Abstract
Most patients contract hypoxic encephalopathy after suffering a cardiac arrest. They usually endure severe neurological sequelae and the temporal profile of the disease progression remains unclear. This case study shows how the effects of hypoxic encephalopathy continue to progress for several years after the initial event. Up to eight years after the hypoxic insult, the patient’s intellect steadily deteriorated, and brain atrophy progressed. As the hypoxic insult on the brain is only transient, the neurological disability seems not to be exacerbated for years. However, our case indicates that this disorder may have a long progression.
Keywords:
Introduction
Recent medical progress has led to an increase in the number of cardiac arrest survivors. According to one estimate, up to 200 people per 1 million inhabitants are saved after cardiac arrest in developed countries (CitationRoine 1996). However, about half of these survivors suffer severe neurological sequelae (CitationBRCT 1991), because the brain is the organ most vulnerable to hypoxia. Focal neurological deficit such as sensorimotor impairment is rare (CitationRoine 1996), but neuropsychological symptoms are common, which can be attributed to cortical as well as diffuse white matter injury (CitationRoine et al 1993a).
The clinical features and their influence on the eventual mortality rate have been investigated extensively (CitationLongstreth et al 1983; CitationLevy et al 1985). However the temporal profile of disease progression, and the clinical characteristics of the chronic stage have not yet been fully elucidated. Even after an intensive search, we could find only a few reports in which radiological exacerbation was demonstrated for no more than a few months (CitationSawada et al 1990; CitationTakahashi et al 1993). Thus whether or not the neurological disability continues to progress for years in hypoxic encephalopathy is still unclear. In this report, we describe a case of hypoxic encephalopathy with years of slow progression, both clinically and radiologically. The hypoxic insult may be only transient, but the progression of hypoxic encephalopathy may be long-lasting.
Case report
A 53-year-old woman with no remarkable disease history was referred to our hospital because of abdominal pain and liver dysfunction. Choledocholithiasis was suspected, and endoscopic retrograde cholangiopantreatography (ERCP) was carried out on December 9, 1999. Papillotomy was simultaneously performed, and the stones in the duct were removed. Just after the procedure, the physician noticed that the patient was not breathing and was pulseless. She had ventricular fibrillation, and intensive resuscitation was immediately attempted. Defibrillation successfully achieved regular heart sinus rhythm. Blood pressure was maintained using catecholamines. She was intubated and mechanically ventilated, and the arterial blood gas was kept within the appropriate range thereafter. The duration of hypoxia suffered by her brain is uncertain, but was estimated to be less than 10 minutes because a nurse confirmed that the patient was alert several minutes before the physician noticed her collapse.
During resuscitation, the physician confirmed that the patient’s bilateral pupils were dilated to 8 mm in diameter. Three hours later, this dilation decreased to 3 mm in diameter. The patient was comatose for the first several hours, but could open her eyes when called 12 hours after resuscitation. Normal respiration and blood pressure were gradually regained. She was extubated at 5 days and catecholamines were discontinued at 6 days.
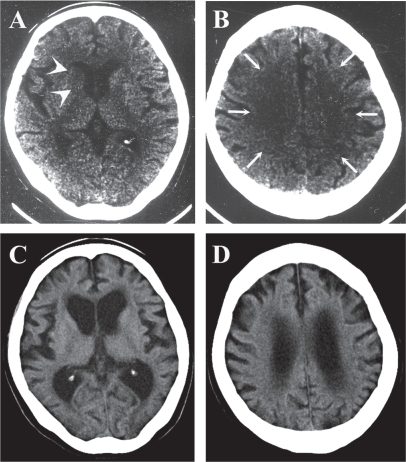
On December 15, when her cardiopulmonary state became stable, she was examined by our neurology department. She was awake and could obey only simple requests such as hand grasping. Precise verbal communication was difficult for her. Although obeying complex requests was difficult at that time, no definite sensorimotor neurological deficits were confirmed. A electroencephalogram revealed theta waves of 5 to 7 Hz bilaterally, with no paroxysms. Computed tomography (CT) ( and ) showed decreased density at the right caudate head (, arrowheads) and diffuse low density in the bilateral cerebral white matter (, arrows). Marked brain edema was not observed however ( and ).
