Abstract
Objective:
Assess the efficacy of duloxetine 60/120 mg (N = 162) once daily compared with placebo (N = 168) in the treatment of patients with fibromyalgia, during six months of treatment.
Methods:
This was a phase-III, randomized, double-blind, placebo-controlled, parallel-group study assessing the efficacy and safety of duloxetine.
Results:
There were no significant differences between treatment groups on the co-primary efficacy outcome measures, change in the Brief Pain Inventory (BPI) average pain severity from baseline to endpoint (P = 0.053) and the Patient’s Global Impressions of Improvement (PGI-I) at endpoint (P = 0.073). Duloxetine-treated patients improved significantly more than placebo-treated patients on the Fibromyalgia Impact Questionnaire pain score, BPI least pain score and average interference score, Clinical Global Impressions of Severity scale, area under the curve of pain relief, Multidimensional Fatigue Inventory mental fatigue dimension, Beck Depression Inventory-II total score, and 36-item Short Form Health Survey mental component summary and mental health score. Nausea was the most common treatment-emergent adverse event in the duloxetine group. Overall discontinuation rates were similar between groups.
Conclusions:
Although duloxetine 60/120 mg/day failed to demonstrate significant improvement over placebo on the co-primary outcome measures, in this supportive study, duloxetine demonstrated significant improvement compared with placebo on numerous secondary measures.
Introduction
Fibromyalgia is a chronic pain disorder characterized by widespread pain, tenderness, fatigue, sleep difficulties, and stiffness.Citation1–Citation3 Fibromyalgia occurs in about 2% of the general population of the United States and is more prevalent in women (estimated at 3.4% to 10.5%) than in men (0.5%).Citation4,Citation5
Serotonin and norepinephrine have been implicated in the mediation of endogenous analgesic mechanisms via the descending inhibitory pain pathways in the brain and spinal cord.Citation6,Citation7 An imbalance in these inhibitory mechanisms may contribute to central sensitization and hyperexcitability of the spinal and supraspinal pain-transmitting pathways. This imbalance may manifest as persistent pain.Citation8 Dysfunction in both serotonin and norepinephrine systems has been implicated in the etiology of fibromyalgia.Citation9–Citation12 Drugs often administered to treat fibromyalgia include analgesics,Citation13 nonsteroidal anti-inflammatory agents,Citation14 sedatives,Citation15 antidepressants,Citation16 and selective serotonin reuptake inhibitors.Citation17 In a 12-week trial, the serotonin and norepinephrine reuptake inhibitor milnacipran was shown to relieve fibromyalgia symptoms at a dose of 200 mg.Citation18
Duloxetine hydrochloride (Cymbalta®) is a selective serotonin and norepinephrine reuptake inhibitor that is relatively balanced in its affinity for both serotonin and norepinephrine reuptake inhibition. Preclinical models of central sensitization suggest that duloxetine will have efficacy in the treatment of persistent/chronic pain. In rodents, duloxetine has demonstrated efficacy in the formalin and capsaicin models of persistent pain, as well as in the partial sciatic nerve ligationCitation19 and L5/L6 spinal nerve ligation models of neuropathic pain.Citation20 Patients with fibromyalgia have quantitatively altered nociception compared with pain-free patients, suggesting that people with fibromyalgia process sensory information differently, most likely due to changes in the central processing of pain at the spinal level.Citation21 Current knowledge of fibromyalgia as a persistent pain condition due to central sensitization, along with the efficacy of duloxetine in preclinical analgesic models of central sensitization, suggests that duloxetine will have efficacy in the treatment of fibromyalgia.
Duloxetine is efficacious in the treatment of the painful physical symptoms associated with depressionCitation22 and the pain associated with diabetic neuropathy in nondepressed patients.Citation23,Citation24 Two double-blind, placebo-controlled, 12-week clinical trials of duloxetine have demonstrated its efficacy in patients with fibromyalgia with and without major depressive disorder (MDD). The first studyCitation25 examined the safety and efficacy of duloxetine 60 mg twice daily (BID) in male and female patients and the second studyCitation26 evaluated duloxetine 60 mg once daily (QD) and 60 mg BID in female patients.Citation26 The first study showed a significant effect on reduction of pain in women but not in men. The small number of men in the study (N = 25) may explain the unexpected discrepancy between results in men and women.
On the basis of the evidence that duloxetine (60 mg QD and/or 60 mg BID) was safe and efficacious in the treatment of fibromyalgia for 12 weeks,Citation25,Citation26 the following trial was conducted to assess the efficacy in pain reduction, as well as safety, of duloxetine 60/120 mg QD compared with placebo during six months of therapy.
Methods
Overview
This multicenter study (F1J-MC-HMEF) was conducted in 36 study centers in Germany, Spain, Sweden, the United Kingdom, and the United States. The study settings included outpatients from private practices and university clinics. Enrollment began in September 2005, and the study was completed in December 2006. Each site’s Institutional Review Board approved the protocol, and all patients provided written informed consent before study procedures were initiated.
