcus aureus bacteraemiaJ Antimicrob Chemother6117718217999973
- LaueHWeissLBernardiA2007In vitro activity of the novel diaminopyrimidine, iclaprim, in combination with folate inhibitors and other antimicrobials with different mechanisms of actionJ Antimicrob Chemother601391417962215
- LeachKLSwaneySMColcaJR2007The site of action of oxazolidinone antibiotics in living bacteria and in human mitochondriaMol Cell2639340217499045
- LeuthnerKDCheungCMRybakMJ2006Comparative activity of the new lipoglycopeptide telavancin in the presence and absence of serum against 50 glycopeptide non-susceptible staphylococci and three vancomycin-resistant Staphylococcus aureusJ Antimicrob Chemother583384316787952
- LinGCreditoKEdnieLM2005Antistaphylococcal activity of dalbavancin, an experimental glycopeptideAntimicrob Agents Chemother49770215673763
- LivermoreDM2006Can beta-lactams be re-engineered to beat MRSA?Clin Microbiol Infect12Suppl 2111616524423
- LivermoreDM2005Tigecycline: what is it, and where should it be usedJ Antimicrob Chemother566111416120626
- LivermoreDMWarnerMMushtaqS2007In vitro activity of the oxazolidinone RWJ-416457 against linezolid-resistant and -susceptible staphylococci and enterococciAntimicrob Agents Chemother5111121417210773
- LodiseTPJrPypstraRKahnJB2007Probability of target attainment for ceftobiprole as derived from a population pharmacokinetic analysis of 150 subjectsAntimicrob Agents Chemother5123788717387149
- McAleeseFWuSWSieradzkiK2006Overexpression of genes of the cell wall stimulon in clinical isolates of Staphylococcus aureus exhibiting vancomycin-intermediate- S. aureus-type resistance to vancomycinJ Bacteriol18811203316428416
- McKinnonPSSorensenSVLiuLZ2006Impact of linezolid on economic outcomes and determinants of cost in a clinical trial evaluating patients with MRSA complicated skin and soft-tissue infectionsAnn Pharmacother4010172316720705
- MeagherAKAmbrosePGGraselaTH2005The pharmacokinetic and pharmacodynamic profile of tigecyclineClin Infect Dis41Suppl 5S33334016080071
- MekaVGGoldHS2004Antimicrobial resistance to linezolidClin Infect Dis3910101515472854
- MerouehSOBenczeKZHesekD2006Three-dimensional structure of the bacterial cell wall peptidoglycanProc Natl Acad Sci USA1034404916537437
- MicekST2007Alternatives to vancomycin for the treatment of methicillin-resistant Staphylococcus aureus infectionsClin Infect Dis45Suppl 3S18419017712745
- MohrJFMurrayBE2007Point: Vancomycin is not obsolete for the treatment of infection caused by methicillin-resistant Staphylococcus aureusClin Infect Dis4415364217516395
- Moise-BroderPAForrestABirminghamMC2004aPharmacodynamics of vancomycin and other antimicrobials in patients with Staphylococcus aureus lower respiratory tract infectionsClin Pharmacokinet439254215509186
- Moise-BroderPASakoulasGEliopoulosGM2004bAccessory gene regulator group II polymorphism in methicillin-resistant Staphylococcus aureus is predictive of failure of vancomycin therapyClin Infect Dis381700515227615
- Munoz-PriceLSLolansKQuinnJP2006Four cases of invasive methicillin-resistant Staphylococcus aureus (MRSA) infections treated with tigecyclineScand J Infect Dis381081417148081
- MurrayBE1990The life and times of the EnterococcusClin Microbiol Rev346652404568
- MurthyBSchmitt-HoffmannA2008Pharmacokinetics and pharmacodynamics of ceftobiprole, an anti-MRSA cephalosporin with broad-spectrum activityClin Pharmacokinet47213318076216
- MushtaqSWarnerMGeY2007In vitro activity of ceftaroline (PPI-0903M, T-91825) against bacteria with defined resistance mechanisms and phenotypesJ Antimicrob Chemother603001117548456
- MwangiMMWuSWZhouY2007Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencingProc Natl Acad Sci USA1049451617517606
