Abstract
Background
Toll-like receptors (TLRs) represent a conserved family of innate immune recognition receptors. Among TLRs, TLR4 is important for the recognition of Gram-negative bacteria, whereas TLR2 recognizes cell wall constituents of Gram-positive microorganisms, such as peptidoglycan (PGN).
Methods
To evaluate the role of TLR4 in the pathogenesis of acute lung injury induced by Escherichia coli endotoxin (lipopolysaccharide; LPS) or PGN, we compared inflammatory cell accumulation in bronchoalveolar lavage (BAL) fluid and lung pathology between C3H/HeJ (TLR4 mutant) and wild-type C3H/HeN mice. The levels of proinflammatory cytokines and chemokines in plasma and BAL fluid and nuclear factor-κB (NF-κB) translocation in the lung were also evaluated.
Results
In C3H/HeJ mice, LPS-induced neutrophil emigration was significantly decreased compared with C3H/HeN mice, whereas PGN-induced neutrophil emigration did not differ. Differential cell count in BAL fluid revealed comparable neutrophil recruitment in the alveolar space. In TLR4 mutant mice, LPS-induced upregulation of tumor necrosis factor-alpha (TNF-α), KC, and CXCL10 in plasma and BAL fluid was attenuate, which was not different after PGN. NF-κB translocation in the lung was significantly decreased in C3H/HeJ compared with C3H/HeN mice, whereas PGN-induced NF-κB translocation was not different.
Conclusion
These results suggest that TLR4 mediates inflammatory cascade induced by Gram-negative bacteria that is locally administered.
Keywords:
Introduction
Lung infection is one of the leading causes of illness and death worldwide. Bacteria or their constituents in the lungs induce the expression of multiple genes of inflammatory mediators and adhesion molecules, leading to neutrophil margination within the microvasculature, adhesion to endothelial cells, and transmigration between endothelial cells into the alveolar space (CitationHogg and Doerschuk 1995; CitationDoerschuk et al 1999). The transcription of these genes, including chemokines, adhesion molecules, and early response cytokines, is regulated at least in part by the nuclear factor-κB (NF-κB) family and other transcription factors (CitationPahl 1999; CitationStrieter et al 2002; CitationCiesielski et al 2002).
Toll-like receptors (TLRs) represent a conserved family of innate immune recognition receptors that play key roles in detecting microbes, initiating innate immune responses, and linking innate and adaptive immunity (CitationAkira et al 2006). Over the last few years, it has become evident that both the recognition and the subsequent response to pathogens are mainly transferred by TLRs (CitationTakeuchi et al 1999; CitationYamamoto et al 2004; CitationDing et al 2005; CitationAkira et al 2006). Ten TLRs have been described so far, among which TLR2 and TLR4 are the family members that have been investigated to the greatest extent (CitationTakeuchi et al 1999; CitationYamamoto et al 2004).
TLR4, the endotoxin (lipopolysaccharide; LPS) receptor, is important for the recognition of Gram-negative bacteria, whereas TLR2 has been designated the major receptor for Gram-positive bacteria by virtue of its capacity to recognize major cell wall constituents of Gram-positive microorganisms, such as peptidoglycan (PGN), lipoteichoic acid, and lipoproteins (CitationTakeuchi et al 1999; CitationSchwartz 2001; CitationBranger et al 2004).
The role of TLRs in the inflammatory process has been the subject of several recent investigations. However, most of the studies evaluated cellular response in vitro or systemic reaction such as survival rate, and a few studies have focused on the role of TLRs in lung inflammatory change following locally administered stimuli (CitationFan et al 2003; CitationFaure et al 2004; CitationSchurr et al 2005). In the present series of experiments, we sought to determine the role of TLR4 in acute inflammatory response induced by bacterial components of gram-positive and gram-negative bacteria. We first evaluated LPS- or PGN-induced neutrophil emigration within the lungs, comparing C3H/HeJ (Tlr4Lps-d) mice that carry a defective allele of TLR4 gene and wild-type C3H/HeN. Differential cell counts and levels of proinflammatory cytokines (TNF-α, IFN-γ) and chemokines (KC, CXCL10) were examined in bronchoalveolar lavage (BAL) fluid. We also evaluated the activation of NF-κB after intratracheal challenge of LPS or PGN using lung homogenate.