Entry criteria
Patients were eligible for the study if they were ≥18 years of age and met criteria for fibromyalgia as defined by the American College of Rheumatology (ACR),Citation1 with or without MDD. Exclusion criteria included: current or previous treatment with duloxetine; any current primary Axis I diagnosis other than MDD (defined by the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition); pain symptoms related to traumatic injury, structural rheumatic disease, or regional rheumatic disease (such as osteoarthritis, bursitis, tendonitis); regional pain syndrome; multiple surgeries or failed back syndrome; confirmed current or previous diagnosis of rheumatoid arthritis, inflammatory arthritis, infectious arthritis, or an autoimmune disease; and serious medical illness.
Study design
This was a phase-III, parallel, double-blind, placebo-controlled study of patients who met the ACR criteria for fibromyalgia.Citation1 The study design is illustrated in . Patients were randomly assigned to either duloxetine 60 mg QD or placebo in a 1:1 ratio. Assignment to treatment groups was determined by a computer-generated random sequence within each study center, stratified by MDD status (yes, no).
Figure 1 Study design.
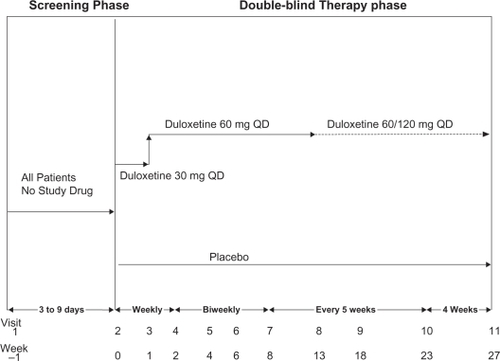
Following the one-week screening phase, patients were treated in a double-blind manner for 27 weeks. Patients randomly assigned to the duloxetine 60 mg QD treatment group underwent a titration in which they received duloxetine 30 mg QD for one week before receiving duloxetine 60 mg QD for 12 weeks. If at Visit 8 (Week 13) the patient did not have ≥50% reduction in the Brief Pain Inventory-Modified Short Form (BPI)Citation27 average pain score, the patient was blindly escalated to 120 mg QD. If the patient could not tolerate this dose, the patient was allowed to return to the 60-mg dose. Patients were allowed to increase their dose to 120 mg any time between Visits 8 and 10 (Weeks 13 and 23), based upon whether they had ≥50% reduction in their BPI average pain score. If at any time between Visits 9 and 11 (Weeks 18 and 27) the patient had tolerability issues with the higher dose (120 mg QD), the patient was allowed to go back to the lower dose (60 mg QD).
Outcome measures
Co-primary efficacy outcome measures
Reduction of pain severity was measured by the change in the average pain item of the BPICitation27 from baseline to endpoint; patient-reported improvement was assessed by the Patient’s Global Impressions of Improvement (PGI-I) questionnaireCitation28 at endpoint.
Secondary efficacy measures
The Fibromyalgia Impact Questionnaire (FIQ)Citation29 is a self-administered questionnaire that measures fibromyalgia patient status, progress, and outcomes over the past week. The total score ranges from 0 to 80. A higher score indicates a more negative impact. The Clinical Global Impressions of Severity (CGI-Severity)Citation28 scale evaluated the severity of illness at the time of assessment. The score ranges from 1 (normal, not at all ill) to 7 (among the most extremely ill patients). The CGI-Severity is administered by a study physician (blinded to study treatment) in the presence of the patient. The tender point pain thresholdCitation30 was assessed using a dolorimeter for all 18 tender points by a study physician or qualified study personnel (blinded to study treatment), and the mean of the thresholds and the number of tender points with a low threshold (≤4 kg/cm2) were evaluated. The area under the curve (AUC) of pain relief was derived from the BPI average pain score.Citation27 The BPI severity score (worst pain, least pain, average pain in the past 24 hours, pain right now) and average interference pain score are self-reported subscales that measure the severity of pain and the interference of pain on function. The severity scores range from 0 (no pain) to 10 (pain as bad as you can imagine). The interference scores range from 0 (does not interfere) to 10 (completely interferes) and include seven questions assessing the interference of pain in the past 24 hours for general activity, mood, walking ability, normal work, relations with other people, sleep, and enjoyment of life. The Multidimensional Fatigue Inventory (MFI)Citation31 is a 20-item, self-report instrument designed to collect data on the following dimensions: general fatigue, physical fatigue, mental fatigue, reduced motivation, and reduced activity. Each dimension is calculated as the sum of four statements regarding fatigue on a 0 to 5 scale, and thus, the score ranges between 0 and 20, with a higher score indicating a higher degree of fatigue. The 17-item Hamilton Depression Rating Scale (HAMD17)Citation32,Citation33 is a widely used observational rating measure of depression severity and its improvement during the course of therapy. The HAMD17 total score ranges from 0 (not at all depressed) to 52 (severely depressed). The Beck Depression Inventory-II (BDI-II)Citation34 is a 21-item patient-completed questionnaire designed to assess characteristics of depression. Each item is rated on a four-point scale (0 = not present; 3 = present in the extreme). The total score ranges from 0 to 63; the higher the score, the more severe the depressive symptoms.