- Navon-VeneziaSLeavittACarmeliY2007High tigecycline resistance in multidrug-resistant Acinetobacter baumanniiJ Antimicrob Chemother59772417353223
- NoelGJBushKBagchiP2008aA randomized, double-blind trial comparing ceftobiprole medocaril with vancomycin plus ceftazidime for the treatment of patients with complicated skin and skin-structure infectionsClin Infect Dis466475518225981
- NoelGJStraussRSAmslerK2008bResults of a double-blind, randomized trial of ceftobiprole treatment of complicated skin and skin structure infections caused by gram-positive bacteriaAntimicrob Agents Chemother52374417954698
- NovielloSIannielloFEspositoS2001In vitro activity of LY333328 (oritavancin) against Gram-positive aerobic cocci and synergy with ciprofloxacin against enterococciJ Antimicrob Chemother48283611481302
- OranjeAPChosidowOSacchidanandS2007Topical retapamulin ointment, 1%, versus sodium fusidate ointment, 2%, for impetigo: a randomized, observer-blinded, noninferiority studyDermatology (Basel, Switzerland)21533140
- PaceJLYangG2006Glycopeptides: Update on an old successful antibiotic classBiochem Pharmacol719688016412985
- PageMG2006Anti-MRSA beta-lactams in developmentCurr Opin Pharmacol648048516899405
- PatersonDL2006Clinical experience with recently approved antibioticsCurr Opin Pharmacol64869016904377
- PillaiSKSakoulasGWennerstenC2002Linezolid resistance in Staphylococcus aureus: characterization and stability of resistant phenotypeJ Infect Dis1861603712447736
- PillarCMAranzaMKShahD2008In vitro activity profile of ceftobiprole, an anti-MRSA cephalosporin, against recent Gram-positive and Gram-negative isolates of European originJ Antimicrob Chemother6159560218218646
- QueenanAMShangWKaniaM2007Interactions of ceftobiprole with {beta}-lactamases from molecular classes A to DAntimicrob Agents Chemother5130899517591851
- RoseWELeonardSNRossiKL2008Relationships between vancomycin pharmacodynamics and the emergence of vancomycin-intermediate Staphylococcus aureus (VISA) from heterogeneous VISA in an in vitro pharmacodynamic modelJ Antimicrob Chemotherdoi:10.1093/jac/dkn037.
- RoseWERybakMJ2006Tigecycline: first of a new class of antimicrobial agentsPharmacotherapy26109911016863487
- RoseWERybakMJKaatzGW2007aEvaluation of daptomycin treatment of Staphylococcus aureus bacterial endocarditis: an in vitro and in vivo simulation using historical and current dosing strategiesJ Antimicrob Chemother603344017540670
- RoseWERybakMJTsujiBT2007bCorrelation of vancomycin and daptomycin susceptibility in Staphylococcus aureus in reference to accessory gene regulator (agr) polymorphism and functionJ Antimicrob Chemother591190317434881
- RossoliniGM2007Redesigning beta-lactams to combat resistance: summary and conclusionsClin Microbiol Infect13Suppl 230317488374
- RovetaSMarcheseASchitoGC2008Activity of daptomycin on biofilms produced on a plastic support by Staphylococcus sppInt J Antimicrob Agents31321818201873
- RubinsteinECammarataSOliphantT2001Linezolid (PNU-100766) versus vancomycin in the treatment of hospitalized patients with nosocomial pneumonia: a randomized, double-blind, multicenter studyClin Infect Dis324021211170948
- RuheJJMonsonTBradsherRW2005Use of long-acting tetracyclines for methicillin-resistant Staphylococcus aureus infections: case series and review of the literatureClin Infect Dis4014293415844065
- RybakMJ2006The pharmacokinetic and pharmacodynamic properties of vancomycinClin Infect Dis42Suppl 1S353916323118
- SabathLDFinlandM1962Inactivation of methicillin, oxacillin and ancillin by Staphylococcus aureusProc Soc Exp Biol Med1115475013975861
- SaderHSFritscheTRJonesRN2008aAntimicrobial activity of ceftaroline and ME1036 tested against clinical strains of community-acquired Methicillin-resistant Staphylococcus aureus (CA-MRSA)Antimicrob Agents Chemotherdoi:10.1128/AAC.01351-07.
