Abstract
Mitochondria are semi-autonomously reproductive organelles within eukaryotic cells carrying their own genetic material, called the mitochondrial genome (mtDNA). Until some years ago, mtDNA had primarily been used as a tool in population genetics. As scientists began associating mtDNA mutations with dozens of mysterious disorders, as well as the aging process and a variety of chronic degenerative diseases, it became increasingly evident that the information contained in this genome had substantial potential applications to improve human health. Today, mitochondria research covers a wide range of disciplines, including clinical medicine, biochemistry, genetics, molecular cell biology, bioinformatics, plant sciences and physiology. The present review intends to present a summary of the most exiting fields of the mitochondrial research bringing together several contributes in terms of original prospective and future applications.
Mitochondrial biology and energy
The mitochondrial DNA, an extremely peculiar genome
About three billion years ago, bacteria with unique energy-producing capabilities took residence in a proto-eukaryotic cell, developing a symbiotic relationship: the host cell provided physical space and materials to the bacterium, which supplied energy to the host. During the evolution of this symbiotic relationship, the bacterium transferred many of its genes (about 1500) to the host nucleus, creating the modern nuclear DNA (nDNA) genome (CitationWallace 2005a). Thus, the modern mitochondrial chromosome is a circular DNA (mtDNA) molecule, which consists of 16,569 base pairs that retain 37 genes critical for producing energy. The entire mtDNA is almost exclusively transcribed (it contains very small noncoding areas) and is arranged in nucleoids (CitationHolt et al 2007).
Because the mitochondria produce most of the cell’s reactive oxygen species (ROS), mtDNA is constantly under mutagenic stress (about 100 times more than the stress experienced by nDNA). Moreover, being maternally inherited and outside the nucleus, mtDNA can change through the sequential accumulation of mutations along radiating maternal lineages (CitationSchapira 2006).
Mitochondrial energy, “beyond the ATP”
The mitochondrial genome controls many cell functions, critical for producing the cell’s energy. The most important of these functions are: 1) OXPHOS (oxidative phosphorylation), the complex mechanism by which the mitochondria’s respiratory machinery strips electron energy from carbohydrates and fats and harnesses it to phosphorylate ADP to make ATP; 2) the production of most of the cell’s ROS; and 3) regulation of apoptosis by activating the mitochondrial permeability transmission pore (CitationWallace 2005b).
Mitochondrial genetics and medicine
Mitochondrial disease and “Heteroplasmy”
Mitochondrial diseases range in severity from almost nondiagnosable to fatal. They also are a function of two types of mtDNA mutations: those inherited through the female germ-line and those that accumulate with age in somatic cells. Because mitochondria of a sexually-reproducing species are inherited maternally, germ-line mtDNA genetic defects can affect both males and females, but can only be transmitted to offspring by females. Somatic mutations accumulate when food calories exceed a cell’s respiratory capacity, resulting in excessive production of ROS, which damages mitochondrial membranes, proteins and DNA (CitationSchapira 2006).
Since there are hundreds or even thousands of mtDNA copies in every eukaryotic cell, mutations may either be present in all copies (homoplasmy) or affect only a fraction of them (heteroplasmy). As they replicate, the populations of their normal and mutant mtDNAs randomly segregate among the daughter cells, the percentages of each changing with each replication. When tissues accumulate a certain ratio of mutant versus wild-type mtDNA (threshold effect), cells produce less energy, more ROS, have an increased apoptosis rate and disease presents itself. Mutations can cause several different diseases depending upon the severity of the problem in the mitochondria and the tissue affected. The most susceptible tissues include those with a high energetic demand, such as brain, heart, skeletal muscle, endocrine, blood, and kidney tissues.
Some diseases are observable at birth while others do not show themselves until late adulthood. The clinical variability of these disorders varies from person to person (depending upon metabolism requirements and the effects of the specific mutation) and makes the diagnosis very challenging. Mitochondrial diseases caused by mtDNA abnormalities are very numerous and diverse, and because they are due to age-related decline in OXPHOS, many of these diseases are late-onset and progressive. In addition, it is suspected that many other diseases are caused in part by dysfunction of mitochondria, including diabetes mellitus, some forms of cardiovascular disease, Alzheimer’s and Parkinson’s diseases, and many more (CitationDiMauro et al 2006a).