Health outcome and quality of life measures
These included the patient-rated Sheehan Disability Scale (SDS),Citation35 the 36-item Short Form Health Survey (SF-36)Citation36 and the European Quality-of-Life Questionnaire – 5 Dimensions (EQ-5D).Citation37
Safety
The safety and tolerability of duloxetine were assessed by evaluation of spontaneously reported adverse events, reasons for discontinuation, laboratory tests (hematology, clinical chemistry, lipids), vital signs, weight, and electrocardiograms (ECG).
Statistical analysis
This study required the enrollment of 320 patients to have at least 80% power to detect a treatment group difference of −1.2 points in the baseline-to-endpoint mean change in BPI average pain score between duloxetine 60/120 mg QD and placebo, assuming a common standard deviation (SD) of 2.90, and the treatment group difference of 0.69 with a common SD of 1.68 for the endpoint PGI-I score. In the sample size calculation, a two-sided test with a significance level of 0.05 and a discontinuation rate of 40% were used.
All analyses were conducted on an intent-to-treat (ITT) basis. An ITT analysis is an analysis of data by the groups to which patients were assigned by random allocation, even if the patient did not take the assigned treatment, did not receive the correct treatment, or otherwise did not follow the protocol. All analyses, except response at endpoint based on ≥30% reduction in the BPI average pain score, were defined a priori (before unblinding) in the protocol or statistical analysis plan. Treatment effects were evaluated based on a two-sided significance level of 0.05 and interaction effects at a significance level of 0.10. No adjustments for multiple comparisons were made. Unless otherwise specified, when a total score was calculated from individual items, it was considered missing if any of the individual items were missing. For the SDS item of “work,” if a patient had not worked/studied at all during the past week for reasons unrelated to the disorder, this item was imputed by using the mean score from the other two items of the SDS for that patient. When a mean score was computed from individual items, it was calculated from existing values.
An analysis-of-covariance (ANCOVA) model was the primary analytic method used to analyze continuous efficacy variables, overall and within subgroups, where the model contained the main effects of treatment and investigator, with the baseline score as a covariate (Patient’s Global Impressions of Severity [PGI-S] for the analysis of the PGI-I). The treatment-by-investigator interaction was tested using a separate ANCOVA. When the interaction was statistically significant, the nature of the interaction was explored, but the treatment effect was evaluated using the model without the interaction term. The consistency of the effect of duloxetine compared with placebo on the BPI average pain score in patient subgroups of age (<65, ≥65), sex, race (Caucasian, other), major depressive disorder (yes, no), secondary diagnosis of anxiety (yes, no), and previous antidepressant use (yes, no) was investigated by adding the relevant subgroup and treatment-by-subgroup interaction terms to the ANCOVA model. Continuous demographic and baseline data, vital signs, ECG intervals and heart rate, and ranked laboratory data were analyzed using an analysis-of-variance (ANOVA) model that contained the main effects of treatment and investigator. Type II sums-of-squares for the least-squares (LS) mean were used for the statistical comparison using ANOVA or ANCOVA. Categorical variables were compared between treatment groups using Fisher’s exact test.
Some efficacy variables measured repeatedly over time were analyzed using a likelihood-based mixed-effects model repeated measures (MMRM) approach,Citation38 to better understand the time profile of response. The MMRM analyses use likelihood-based estimation, and subject-specific effects. Correlations between repeated measures are developed through the within-subject error correlation structure. The model included the fixed categorical effects of treatment, investigator, visit, and treatment-by-visit interaction, as well as the continuous fixed covariates of baseline score and baseline score-by-visit interaction. Type III sums-of-squares for the LS mean were used.
Treatment-emergent adverse events (TEAEs) were defined as events that first occurred or worsened after randomization as compared with the maximum prerandomization severity. Events were reported using preferred terms for Version 9.1 of MedDRA® (Medical Dictionary for Regulatory Activities) terminology.Citation39
Unless otherwise specified, “baseline” refers to the last nonmissing observation at or before the randomization visit (Visit 2, Week 0), and “endpoint” refers to the last nonmissing observation in the time period of analysis. The baseline used for determination of elevated blood pressure (BP) was the maximum prerandomization observation. When the investigator sites were used in the analyses, the sites having fewer than eight randomly assigned patients with a nonmissing value for baseline-to-endpoint change in the BPI average pain score were pooled and considered a single site. If a pooled site still had fewer than eight randomly assigned patients, these patients were pooled with the smallest remaining site. This pooling procedure continued until every site used in the analyses had at least eight patients with data for change in the BPI average pain score.
All statistical analyses were performed using SAS®, Version 9.1.3 (SAS Institute Inc., Cary, NC, USA), running on a UNIX® system using SAS® Drug Development. Throughout this article, the term “significant” indicates statistical significance, and “mean” refers to LS mean, except for demographic and clinical characteristics at baseline and for laboratory tests, for which it refers to the arithmetic mean.