- SaderHSFritscheTRKanigaK2005Antimicrobial activity and spectrum of PPI-0903M (T-91825), a novel cephalosporin, tested against a worldwide collection of clinical strainsAntimicrob Agents Chemother4935011216048970
- SaderHSWattersAAFritscheTR2008bActivity of daptomycin and selected antimicrobial agents tested against Staphylococcus aureus from patients with bloodstream infections hospitalized in European medical centersJ Chemother (Florence, Italy)202832
- SaderHSWattersAAFritscheTR2007Daptomycin antimicrobial activity tested against methicillin-resistant staphylococci and vancomycin-resistant enterococci isolated in European medical centers (2005)BMC Infect Dis72917442104
- SakoulasGEliopoulosGMFowlerGMJr2005Reduced susceptibility of Staphylococcus aureus to vancomycin and platelet microbicidal protein correlates with defective autolysis and loss of accessory gene regulator (agr) functionAntimicrob Agents Chemother4926879215980337
- SakoulasGEliopoulosGMMoelleringRCJr2003Staphylococcus aureus accessory gene regulator (agr) group II: is there a relationship to the development of intermediate-level glycopeptide resistanceJ Infect Dis1879293812660939
- SakoulasGMoelleringRCJrEliopoulosGM2006Adaptation of methicillin-resistant Staphylococcus aureus in the face of vancomycin therapyClin Infect Dis42Suppl 1S405016323119
- SakoulasGMoise-BroderPASchentagJ2004Relationship of MIC and bactericidal activity to efficacy of vancomycin for treatment of methicillin-resistant Staphylococcus aureus bacteremiaJ Clin Microbiol42239840215184410
- SchneiderPHawserSIslamK2003Iclaprim, a novel diaminopyrimidine with potent activity on trimethoprim sensitive and resistant bacteriaBioorgMed Chem Lett13421721
- SchwaberMJWrightSBCarmeliY2003Clinical implications of varying degrees of vancomycin susceptibility in methicillin-resistant Staphylococcus aureus bacteremiaEmerg Infect Dis96576412781004
- SeltzerEDorrMBGoldsteinBP2003Once-weekly dalbavancin versus standard-of-care antimicrobial regimens for treatment of skin and soft-tissue infectionsClin Infect Dis37129830314583862
- ShawJPSeroogyJKanigaK2005Pharmacokinetics, serum inhibitory and bactericidal activity, and safety of telavancin in healthy subjectsAntimicrob Agents Chemother4919520115616296
- ShorrAFKunkelMJKollefM2005Linezolid versus vancomycin for Staphylococcus aureus bacteraemia: pooled analysis of randomized studiesJ Antimicrob Chemother569232916195255
- SieradzkiKLeskiTDickJ2003Evolution of a vancomycin-intermediate Staphylococcus aureus strain in vivo: multiple changes in the antibiotic resistance phenotypes of a single lineage of methicillin-resistant S. aureus under the impact of antibiotics administered for chemotherapyJ Clin Microbiol4116879312682161
- SieradzkiKRobertsRBHaberSW1999The development of vancomycin resistance in a patient with methicillin-resistant Staphylococcus aureus infectionN Engl J Med3405172310021472
- SievertDMRudrikJTPatelJB2008Vancomycin-resistant Staphylococcus aureus in the United States, 2002–2006Clin Infect Dis466687418257700
- SilvermanJAMortinLIVanpraaghAD2005Inhibition of daptomycin by pulmonary surfactant: in vitro modeling and clinical impactJ Infect Dis19121495215898002
- SimonGLSmithRHSandeMA1983Emergence of rifampin-resistant strains of Staphylococcus aureus during combination therapy with vancomycin and rifampin: a report of two casesRev Infect Dis5Suppl 3S50786635441
- SmithTLPearsonMLWilcoxKR1999Emergence of vancomycin resistance in Staphylococcus aureus. Glycopeptide-Intermediate Staphylococcus aureus Working GroupN Engl J Med34049350110021469
- SorianoAMarcoFMartinezJA2008Influence of vancomycin minimum inhibitory concentration on the treatment of methicillin-resistant Staphylococcus aureus bacteremiaClin Infect Dis4619320018171250
- SteenbergenJNAlderJThorneGM2005Daptomycin: a lipopeptide antibiotic for the treatment of serious Gram-positive infectionsJ Antimicrob Chemother55283815705644
- SteinGESchooleySPeloquinCA2007Linezolid tissue penetration and serum activity against strains of methicillin-resistant Staphylococcus aureus with reduced vancomycin susceptibility in diabetic patients with foot infectionsJ Antimicrob Chemother608192317673476