Human mtDNA haplogroups
As humans migrated out of Africa, a dozen mtDNA lineages (haplogroups) evolved. The conventional wisdom is that the expression of pathogenic mtDNA mutations can be modulated by haplogroups (CitationCrimi et al 2005) or, more generally, certain mtDNA haplogroups may affect OXPHOS, thus predisposing to, or protecting from, disease.
In recent studies, some ancient European mtDNA haplogroups were found to be associated with increased longevity and, moreover, to produce more heat relative to other variants, an advantage in colder northern latitudes (CitationCapri et al 2006). Because these variants maintain their electron transport chains in a more oxidized state than do other variants, they produce less ROS and are less likely to accumulate mitochondrial damage from ROS.
Cancer and mtDNA
Since tumor cells actively metabolize glucose and generate lactate while still consuming oxygen, OXPHOS defects must be important for neoplastic transformation. Recently, certain tumors have been shown to result from mutations in nDNA encoded mitochondrial proteins, at least some of which are likely to result in increased ROS production (CitationWallace 2005a). Since the very first demonstration that the G10398A mtDNA polymorphism is associated with increase risk for breast cancer in African-American women (CitationCanter et al 2005), mtDNA mutations have also been associated with cancer, including incidence in renal adenocarcinoma, colon cancer cells, head and neck tumors, astrocytic, thyroid, breast, and prostate tumors. Meta-analysis of “tumor-specific somatic mtDNA mutations” has revealed that cancer mtDNA mutations fall into two major classes: those that partially inhibit OXPHOS thus increasing ROS production and those that alter the mitochondrial OXPHOS circuitry to permit the tumor cells to adapt to new environmental constraints such as alterations in available nutrients and oxygen tension (CitationBrandon et al 2006).
Genetic classification of mitochondrial diseases
Mutations in mtDNA
Given that the mtDNA genome is very compact, almost any mutation could be deleterious; so far, over 170 known mutations of the mtDNA are responsible for a myriad of diseases. Mutations in mtDNA are classified into three categories (): large-scale rearrangements, point mutations in RNA genes affecting the mitochondrial protein translation, and point mutations in protein coding genes, resulting in specific OXPHOS defects (CitationDiMauro et al 2006b).
Figure 1 Mitochondrial disease evaluation flowchart, from a clinical suspect to the molecular diagnosis. When a mitochondrial illness is suspected, laboratory testing is the usual method physicians go about evaluating patients for disorders of energy metabolism. If the suspect a mitochondrial defect is confirmed, more integrated tests are a must and include: the biochemical analysis of the respiratory chain and the screening of the most frequent mtDNA mutations. If necessary, sequencing the so-far-known nuclear genes responsible for mitochondrial diseases and the complete sequence analysis of the entire mitochondrial genome are another chances for selected cases. A scattered line states when a diagnosis could be done.
Abbreviations: AID, aminoglycoside-induced deafness; AS, Alper’s Syndrome; COX, Cytochrome c OXidase; CSF, cerebrospinal fluid; KSS, Kearns-Sayre syndrome; PS, Pearson’s syndrome; LHON, Leber’s hereditary optic neuropathy; LS, Leigh’s syndrome; MDS, MtDNA depletion syndrome; MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke; MERRF, myoclonus epilepsy with ragged red fibers; MIDD, maternal inherited diabetes and deafness; MM, mitochondrial myopathy; MMC, maternal myopathy and cardiomyopathy; OA, optic atrophy; MNGIE, mitochondrial neurogastrointestinal encephalopathy; MRI, magnetic resonance imaging; NARP, neurogenic weakness, ataxia and retinitis pigmentosa; PEO, progressive external ophthalmoplegia; RRF, ragged red fibers; SANDO, sensory ataxic neuropathy, dysarthria, and ophthalmoparesis.