Results
Patient disposition
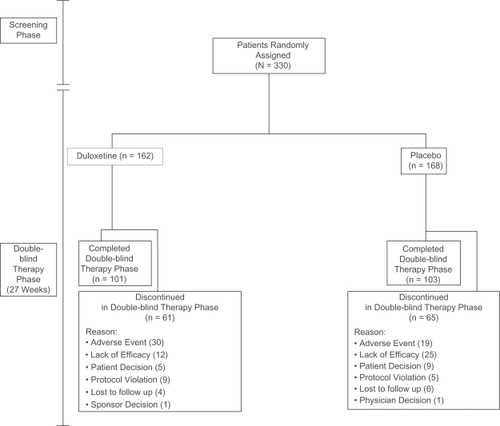
A total of 330 patients who met the entry criteria were randomly assigned to duloxetine 60 mg QD (N = 162) or placebo (N = 168) (). A significant difference was seen in rates of patients discontinuing due to lack of efficacy (duloxetine, 12 [7.4%], placebo 25 [14.9%], P = 0.036).
Figure 2 Patient disposition.
Baseline demographic and clinical characteristics
The majority of the patients were female (93.3%) and Caucasian (90.9%), followed by Hispanic (7.6%), and African descent (0.9%). The mean (SD) age of the enrolled patients was 50.5 (10.7) years, and 22.4% of the enrolled patients had current MDD. No significant differences among treatment groups were observed in any of the patient demographics or clinical characteristics ().
Table 1 Demographic and clinical characteristics at baseline
Efficacy
Although the co-primary efficacy measures of mean change in the BPI average pain score from baseline to endpoint and the mean PGI-I at endpoint showed greater numerical improvement for duloxetine- than placebo-treated patients, the differences between the treatment groups were not statistically significant (). For these, and several other variables, a significant treatment-by-investigator interaction was observed (). The nature of the interaction was investigated, and it could not be clearly explained by country-specific or geographic region (United States vs Europe) effects. The duloxetine group improved significantly more than the placebo group at all visits through Week 8 on both co-primary measures (BPI average pain score: Week 1 P < 0.001, Week 2 P < 0.001, Week 4 P = 0.001, Week 6 P = 0.002, Week 8 P = 0.015; PGI-I: Week 1 P = 0.043, Week 2 P = 0.005, Week 4 P = 0.014, Week 6 P = 0.003, Week 8 P = 0.007) as well as at Week 18 (P = 0.014) for the BPI average pain score, and Weeks 18 (P = 0.008), 23 (P = 0.002), and 27 (P = 0.003) for the PGI-I, from the repeated measures analyses. Duloxetine-treated patients had significantly greater AUC of pain relief (mean AUC = 270.07) than placebo-treated patients (mean AUC = 185.25; P = 0.010) (). Compared with the placebo group, the duloxetine group had a significantly greater improvement from baseline to endpoint in the BPI least pain score (mean change: duloxetine −1.22, placebo −0.73, P = 0.046) and average interference score (mean change: duloxetine −1.69, placebo −1.03, P = 0.009). In addition, significant between-group differences were observed in the FIQ pain score (mean change: duloxetine −1.69, placebo −1.06, P = 0.030), CGI-S (mean change: duloxetine −0.57, placebo −0.28, P = 0.011), MFI mental fatigue score (mean change: duloxetine −0.99, placebo −0.03, P = 0.023), and BDI-II total score (mean change: duloxetine −3.42, placebo −1.45, P = 0.017) ().
Table 2 Least-squares (LS) mean change from baseline to endpoint or at endpoint: efficacy measures
Analysis of the BPI average pain severity score response rates (defined as ≥50% reduction from baseline to endpoint) revealed no significant differences for the duloxetine group (29.1% [46/158], P = 0.455) compared with the placebo group (25.1% [42/167]). Analysis of the response rates at endpoint, based on a ≥30% reduction in the BPI average pain severity score, also revealed no significant difference between the duloxetine (38.0% [60/158]; P = 0.355) and placebo groups (32.9% [55/167]). Sustained response was defined as ≥50% reduction from baseline to endpoint in the BPI average pain severity, with a ≥50% reduction from baseline at an earlier visit, and with at least a 30% reduction from baseline at every visit with data in between. Analysis of the BPI average pain severity score sustained response showed no significant difference for the duloxetine group (23.4% [37/158], P = 0.272) compared with the placebo group (18.0% [30/167]).
For therapy-by-subgroup evaluation of the mean change in the BPI average pain score from baseline to endpoint, there was no significant interaction by age (P = 0.523), sex (P = 0.929), race (P = 0.382), diagnosis of MDD (P = 0.496) and secondary diagnosis of anxiety (P = 0.152). Among patients with a diagnosis of MDD, the mean change (SE) from baseline for duloxetine-treated patients was −1.34 (0.42) and in placebo-treated patients was −0.48 (0.46). For patients who did not have a diagnosis of MDD, the mean change (SE) from baseline for duloxetine-treated patients was −1.60 (0.23) and in placebo-treated patients was −1.20 (0.22). There was a significant therapy-by-previous antidepressant use interaction (P = 0.029). Among patients with previous antidepressant use, the mean change (SE) in the BPI average pain score for duloxetine-treated patients was −1.85 (0.29) and in placebo-treated patients was −0.65 (0.27). For patients without previous antidepressant use, the mean change (SE) for duloxetine-treated patients was −1.56 (0.29) and in placebo-treated patients was −1.51 (0.27).