Figure 1 Computed tomography (CT) images in December 2000 showed decreased density at the right caudate head (A, arrowheads) and in the bilateral cerebral white matter (B, arrows). Those in November 2007 (C, D) showed marked cortical atrophy and dilatation of ventricles.
Her mental state gradually improved. Two weeks later when she visited the neurology department for the second time, she could verbally express her distress. On the other hand, her recent memory was still severely impaired. Furthermore, motor clumsiness and intention tremor became apparent, which may only have been revealed as a result of our extensive neurological examination. Nevertheless, the patient could walk independently. She was discharged and returned home on February 5, 2000. Although dependent, she could live in her own home with no extreme difficulty. Magnetic resonance imaging (MRI) on February 9 () revealed scattered foci of high intensity lesions in the cerebral cortex of the T1-weighed image ( and , arrowheads). The right caudate head, which showed decreased density by CT in the acute stage (), showed high intensity in the T1-weighed image (, arrow). In addition, slight brain atrophy was suspected when the patient’s age was taken into account. The T-2 weighed image, however, showed no abnormal intensity at this time point ().
Figure 2 Serial changes of the brain magnetic resonance imaging (MRI) (A–F, February, 2000; G–L, May, 2004; M–R, November, 2007). In 2000, scattered high intensity lesions were confirmed both in the caudate (B, arrow) and cerebral cortex (A and C, arrowheads) by T1-weighed image, though T2-weighed images (D–F) revealed no definite abnormalities. In 2004, the cerebral sulci became wider (H, arrowheads, and I, arrow) and the ventricles became larger (H, arrows). T2-weighed image showed high intensity lesions in the subcortical white matter (L, arrow). The brain became further atrophic in 2007, with widening of cerebral sulci (N, arrowheads) and dilatation of ventricles (M, N, and O, arrows). High intensity lesions were confirmed in the subcortical white matter of the occipital (Q, arrows) and parietal (R, arrows) lobes.
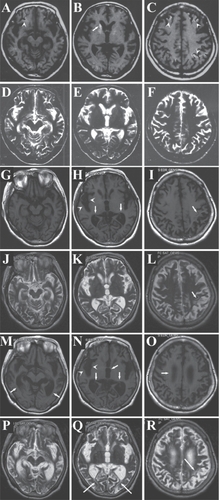
Her neurological findings seemed stable. Dementia as well as clumsiness and intention tremor continued, but no exacerbation was apparent for a while. By April 2002, on the Revised Hasegawa Dementia Scale, an analogue of Mini-Mental Status Examination in Japan (CitationJeong et al 2007), the patient had gained 18 points. Disturbance of calculation and recent memory markedly decreased the total score, but both spatial and temporal orientation had been spared any damage. Nevertheless, her family felt that her mentality began to deteriorate from that time. They insisted that the patient had become more dependent in daily activities, such as excretion and bathing. By 2003, we also noticed that she needed more support in her daily activities. Her disattentive state worsened, though motor function seemed stable. Her score on the Revised Hasegawa Dementia Scale at May 21, 2004 was 8 points, with marked disorientation as well as memory disturbance. MRI examined on the same day () revealed that the cerebral sulci had become wider ( arrowhead, arrow), suggesting that the cerebral cortex had degenerated further. Dilatation of the lateral ventricles (, arrows) indicated that cerebral white matter had decreased in volume. In some subcortical white-matter regions, high intensity lesions were newly discernible by T2-weighed image (, arrow).
Her mentality continued to deteriorate thereafter. Her ability to move worsened, so that from late 2004, she could no longer walk independently. Because of the difficulty in coming to hospital, her care was taken over by a physician and she did not visit our neurology department for more than 3 years. When she visited us in November 2007, establishing a score on the Revised Hasegawa Dementia Scale was difficult because of her severe dementia: she was unable to answer our questions. Evaluating precise motor function was also difficult, but intention tremor was still evident. CT showed marked cortical atrophy and dilatation of ventricles ( and ). MRI (–R) revealed further widened cerebral sulci (, arrowheads) and pronounced lateral ventricle dilatation (–O, arrows). The third ventricle was also dilated (, arrow). The T2-weighed image showed scattered high-intensity foci in the subcortical white matter of the occipital (, arrows) and parietal (, arrow) lobes. It was suggested that cortical as well as subcortical structures were affected in a delayed manner.