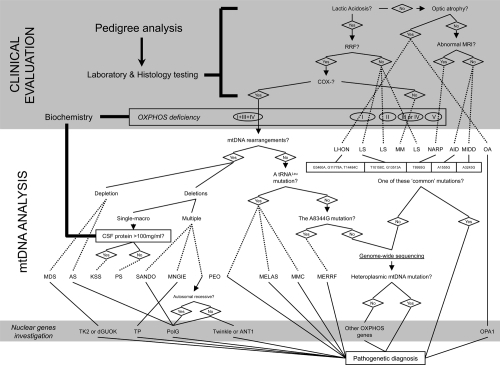
Large-scale mtDNA rearrangements occur as multiple short deletions or a single macro-deletion with unknown mechanisms. They have been commonly associated with progressive external ophthalmoplegia (PEO), a paralysis of the extra-ocular muscles. PEO may manifest as part of a more severe and fatal multi-system disorder termed Kearns-Sayre syndrome (KSS). Rearrangements of mtDNA have also been identified in patients with syderoblastic anemia, known as Pearson’s syndrome. Recent research reveals that sustanatia nigra neurons of the elderly show high concentrations of clonally expanded deleted mtDNAs that lead to functional impairment of the respiratory chain (CitationKraytsberg 2006). With more than 60 different mutated nucleotides already reported, RNAs are the most common class of pathogenic mtDNA point mutations that occur in mitochondrial tRNA genes. These mutations cause a variety of metabolic syndromes, including mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELAS), myoclonus epilepsy and ragged-red fibers (MERRF), and several other combinations of symptoms involving different tissues. The most common mtDNA mutation in this group is the A3243G transition in the tRNALeu(UUR) gene, disclosed in the majority of patients with the clinical features of MELAS. This same mutation also accounts for a large number of PEO patients that do not have large-scale deletions. Point mutations in the mitochondrial tRNALys gene (A8344G and T8356C) have been found in most patients with MERRF.
Several point mutations in protein coding genes have been described in patients with Leber’s hereditary optic neuropathy (LHON), specifically the transition G11778A accounts for approximately 50% of all cases. Neuropathy, ataxia, and retinitis pigmentosa (NARP) is associated with a missense G8993T mutation in the ATPase 6 gene. The same mtDNA mutation can also cause a severe infantile metabolic encephalomyopathy known as maternal inherited Leigh’s syndrome (MILS).
New mtDNA mutations, especially in protein-coding genes, are still being identified in very heterogeneous clinical pictures where the heteroplasmic degree usually correlates with the phenotypical severity. Thus, heteroplasmy was included among the canonical criteria for pathogenicity (CitationSchapira 2006). In addition, the ‘candidate’ mutation ought to: (i) be absent in normal subjects of the same ethnic background; (ii) alter a functionally important and evolutionarily conserved site; (iii) cause a functional alteration, demonstrable with a specific biochemical defect or as a correlation between mutation burden and RRF or COX-negative fibers (single fiber PCR).
Unlike heteroplasmic mutations that are always pathogenic, homoplasmic variants could be not-neutral. There are several possible factors conditioning the phenotypic expression of a homoplasmic mtDNA mutation: environmental factors, mtDNA haplotype, nDNA background, and tissue-specific expression of interacting genes (CitationLimongelli et al 2004).
Mutations in nDNA
Mitochondrial diseases resulting from mutations in nDNA-encoded mitochondrial genes fall into three other major categories: those affecting structural genes, those affecting mtDNA maintenance and expression, and those regulating mitochondrial antioxidant defenses (CitationDiMauro 2006c).
Identification of the disorders due to defects of inter-genomic signaling have progressed significantly after the discovery of thymidine phosphorylase (TP), the gene responsible for the mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). Mutations in TP attracted attention to the nucleotide pool in the pathogenesis of multiple deletions and depletion of mtDNA (with the mtDNA copy number per mitochondrion reduced as much as to less than 5% of normal), as both alterations coexist in muscle from patients with MNGIE. Moreover, mutations in ANT1, encoding one adenine nucleotide transporter, and in Twinkle, a helicase, have been identified in patients with PEO and multiple mtDNA deletions. Mutations in other genes controlling the nucleotide pool, as dGK (encoding deoxyguanosine kinase) and TK2 (a thymidine kinase), are associated with the hepatocerebral and myopathic form of mtDNA depletion.