Health outcomes and quality of life
Duloxetine-treated patients had significantly greater improvements, compared with placebo-treated patients in the SF-36 mental component summary score (mean change: duloxetine 3.37, placebo 0.79, P = 0.026), and SF-36 mental health score (mean change: duloxetine 6.63, placebo 1.19, P = 0.005) ().
Table 3 Least-squares (LS) mean change from baseline to endpoint: health outcome and quality-of-life measures
Safety
For the 117 duloxetine-treated patients who continued past Week 13, the number of patients who stayed on duloxetine 60 mg QD were 19 (16.2%) and those who escalated were 98 (83.8%). The mean (SD) average prescribed daily dose between Weeks 13 and 27 for patients who escalated (N = 97) was 113.4 (12.6) mg.
Of the 330 randomly assigned patients, 145/162 (89.5%) duloxetine- and 137/168 (81.5%) placebo-treated patients reported at least 1 TEAE (P = 0.043). TEAEs that occurred in ≥5% of duloxetine- and twice the rate of placebo-treated patients are shown in . There were no TEAEs that occurred at a significantly higher rate in the placebo- compared with the duloxetine-treated group. No deaths occurred during the study. No significant difference between treatment groups was observed in the percentage of patients with at least 1 serious adverse event (SAE). During the study, 4 patients (2.5%) in the duloxetine-treated group experienced the SAEs of arthralgia, gait disturbance, pseudomonal lung infection, muscular weakness, paraesthesia, and pseudoneurologic symptom (1 [0.6%] for each event), and 4 (2.4%) patients in the placebo-treated group experienced the SAEs of arthralgia, abdominal pain upper, cystocele, and noncardiac chest pain (1 [0.6%] for each event). A total of 49 (14.8%) patients discontinued during the therapy phase due to an adverse event, with no significant difference between the duloxetine- and placebo-treated groups (duloxetine, 30 [18.5%]; placebo, 19 [11.3%]; P = 0.088). The most common (≥1%) adverse events reported as reasons for discontinuation in the duloxetine-treated group were nausea (3 [1.9%]), dizziness, diarrhea, lethargy, somnolence, and vomiting (2 [1.2%] for each event), and in the placebo-treated group, dizziness and irritability (2 [1.2%] for each event).
Table 4 Treatment-emergent adverse events that occurred in ≥5% of duloxetine patients and at least twice the rate in placebo patients
There were significant differences between the duloxetine-and the placebo-treated groups for mean change in alkaline phosphatase (mean change [SD] units/liter: duloxetine = 2.38 [12.02], placebo = −2.45 [9.94]; P < 0.001), alanine transaminase/serum glutamate pyruvate transaminase (mean change [SD], units/liter: duloxetine, 6.97 [50.29]; placebo, −0.93 [5.70]; P = 0.038), total bilirubin (mean change [SD], micromole/liter: duloxetine, −0.35 [2.97]; placebo, 0.27 [2.54]; P = 0.020), cholesterol (mean change [SD], micromole/liter: duloxetine −0.04 [0.71]; placebo, −0.23 [0.75]; P = 0.016), and uric acid (mean change [SD], micromole/liter: duloxetine, −19.51 [43.50]; placebo, 3.91 [42.92]; P < 0.001).
Significant treatment group differences were observed for mean change in sitting pulse rate (mean change [SE], beats/minute: duloxetine, 1.08 [0.87]; placebo, −1.64 [0.84]; P = 0.016) and diastolic BP (mean change [SE], mmHg: duloxetine, 1.68 [0.85]; placebo, −1.46 [0.82]; P = 0.004). Three patients in each group experienced sustained elevation of BP (defined as systolic BP ≥140 and ≥10 mmHg increase for at least 3 consecutive visits or diastolic BP ≥90 and ≥10 mmHg increase for at least 3 consecutive visits), and the between-group difference was not significant.
No significant differences were observed between treatment groups in mean change for corrected QT intervals using either Fridericia’s (QTcF), Bazett’s (QTcB), or the regression correction, and in the QRS interval. Significant differences were observed between treatment groups in mean change in the PR interval (millisecond) from baseline to endpoint (mean change [SE]: duloxetine, −4.51 [1.20]; placebo, 1.97 [1.14]; P < 0.001), QT interval (mean change [SE]: duloxetine, −4.27 [1.98]; placebo, 3.79 [1.87]; P = 0.002), RR interval (mean change [SE]: duloxetine, −36.00 [10.73]; placebo, 14.51 [10.15]; P < 0.001), and heart rate (mean change [SE] beats/minute: duloxetine, 2.87 [0.83]; placebo, −0.99 [0.79]; P < 0.001).