The patient continues to be cared for in her home, because her family strongly prefers it. A helper visits frequently, and monitors her general condition carefully.
Discussion
Hypoxic encephalopathy can be caused by various disorders, but the most common etiology is cardiac arrest (CitationRoine 1996). Coma and seizure are commonly observed in the first several hours to days. About half of these patients die, but others take the course of gradual recovery or persistent vegetative state. The determinants of neurological outcome are not fully elucidated, but experimental studies suggest that brain temperature and blood glucose level influence the prognosis (CitationLeonov et al 1990; CitationStrong et al 1990). Clinical studies show that long duration of initial unconsciousness (CitationLongstreth et al 1983), and the occurrence of generalized myoclonus, (CitationWijdicks et al 1994) may be ominous indicators of a poor outcome. Of interest is that delayed resuscitation does not necessarily mean severe neurological sequelae (CitationRoine et al 1993a); some neurons may degenerate only when post-ischemic activity is regained early (discussed below).
Apart from the above mentioned clinical and experimental studies, no reports were found of the investigation of disease progression over several years. CitationRoine and colleagues (1993a) followed neuropsychological sequelae after hypoxic encephalopathy, and showed that patients with severe cognitive impairment gradually declined. However, we should note that their patients were studied for only up to 12 months. Some patients with mild or moderate cognitive impairment could possibly show cognitive decline several years later. We found no studies that followed neurological sequelae of hypoxic encephalopathy for more than 1 year. This may be, at least in part, because patients suffering from hypoxic encephalopathy are not followed by the same doctor for very long.
Our case showed not only clinical but also radiological exacerbation in a delayed manner ( and ). Early CT features of hypoxic encephalopathy include decreased density in the basal ganglia, cerebral cortex, and cerebral white matter especially at the watershed area (CitationKjos et al 1983; CitationTippin et al 1984). Our case also showed these features, though decreased density in the cerebral cortex was obscure ( and ). A previous report showed that the CT number in the putamen correlated with good prognosis (CitationYanagawa et al 2005). If this rule had been applied to our case, the prognosis would have been very poor, for example, an early demise. However, our patient at one stage recovered to the extent of being able to walk. Decreased density in CT does not necessarily mean neuronal loss, but rather suggests brain edema. Severe brain edema is caused by severe energy failure, massive production of radical oxygen species, or copious release of excitatory amino acids (CitationHossman 2006). Our patient’s brain may have suffered these biochemical insults at the acute stage, but fortunately these were resolved promptly. This may be, as discussed later, one of the causes of this case’s unique temporal profile of disease progression.
Early MRI findings in our case were also compatible with previous reports (CitationSawada et al 1990; CitationRoine et al 1993b), scattered high intensity spots by T1-weighed image were confirmed at the cerebral cortex and putamen (). Those in the cerebral cortex reflect cortical laminar necrosis, because some neuronal populations show selective vulnerability against hypoxia (CitationSawada et al 1990; CitationRoine 1996). On the other hand, the T2-weighed image at that time () showed no definite abnormalities. A previous report demonstrated that high intensity lesions in the T2-weighed image were confirmed as early as several days after the hypoxic insult (CitationTakahashi et al 1993), but another showed that they appeared only several months after the insult (CitationSawada et al 1990). Our case results were similar to those of the latter report, that is, high intensity subcortical lesions in the T2-weighed image appeared only in the chronic stage (, and ). Of interest is that subcortical high intensity lesions in the T2-weighed image should be the cause of delayed exacerbation; even though it was investigated only for several months, cases with subcortical high intensity by T2-weighed image showed progression of brain atrophy (CitationTakahashi et al 1993). If we can clarify what is occurring in this T2-weighed high intensity area, we may be able to understand the mechanisms of delayed exacerbation in hypoxic encephalopathy.