The focus now lies on POLG, the gene encoding the mitochondrial polymerase γ, not only because mutations have been found in families with PEO, but also because a variety of human diseases (Alper’s syndrome, Parkinson’s disease, etc) are associated with it (CitationDiMauro 2006c).
Recently, it was demonstrated that two human diseases have been associated with impaired mitochondrial fusion, a process which requires several proteins, including some GTPases, like mitofusin (MFN) and OPA-1. A form of autosomal dominant optic atrophy, an important cause of blindness in young adults, has documented mutations in the OPA-1 gene; moreover, mutations in the MFN2 gene have been found in CMT patients (CitationDiMauro et al 2006b).
Detection of pathogenic mtDNA mutations
Several different mutations may present themselves as the same disease. This almost patient-specific characterization of mitochondrial diseases makes them very hard to accurately diagnose and trace ().
Most mtDNA rearrangements are easily detected by Southern blot analysis. Total DNA used for these studies should preferably be purified from a post-mitotic tissue, such as muscle. Restriction enzymes recognizing one unique restriction site (such as PvuII) can reveal anomalous bands if rearrangements are present (CitationWong and Boles 2005).
A number of established techniques have been used to screen selected regions of mtDNA for the identification of pathogenic mutations. If a specific mtDNA heteroplasmic mutation is suspected, RFLP-PCR can easily reveal the presence of mutated mtDNAs. Otherwise, with the advances in DNA sequencing techniques, it is now relatively simple to sequence the mtDNA in its entirety.
Denaturing high performance liquid chromatography (DHPLC) is a method that separates heteroduplex DNA molecules in a stationary phase under partially denaturing conditions. As pathogenic mtDNA mutations are mostly heteroplasmic, DHPLC could detect these different species and therefore act as a rapid screening test for mtDNA mutations. By eliminating secondary amplification and excessive manipulation prior to sequencing, DHPLC is a sensitive, rapid and cost-effective method to streamline the analysis of mitochondrial heteroplasmy (CitationBiggin et al 2005).
Other mtDNA genome-wide sequencing technologies, encompassing SNPs microarray, are still dismally limited and the sensitivity of heteroplasmy detection is debated (CitationCrimi et al 2005).
Mitochondrial therapeutic strategies
“Classical” approaches
Heteroplasmy is the biggest problem to overcome in devising realistic therapies; consequently, progress has been slow. To date, therapy regimes remains woefully inadequate, as we still treat patients with OXPHOS disorders adding extra vitamins and co-factors (such as the coenzyme Q10), even if there is some doubt to their effectiveness among clinicians (CitationMarriage 2003). The use of these compounds could improve the production of energy by the increasing of enzyme activity and delaying the disease progression as antioxidant action (CitationDiMauro 2007). In addition, repetitive physical exercise could be a suitable alternative by causing sufficient muscle damage to result in the sustained recruitment of satellite cells (CitationTaivassalo and Haller 2005).
Gene therapy
The inadequacy of current pharmacotherapy for mitochondrial disorders has stimulated the development of genetic treatments for mtDNA-related diseases.
MtDNA gene therapy may hold promise for ameliorating symptoms of patients with mitochondrial disease by taking two major approaches: direct genetic modification of a mutant mtDNA gene or insertion of a modified mitochondrial protein gene into the nucleus.
First experimental issues come from the direct genetic modification that encompasses the delivery of restriction enzymes into mitochondria, where only the mutant mtDNA, but not the wild-type mtDNA, is selectively digested by the enzyme. Manipulation of mtDNA heteroplasmy by targeting specific enzymes into mitochondria could, in theory, lead to the exclusive elimination of the mutant mtDNA and repopulation by the wild-type mtDNA, resulting in restoration of both the intracellular ATP level and a normal clinical phenotype (CitationFlierl et al 2003; CitationBayona-Bafaluy et al 2005).
These approaches are exclusively somatic and will help people harboring pathogenic mtDNA mutations but do not assure mothers that their children will have normal health. Only germ-line therapy, breaking the maternal transmission of mutant mtDNAs, could eliminate maternally-inherited pathogenic mutations.