Discussion
In this phase-III, parallel, double-blind, 27-week, placebo-controlled trial, no statistically significant differences were observed between the treatment groups for the co-primary efficacy measures (mean change in BPI average pain score from baseline to endpoint and mean PGI-I score at endpoint). For these, and several other variables, there was a statistically significant treatment-by-investigator interaction (shown in and ); therefore, the overall effect must be interpreted with caution. The nature of the interaction was investigated, and it could not be clearly explained by country-specific or geographic region (United States vs Europe) effects. The duloxetine group improved significantly more than the placebo group at all visits through Week 8, as well as at Week 18, for the BPI average pain score and at all visits through Week 8, as well as Weeks 18, 23, and 27 for the PGI-I.
The magnitude of the treatment benefit reported in the current study for the mean change in the BPI average pain score from baseline to endpoint and the PGI-I score at endpoint are not consistent with those reported in the previous clinical trials evaluating duloxetine in the treatment of fibromyalgia.Citation25,Citation26 Most previous pain studies were 12 weeks or less in duration.Citation23–Citation26 In the previous phase-II, 12-week, randomized, placebo-controlled trial of duloxetine in which male and female patients with ACR-defined primary fibromyalgia were randomized to duloxetine 60 mg BID or placebo,Citation25 duloxetine was superior to placebo on the BPI average pain score and PGI-I score. The previous phase-III study of female patients randomized to duloxetine 60 mg QD, 60 mg BID, or placeboCitation26 also showed superiority of duloxetine compared with placebo on the BPI average pain score and the PGI-I score. The treatment effects for duloxetine compared with placebo for the co-primary efficacy variables of the BPI average pain score and the PGI-Improvement in this study were smaller than were observed in three other placebo-controlled studies of duloxetine. Also, the treatment effects for almost all secondary measures were smaller than in the previous studies. Rater training and other aspects of study conduct were similar for all the studies. One difference between this and previous studies is the statistically significant treatment-by-investigator interactions for the mean change in both co-primary variables and for many secondary measures in this study. Eighteen of the 36 investigators had small numbers of patients with data and were pooled into one investigator for analysis, for which the results were numerically in favor of duloxetine compared with placebo for both the BPI average pain score and the PGI-Improvement. However, for the remaining 18 investigators, the results were numerically in favor of placebo compared with duloxetine for 10 investigators for the BPI and for seven investigators for the PGI-Improvement. This was the first study to include patients in the United States and also elsewhere (Germany, Spain, Sweden, and the United Kingdom). The interaction did not appear to be attributable to country or region (United States vs Europe). In spite of the treatment reversals for a large number of investigators, which dilutes the overall treatment effect, there were large benefits for duloxetine compared with placebo for some investigators, resulting in an overall numerical advantage for duloxetine for both the BPI average pain score and the PGI-Improvement, indicating the supportive nature of this study for a treatment benefit for duloxetine in the treatment of patients with fibromyalgia.
Compared with placebo-treated patients, patients treated with duloxetine had significantly greater AUC of pain relief and experienced greater improvements in the BPI least pain score and average interference score. In addition, duloxetine-treated patients experienced significantly greater improvements, compared with placebo, in the FIQ pain item, MFI mental fatigue dimension, CGI-S, and BDI-II total scores. Approximately 22% of all patients had MDD at baseline, which is consistent with the prevalence of depression concurrent with fibromyalgia (22%–45%).Citation4,Citation40–Citation43 No treatment-by-MDD interaction was observed for mean change in the BPI average pain score. In the present study there was no significant therapy-by-sex interaction, suggesting that the effect of duloxetine compared with placebo was similar in males and females. A previous clinical trial evaluating duloxetine in the treatment of fibromyalgia demonstrated a significant effect on reduction of pain in women but not in men,Citation25 although this discrepancy might have been be due to the small number of men enrolled in the study.
Generalizability of these results is limited by the fact that patients in this study were carefully selected to exclude psychiatric and medical co-morbidities, and could be less severely ill than fibromyalgia patients in the general population.
In summary, in patients with fibromyalgia, with or without MDD, duloxetine 60/120 mg/day improved average pain severity and self-reported global improvement through week 8 (and some subsequent visits), but not at endpoint, relative to placebo. Consistent with earlier trials of duloxetine in the treatment of fibromyalgia,Citation25,Citation26 in this study, duloxetine was safely administered, and relatively well tolerated (considering the length of the trial) by most patients. Although duloxetine failed to demonstrate significant improvement over placebo on the co-primary outcome measures, in this supportive study, duloxetine demonstrated significant improvement compared with placebo on a number of secondary measures that are important in assessing treatment efficacy in patients with fibromyalgia.