The molecular mechanisms of delayed exacerbation seen in this case are difficult to explain. After transient global ischemia in the brain, the phenomenon of “delayed neuronal cell death (DND)” has gained much attention. DND of the hippocampal pyramidal cells in rodent models is well known, but some cortical as well as caudate neurons also show such degeneration (CitationPulsinelli et al 1982; CitationKirino and Sano 1984). However, this kind of neuronal loss occurs only several days after transient global ischemia. Whether neuronal cell death occurs years after transient ischemic insult has not yet been investigated, because it is impossible to use rodents as model animals for such studies.
Our case suffered a sudden cardiopulmonary collapse in the hospital. Intensive resuscitation was quickly started. Cerebral blood flow with appropriate oxygen level was restored promptly. Paradoxically, this procedure might have been the cause of delayed exacerbation in this case. In an experimental study, some neurons degenerated only when the ischemic period was shorter than 10 minutes, but were spared when it was longer than 10 minutes (CitationRoss and Graham 1993). Sustained neuronal activity, established only when the ischemic period is quite short, could lead to delayed neuronal cell death. Although this may be a rare case, the symptoms of some patients who suffer hypoxic encephalopathy may continue to progress over a very long period.
Disclosure
The authors report no conflicts of interest in this work.
References
- [BRCT] Brain Resuscitation Clinical Trial II Study Group1991A randomized clinical study of a calcium-entry blocker (lidoflazine) in the treatment of comatose survivors of cardiac arrestN Engl J Med3241225312014035
- HossmanKA2006Pathophysiology and therapy of experimental strokeCell Mol Neurobiol2610578316710759
- JeongJWKimKWLeeDY2007A normative study of the Revised Hasegawa Dementia Scale: comparison of demographic influences between the Revised Hasegawa Dementia Scale and the Mini-Mental Status ExaminationDement Geriatr Cogn Disord242889317717415
- KjosBOBrant-ZawadzkiMYoungRG1983Early CT findings of global central nervous system hypoperfusionAm J Roentgenol1411227326606321
- KirinoTSanoK1984Selective vulnerability in the gerbil hippocampus following transient ischemiaActa Neuropathol6120186695554
- LeonovYSterzFSafarP1990Moderate hypothermia after cardiac arrest of 17 minutes in dogs. Effect on cerebral and cardiac outcomeStroke21160062237954
- LevyDECaronnaJJSingerBH1985Predicting outcome from hypoxic-ischemic comaJAMA253142063968772
- LongstrethWTDiehrPInuiTS1983Prediction of awaking after out-of-hospital cardiac arrestN Engl J Med3081378846843631
- PulsinelliWABrierleyJBPlumF1982Temporal profile of neuronal damage in a model of transient forebrain ischemiaAnn Neurol1149187103425
- RoineROKajasteSKasteM1993aNeuropsychological sequelae of cardiac arrestJAMA269237428417242
- RoineRORaininkoRErkinjunttiT1993bMagnetic resonance imaging findings associated with cardiac arrestStroke241005148322374
- RoineRO1996Global cerebral ischemiaFisherMBogousslavskyJCurrent Review of Cerebrovascular DiseasePhiladelphiaCurrent Medicine16974
- RossDTGrahamDI1993Selective loss and selective sparing of neurons in the thalamic reticular nucleus following human cardiac arrestJ Cereb Blood Flow Metab13558678314911
- SawadaHUdakaFSeriuN1990MRI demonstration of cortical laminar necrosis and delayed white matter injury in anoxic encephalopathyNeuroradiology32319212234393
- StrongAJFairfieldJEMonteiroE1990Insulin protects cognitive function in experimental strokeJ Neurol Neurosurg Psychiatry53847532266364
- TakahashiSHiganoSIshiiK1993Hypoxic brain damage: cortical laminar necrosis and delayed change in white matter at sequential MRI imagingRadiology189449568210374
- TippinJAdamsHPJrSmokerWR1984Early computed tomographic abnormalities following profound cerebral hypoxiaArch Neurol4110981006477220
- WijdicksEFParisiJESharbroughFW1994Prognostic value of myoclonus status in comatose survivors of cardiac arrestAnn Neurol35239438109907
- YanagawaYUn-noYSakamotoT2005Cerebral density on CT immediately after a successful resuscitation of cardiopulmonary arrest correlates with outcomeResuscitation649710115709219