Stem cells
Transplant of stem cells is a promising approach to the treatment of mitochondrial disorders. In the current year, two patients affected by MNGIE have received allogeneic stem cell transplantation. The stem cells used in this groundbreaking therapy partially restored TP activity, encouraging rapid progresses in this field (CitationHirano et al 2006).
Embryonic stem cell approaches raise important issues about a possible transmission of host cell mitochondrial mutations. Since pathogenic heteroplasmic mtDNAs are transmissible, the potential exists to create heteroplasmic sources of mitochondria in the resultant embryo (CitationHall et al 2006). Thus, the prescreening of female oocyte donors for mtDNA mutations may need to be considered.
New pharmacological trends
Besides genetic alteration, mitochondrial dysfunction has been related to large spectrum of human disorders based on metabolic deficit. The metabolism fall could be of interest in the development of a pharmacological mitochondrial-based approach in the treatment or prevention of these pathologies. In the last years, several studies on the healthy properties of food-derived compounds has greatly developed together with those on the effect of nutritional intake on the lifespan. Resveratrol, a polyphenolic compound mainly present in red wine and grape, has been one of these compounds and it has been shown to positively interfering with several biological and pathological processes such as oxidative stress, cancer development and aging (CitationShankar et al 2007).
Recently, resveratrol has been also demonstrated to act on mitochondria in vivo counteracting the effect of high fat diet in mice and mimicking the biological processes stimulated by caloric restriction. In mice, resveratrol stimulate mitochondria biogenesis and modulate several energy pathways both on signal transduction and transcriptional level with enrichment of OXPHOS and ATP synthesis. These events are also associated with more healthy changes such as the reduction in the plasma levels of insulin and the switch in muscle fibers composition towards more oxidative ones, changes which both positive correlate with the prevention and therapy for diabetes (CitationBaur et al 2006). Moreover the interference of resveratrol with the pathological stress- and insulin-induced signaling pathway, which is strongly associated with aging, highlight new mechanism for several resveratrol healthy properties (CitationLagouge et al 2006).
Mitochondrial research in the new era
Models of mitochondrial diseases
Deleterious mtDNA mutations can cause mitochondrial disease in the mouse with a severity and nature analogous to that seen in humans. Over the past decade, several mouse models (‘mitomice’) have been developed for both mtDNA mutations and nDNA mutations (CitationWallace 2002). Although the mitomice develop phenotypes and inheritance patterns somewhat different from humans, they are revolutionizing our understanding of mitochondrial diseases and can be used to demonstrate the efficacy of new antioxidant drugs.
Mitomice can be created by introducing genetically distinct mtDNAs into the female germ-line, using two basic procedures: (i) fusion of enucleated cell cytoplasts bearing mutant mtDNA to undifferentiated mouse female stem cells, injection of the stem cell cybrids into mouse blastocysts, and implantation of the chimeric embryos into a foster mother, and (ii) fusion of cytoplasts from mutant cells directly to mouse single-cell embryos and implantation of the embryos into the oviduct of pseudo-pregnant females. The former method has permitted the creation of mouse strains bearing deleterious base substitution mutations, while the latter has been used to create mouse strains harboring mtDNA deletions (Inoue et al 2006).
In order to render the mitochondrial DNA polymerase error-prone by eliminating its proofreading activity, a very important mouse model was created in 2004 (CitationTrifunovic et al 2004). The homozygous knock-in mice expressing a proof-reading-deficient version of POLG show dramatic evidence of premature aging. Further studies of this mouse will help in dissecting the pathogenesis of age-related degenerative phenomena, specifically by understanding the contribution of mitochondrial factors.
Microarray applications
The interplay between the mtDNA and nDNA encoded mitochondrial genes and their role in mitochondrial disorders is still largely unclear. One approach for clarifying the "dialogue" between the nuclear and mitochondrial genomes and the pathogenesis of mitochondrial diseases has been to look at changes in the expression of mitochondrial genes in response to specific mitochondrial defects.