Acknowledgements
This work was sponsored by Eli Lilly and Company and Boehringer Ingelheim GmbH. Trial Investigators were: Dr Gabriel Herrero-Beaumont, Fundación Jiménez Díaz; Dr Arturo Rodriguez De La Serna, Hospital Santa Creu I Sant Pau; Dr Cayetano Alegre De Miquel, Ciutat Sanitaria De La Vall De Hebron; Dr Juan Roberto Minguélez Sánchez, Hospital De Mostoles; Dr Javier Rivera Redondo, Instituto De Rehabilitación-Hospital Gregorio Marañón; Dr Roland Woerz (Office); Dr Wolfgang Bolten, Rheumaklinikum Wiesbaden-Zentrum Für Schmerztherapie; Dr Wolfgang Brueckle, Rheumaklinik Bad Nenndorg; Dr Gerhard Mueller-Schwefe (Office); Dr Wolfgang Molt (Practice); Dr Michael Ribbat (Office); Dr Walter Albrecht (Practice), Dr Michael Spaeth (Practice), Dr Olof Zachrisson, Gottfriesmottagningen; Dr Britt-Marie Öberg, Danderyds Sjukhus Rehabiliteringsmedicinska kliniken; Dr Jan Sorensen, Universitetssjukhuset Linkoping Smärt-och rehabcenter Smärtoch Rehabcenter; Dr Brian Hazleman, Addenbrookes Hospital; Dr Anthony D Woolf, Royal Cornwall Hospital; Dr Bruce Kirkham, Guys and St. Thomas Hospital Rheumatology Department; Dr Ashok Bhalla, Royal National Hospital For Rheumatic Diseases; Dr James R. Bellor, Columbia Medical Practice; Dr Laurence Bradley, University of Alabama at Birmingham; Dr Nicholas A. Bertini, Clinical Research Consultants, Inc.; Dr Mildred V. Farmer, Meridien Research; Dr H. S. Eugene Fung, Arthritis and Osteoporosis Clinic Of Central Texas; Dr Ronica Kluge, Clinical Physiology Associates, Clinical Study Center; Dr Andrew F. Leuchter, Neuropsychiatric Institute; Dr Lori Wynstock, Southern California Clinical Lab; Dr C Brendan Montano, Connecticut Clinical Trials Llc; Dr Paul K. Pickrell, Metaclin Research Inc.; Dr Steven Elliot, Medisphere Medical Research Center Llc; Dr Oscar Soto (Private); Dr Harris H. Mcilwain, Tampa Medical Group, P. A; Dr Craig M. Mccarthy, Pivotal Research Centers; Dr Siavash Nael, Neuropsychiatric Center; Dr Timothy R. Smith, Mercy Health Research.
Disclosures
Drs. Chappell, Detke, and D’Souza are employees and stockholders of Eli Lilly and Company. Dr Wiltse is a former employee of Eli Lilly and Company. Dr Spaeth is a consultant to Allergan, Eli Lilly, Jazz, and Pierre Fabre Medicament, and is on the speaker bureaus of Eli Lilly and Pierre Fabre Medicament. Dr Bradley is a consultant for Eli Lilly, Pfizer, and Forest; has received grant/research support from the National Institutes of Health, the Agency for Healthcare Research and Quality, Eli Lilly, Pfizer, and the American Fibromyalgia Syndrome Association; has received honoraria from Eli Lilly, Pfizer, Forest, and the Society for Women’s Health Research; is a member of the speaker/advisory board for Pfizer; and has received royalties from UpToDate Rheumatology.
References
- WolfeFSmytheHAYunusMBThe American College of Rheumatology 1990 criteria for the classification of fibromyalgia. Report of the Multicenter Criteria CommitteeArthritis Rheum1990331601722306288
- HudsonJIPopeHGJrThe relationship between fibromyalgia and major depressive disorderRheum Dis Clin N Am199622285303
- Blackburn-MunroGBlackburn-MunroREChronic pain, chronic stress and depression: coincidence or consequence?J Neuroendocrinol2001131009102311722697
- WolfeFRossKAndersonJThe prevalence and characteristics of fibromyalgia in the general populationArthritis Rheum19953819287818567
- NeumannLBuskilaDEpidemiology of fibromyalgiaCurr Pain Headache Rep2003736236812946289
- BasbaumAIFieldsHLEndogenous pain control systems: brainstem spinal pathways and endorphin circuitryAnnu Rev Neurosci198473093386143527
- ClarkFMProudfitHKThe projections of noradrenergic neurons in the A5 catecholamine cell group to the spinal cord in the rat: anatomical evidence that A5 neurons modulate nociceptionBrain Res19936162002107689410
- CoderreTJKatzJPeripheral and central hyperexcitability: differential signs and symptoms in persistent painBehav Brain Sci199720404419discussion 435–513.10097003
- RussellIJMichalekJEVipraioGAPlatelet 3H-imipramine uptake receptor density and serum serotonin levels in patients with fibromyalgia/fibrositis syndromeJ Rheumatol1992191041091313504
- RussellIJVaeroyHJavorsMCerebrospinal fluid biogenic amine metabolites in fibromyalgia/fibrositis syndrome and rheumatoid arthritisArthritis Rheum1992355505561374252
- YunusMBDaileyJWAldagJCPlasma tryptophan and other amino acids in primary fibromyalgia: a controlled studyJ Rheumatol19921990941556707
- LegangneuxEMoraJJSpreux-VaroquauxOCerebrospinal fluid biogenic amine metabolites, plasma-rich platelet serotonin and [3H]imipramine reuptake in the primary fibromyalgia syndromeRheumatology (Oxford)20014029029611285376