- StevensDLHerrDLampirisH2002Linezolid versus vancomycin for the treatment of methicillin-resistant Staphylococcus aureus infectionsClin Infect Dis3414819012015695
- StevensDLMaYSalmiDB2007Impact of antibiotics on expression of virulence-associated exotoxin genes in methicillin-sensitive and methicillin-resistant Staphylococcus aureusJ Infect Dis1952021117191165
- StreitJMFritscheTRSaderHS2004Worldwide assessment of dalbavancin activity and spectrum against over 6,000 clinical isolatesDiagn Microbiol Infect Dis481374314972384
- StreitJMSaderHSFritscheTR2005Dalbavancin activity against selected populations of antimicrobial-resistant Gram-positive pathogensDiagn Microbiol Infect Dis533071015922534
- StryjewskiMEChuVHO’RiordanWD2006Telavancin versus standard therapy for treatment of complicated skin and skin structure infections caused by gram-positive bacteria: FAST 2 studyAntimicrob Agents Chemother50862716495243
- StryjewskiMEO’RiordanWDLauWK2005Telavancin versus standard therapy for treatment of complicated skin and soft-tissue infections due to gram-positive bacteriaClin Infect Dis401601715889357
- SwensonJMTenoverFC2002In vitro activity of a new cephalosporin, RWJ-54428, against streptococci, enterococci and staphylococci, including glycopeptide-intermediate Staphylococcus aureusJ Antimicrob Chemother498455012003982
- TalbotGHBradleyJEdwardsJEJr2006Bad bugs need drugs: an update on the development pipeline from the Antimicrobial Availability Task Force of the Infectious Diseases Society of AmericaClin Infect Dis426576816447111
- TalbotGHThyeDDasA2007Phase 2 study of ceftaroline versus standard therapy in treatment of complicated skin and skin structure infectionsAntimicrob Agents Chemother5136121617682094
- TenoverFCMoelleringRCJr2007The rationale for revising the Clinical and Laboratory Standards Institute vancomycin minimal inhibitory concentration interpretive criteria for Staphylococcus aureusClin Infect Dis4412081517407040
- VandeneschFNaimiTEnrightMC2003Community-acquired methicillin-resistant Staphylococcus aureus carrying Panton-Valentine leukocidin genes: worldwide emergenceEmerg Infect Dis99788412967497
- VisalliMAMurphyEProjanSJ2003AcrAB multidrug efflux pump is associated with reduced levels of susceptibility to tigecycline (GAR-936) in Proteus mirabilisAntimicrob Agents Chemother47665912543675
- VossAMoutonJWvan ElzakkerEP2007A multi-center blinded study on the efficiency of phenotypic screening methods to detect glycopeptide intermediately susceptible Staphylococcus aureus (GISA) and heterogeneous GISA (h-GISA)Ann Clin Microbiol Antimicrobiol69
- VoyichJMOttoMMathemaB2006Is Panton-Valentine leukocidin the major virulence determinant in community-associated methicillin-resistant Staphylococcus aureus diseaseJ Infect Dis19417617017109350
- WalshTRHoweRAWoottonM2001Detection of glycopeptide resistance in Staphylococcus aureusJ Antimicrob Chemother47357811222571
- WeigeltJItaniKStevensDLinezolid versus vancomycin in treatment of complicated skin and soft tissue infectionsAntimicrob Agents Chemother492260615917519
- WunderinkRGRelloJCammarataSK2003Linezolid vs vancomycin: analysis of two double-blind studies of patients with methicillin-resistant Staphylococcus aureus nosocomial pneumoniaChest12417899714605050
- WysockiMDelatourFFaurissonF2001Continuous versus intermittent infusion of vancomycin in severe Staphylococcal infections: prospective multicenter randomized studyAntimicrob Agents Chemother452460711502515
- YamaokaT2007The bactericidal effects of anti-MRSA agents with rifampicin and sulfamethoxazole-trimethoprim against intracellular phagocytized MRSAJ Infect Chemother13141617593499
- YanKMaddenLChoudhryAE2006Biochemical characterization of the interactions of the novel pleuromutilin derivative retapamulin with bacterial ribosomesAntimicrob Agents Chemother5038758116940066
- ZeckelMLPrestonDAAllenBS2000In vitro activities of LY333328 and comparative agents against nosocomial gram-positive pathogens collected in a 1997 global surveillance studyAntimicrob Agents Chemother441370410770782