Recently, microarray technology has been employed to study mRNAs amount changes in mitochondrial diseases. Gene expression variability has been already described in metabolic pathways (included amino acid catabolism, lipid peroxidation and energy production) and in many transcripts involved in the cell cycle regulation (CitationCrimi et al 2005). These evidences underlie the ability of the cell to access to an alternative energy resource when a compromised OXPHOS occurs. These preliminary microarray studies seem a promising strategy to identify the presence of coordinate changes in mitochondrial gene expression in response to specific mitochondrial defects. In addition, mtDNA mutations are recurrent events in collected body fluids from patients with primary tumor tissues. Thus, mtDNA mutation analysis with nanotechnologies-based sequencing tools may provide a molecular platform for the early and noninvasively detection and prognosis of cancer (CitationJakupciak et al 2006).
Mitochondrial bioinformatics
New mitochondrial genes discovery
Computational and experimental approaches have a symbiotic relationship for identifying mitochondrial proteins (CitationCalvo et al 2006). The “Maestro” program integrates eight different “predictors”, based on both structural and experimental data, to generate scores predicting the likelihood of mitochondrial localization. Using this approach, the authors confidently identified nearly 500 mitochondrial proteins that were previously unidentified, and additionally identified genes associated with several human mitochondrial diseases.
Online resources
Several databases are frequently updated to garner further information on the mitochondria field of research (CitationChuang et al 2006). While online programs are rapidly becoming popular tools for the scientific community and offer unprecedented accessibility to quality information, there are weaknesses inherent the use of these sources by patients and/or people without enough competencies regarding the mitochondria biology and genetics.
Family Tree DNA “DNA Matches” is the starting point before beginning any other searches. The haplogroup and mutation results obtained are very useful for broad comparison with the worldwide databases noted below. Available at: http://www.familytreedna.com
Mitomap: A Human Mitochondrial Genome Database is a compendium of polymorphisms and mutations of the human mitochondrial DNA. Available at: http://www.mitomap.org
Mamit-tRNA provides access to a gene compilation aimed at defining a consensus of mammalian mitochondrial tRNAs structural features. Available at:http://mamit-trna.u-strasbg.fr/Summary.html
mtDB is the Human Mitochondrial Genome Database with 1711 complete sequences and 839 coding region sequences. Available at: http://www.genpat.uu.se/mtDB/
The POLG mutation database lists all known mutations in the coding region of the POLG gene. The POLG mutation database describes the heterogeneous clinical pictures related with several mutations in this gene. Available at: http://dir-apps.niehs.nih.gov/polg
The Human Mitochondrial Protein Database (HMPDb) provides comprehensive data on mitochondrial and human nuclear encoded proteins involved in mitochondrial biogenesis and function. Available at: http://bioinfo.nist.gov/hmpd
MitEURO is the European consortium of laboratories conducting research on mitochondrial biogenesis and disease. Available at: http://www.miteuro.org
MtDNA Test Results Log encourages individuals to submit their own information so that, with time, the database will grow and become a useful tool to genealogists. Available at: http://www.kerchner.com/cgi-kerchner/mtdna.cgi
Mitosearch directs you to the Family Tree DNA database with a free service that permits customers to directly upload their results and to add information about the genealogy of the earliest known ancestor in the female line. Available at: http://www.mitosearch.org
V-MitoSNP is a new web-based visualization interface of mitochondrial Single Nucleotide Polymorphism (mtSNP) that clarifies their strategic influence on the development of human diseases and cancers. Available at: http://bio.kuas.edu.tw/v-mitosnp
Conclusions
The application of mitochondrial genomics to modern medicine has already begun to offer new clues to the development of many ailments and, if speculations on the role of mtDNA in human health prove correct, mitochondrial biology studies can suggest effective approaches to the treatment and prevention of their progression.
Acknowledgements
The Sass Foundation for Medical Research, Inc (Roslyn, New York) and “Centro Dino Ferrari” association (Milan, Italy) are gratefully acknowledged for their financial support. The authors deeply thank Ms Sara Klaus for the stylistic revision. There are no conflicts of interest to report.