- WolfeFZhaoSLaneNPreference for nonsteroidal antiinflammatory drugs over acetaminophen by rheumatic disease patients: a survey of 1,799 patients with osteoarthritis, rheumatoid arthritis, and fibromyalgiaArthritis Rheum20004337838510693878
- YunusMBMasiATAldagJCShort term effects of ibuprofen in primary fibromyalgia syndrome: a double blind, placebo controlled trialJ Rheumatol19891652732Erratum in: J Rheumatol 1989;16:855.2664173
- BennettRMGatterRACampbellSMA comparison of cyclobenzaprine and placebo in the management of fibrositis. A double-blind controlled studyArthritis Rheum198831153515423058130
- GoldenbergDLFelsonDTDinermanHA randomized, controlled trial of amitriptyline and naproxen in the treatment of patients with fibromyalgiaArthritis Rheum198629137113773535811
- WolfeFCatheyMAHawleyDJA double-blind placebo controlled trial of fluoxetine in fibromyalgiaScand J Rheumatol1994232552597973479
- GendreauRMThornMDGendreauJFEfficacy of milnacipran in patients with fibromyalgiaJ Rheumatol2005321975198516206355
- SeltzerZDubnerRShirYA novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injuryPain1990432052181982347
- KimSHChungJMAn experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the ratPain1992503553631333581
- BennettRMEmerging concepts in the neurobiology of chronic pain: evidence of abnormal sensory processing in fibromyalgiaMayo Clin Proc19997438538910221469
- GoldsteinDJLuYDetkeMJEffects of duloxetine on painful physical symptoms associated with depressionPsychosomatics200445172814709757
- GoldsteinDJLuYDetkeMJDuloxetine vs placebo in patients with painful diabetic neuropathyPain200511610911815927394
- WernickeJFPritchettYLD’SouzaDNA randomized controlled trial of duloxetine in diabetic peripheral neuropathic painNeurology2006671411142017060567
- ArnoldLMLuYCroffordLJA double-blind, multicenter trial comparing duloxetine to placebo in the treatment of fibromyalgia patients with or without major depressive disorderArthritis Rheum2004502974298415457467
- ArnoldLMRosenAPritchettYLA randomized, double-blind, placebo-controlled trial of duloxetine in the treatment of women with fibromyalgia with or without major depressive disorderPain200511951516298061
- CleelandCSRyanKMPain assessment: global use of the Brief Pain InventoryAnn Acad Med Singap1994231291388080219
- GuyWECDEU Assessment Manual for Psychopharmacology, Revised Rockville (MD)US Dept of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute of Mental Health, Psychopharmacology Research Branch, Division of Extramural Research Programs1976
- BurckhardtCSClarkSRBennettRMThe fibromyalgia impact questionnaire: development and validationJ Rheumatol1991187287331865419
- FischerAAPressure threshold meter: its use for quantification of tender spotsArch Phys Med Rehabil1986678368383778185
- SmetsEMGarssenBBonkeBThe Multidimensional Fatigue Inventory (MFI) psychometric qualities of an instrument to assess fatigueJ Psychosom Res1995393153257636775
- HamiltonMA rating scale for depressionJ Neurol Neurosurg Psychiatry196023566214399272
- HamiltonMDevelopment of a rating scale for primary depressive illnessBr J Soc Clin Psychol196762782966080235
- BeckATEpsteinNBrownGAn inventory for measuring clinical anxiety: psychometric propertiesJ Consult Clin Psychol1988568938973204199
- SheehanDHarnett-SheehanKRajBThe measurement of disabilityInt Clin Psychopharmacol1996113 Suppl89958923116
- WareJESnowKKKosinskiMSF-36 health survey: manual and interpretation guideBoston, MAThe Health Institute, New England Medical Center1993
- KindPThe EuroQoL instrument: an index of health-related quality of lifeSpilkerBQuality of Life and Pharmacoeconomics in Clinical Trials2nd edPhiladelphia (PA)Lippincott Williams and Wilkins1996191201
- MallinckrodtCHSangerTMDubéSAssessing and interpreting treatment effects in longitudinal clinical trials with missing dataBiol Psychiatry20035374376012706958
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. Medical Dictionary for Regulatory Activities Terminology (MedDRA). 2007Accessed June 8, 2007. Available from: http://meddramsso.com
- CelikerRBormanPOktemFPsychological disturbance in fibromyalgia: relation to pain severityClin Rheumatol1997161791849093801
- AnderbergUMForsgrenTEkseliusLPersonality traits on the basis of the Temperament and Character Inventory in female fibromyalgia syndrome patientsNord J Psychiatry199953353359
- EpsteinSAKayGClauwDPsychiatric disorders in patients with fibromyalgia. A multicenter investigationPsychosomatics19994057639989122
- WhiteKPNielsonWRHarthMChronic widespread musculoskeletal pain with or without fibromyalgia: psychological distress in a representative community adult sampleJ Rheumatol20022958859411908578