References
- BaurJAPearsonKJPriceNLResveratrol improves health and survival of mice on high-calorie diet Nature20064443374217086191
- Bayona-BafaluyMPBlitsBBattersbyBJRapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonucleaseProc Natl Acad Sci USA200510214392716179392
- BigginAHenkeRBennettsBMutation screening of the mitochondrial genome using denaturing high-performance liquid chromatographyMol Genet Metab200584617415639196
- BrandonMBaldiPWallaceDCMitochondrial mutations in cancerOncogene20062546476216892079
- CalvoSJainMXieXSystematic identification of human mitochondrial disease genes through integrative genomicsNat Genet2006385768216582907
- CanterJAKallianpurARParlFFMitochondrial DNA G10398A polymorphism and invasive breast cancer in African-American womenCancer Res20056580283316140977
- CapriMSalvioliSSeviniFThe genetics of human longevityAnn N Y Acad Sci200610672526316803995
- ChuangLYYangCHChengYHV-MitoSNP: visualization of human mitochondrial SNPsBMC Bioinformatics2006157379
- CrimiMDel BoRGalbiatiSMitochondrial A12308G polymorphism affects clinical features in patients with single mtDNA macrodeletionEur J Hum Genet200311896814571278
- CrimiMO’HearnSFWallaceDCMolecular research technologies in mitochondrial diseases: the microarray approachIUBMB Life200557811816393784
- DiMauroSDavidzonGHiranoMA polymorphic polymeraseBrain20061291637916803839
- DiMauroSHiranoMSchonEAApproaches to the treatment of mitochondrial diseasesMuscle Nerve2006342658316810684
- DiMauroSMancusoMMitochondrial diseases: therapeutic approachesBiosci Rep2007271253717486439
- DiMauroSMitochondrial myopathiesCurr Opin Rheumatol2006186364117053512
- FlierlAJacksonCCottrellBTargeted delivery of DNA to the mitochondrial compartment via import sequence-conjugated peptide nucleic acidMol Ther20037550712727119
- HallVJStojkovicPStojkovicMUsing therapeutic cloning to fight human disease: a conundrum or reality?Stem Cells20062416283716556706
- HiranoMMartiRCasaliCAllogeneic stem cell transplantation corrects biochemical derangements in MNGIENeurology20066714586016971696
- HoltIJHeJMaoCCMammalian mitochondrial nucleoids: organizing an independently minded genomeMitochondrion200773112117698423
- InoueKNakadaKOguraAGeneration of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotesNat Genet2000261768111017072
- JakupciakJPDakuboGDMaraghSAnalysis of potential cancer biomarkers in mitochondrial DNACurr Opin Mol Ther20068500617243485
- KraytsbergYKudryavtsevaEMcKeeACMitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neuronsNat Genet2006385182016604072
- LagougeMArgmannCGerhart-HinesZResveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1aCell200612711092217112576
- LimongelliASchaeferJJacksonSVariable penetrance of a familial progressive necrotising encephalopathy due to a novel tRNA(Ile) homoplasmic mutation in the mitochondrial genomeJ Med Genet200441342915121771
- MarriageBClandininMTGlerumDMNutritional cofactor treatment in mitochondrial disordersJ Am Diet Assoc200310310293812891154
- SchapiraAHMitochondrial diseaseLancet2006368708216815381
- ShankarSSinghGSrivastavaRKChemoprevention by resveratrol: molecular mechanisms and therapeutic potentialFront Biosci20071248395417569614
- TaivassaloTHallerRGExercise and training in mitochondrial myopathiesMed Sci Sports Exerc200537209410116331135
- TrifunovicAWredenbergAFalkenbergMPremature ageing in mice expressing defective mitochondrial DNA polymeraseNature20044294172315164064
- WallaceDCAnimal models for mitochondrial diseaseMethods Mol Biol200219735412013805
- WallaceDCA mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicineAnnu Rev Genet2005a3935940716285865
- WallaceDCThe mitochondrial genome in human adaptive radiation and disease: on the road to therapeutics and performance enhancementGene2005b3541698016024186
- WongLJBolesRGMitochondrial DNA analysis in clinical laboratory diagnosticsClin Chim Acta200535412015748595