Materials and methods
Animals and reagents
Female C3H/HeN and C3H/HeJ mice were purchased from CLEA Japan (Tokyo, Japan). All mice were housed in the Keio University Animal Resource Center, and all experiments received institutional approval, conforming to NIH guidelines. The animals were provided water and rodent chow ad libitum for at least 3 days prior to the experiment. Escherichia coli endotoxin (serotype B:55) was purchased from Sigma Chemicals (St. Louis, MO). PGN from Staphylococcus aureus was purchased from Fluka (Buchs, Switzerland).
Experimental protocol for acute lung injury in mice
Eight-week old mice weighing approximately 22 g were anesthetized using ketamine hydrochloride (80–100 mg/kg i.m.) and acepromazine maleate (5–10 mg/kg i.m.). Either E. coli LPS (3.0 mg/kg given as a solution of 1.2 mg LPS/ml phosphate buffer solution [PBS]) or PGN (1.0 mg/kg given as a solution of 0.4 mg PGN/ml PBS) was instilled intratracheally, and control mice received PBS (2.5 ml/kg). After 6 h, the lungs were removed and fixed using intratracheal instillation of 6% glutaraldehyde at 22 cm-H2O or lavaged with three separate 1.0 ml volumes of PBS containing 0.6 mM EDTA, each volume being instilled and withdrawn three times. Blood samples were obtained from the inferior vena cava and subjected to differential count of circulating leukocytes.
Inflammatory cell accumulation in the lung
Pulmonary neutrophils were quantified by morphometric analysis in histological sections as previously described (CitationTasaka et al 2003). Paraffin-embedded histologic 5-μm sections of lungs were cut and stained with hematoxylin and eosin. Neutrophil emigration was quantitated by counting the number of neutrophils in 200 randomly selected alveoli and was expressed as the number of neutrophils per 100 alveoli. Samples were also taken from the control mice that received PBS instillation. After the cell counting, the BAL fluid was centrifuged, and the supernatants were stored at −80 °C until cytokine measurement. The levels of murine TNF-α, IFN-γ, and KC in the plasma and BAL fluid were quantitated using a suspension array system (Bio-Plex; Bio-Rad, Hercules, CA). The murine CXCL10 was measured using an ELISA kit (R&D Systems, Minneapolis, MN) according to the manufacturer’s instruction. The minimum detectable dose of CXCL10 was 2.2 pg/mL.
Nuclear protein extraction from the lungs
Mice were anesthetized as above and received intratracheal instillation of PBS, LPS (3.0 mg/kg), or PGN (1.0 mg/kg). After 3 h, the lungs were removed and frozen immediately in liquid nitrogen. Nuclear extracts were prepared as previously described (CitationMizgerd et al 2002; CitationTasaka et al 2003). Briefly, the isolated lungs were homogenized in 3 ml of ice-cold Buffer A (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.5 mM PMSF), with a 0.1% volume of Nonidet P-40 and protease inhibitor cocktail (1 mg/ml leupeptin, 1 mg/ml aprotinin, 10 mg/ml soy bean trypsin inhibitor, 1 mg/ml pepstatin). Following 10-min incubation on ice, the homogenates were centrifuged at 850 × g for 10 min at 4 °C. The pellets were resuspended in 3 ml of Buffer A and centrifuged at 1,400 × g for 10 min at 4 °C. The crude nuclear pellets were resuspended in 50 μl of Buffer B (20 mM HEPES, 1.5 mM MgCl2, 0.42 M NaCl, 0.2 mM EDTA, 25% vol/vol glycerol, 0.5 mM DTT, 0.5 mM PMSF) with protease inhibitor cocktail (as described above) and incubated for 30 min on ice. Nuclear extracts were recovered following centrifugation at 20,000 × g for 15 min at 4 °C and stored at −80 °C. The protein concentration of the nuclear extracts was determined by bicinchoninic acid assay. The regression curve was obtained using bovine serum albumin as the standard.
Electrophoretic mobility shift assay and supershift assay
Electrophoretic mobility shift assay (EMSA) was performed following the manufacturer’s protocol (Gel Shift Assay Systems; Promega, Madison, WI) with several modifications (CitationTasaka et al 2003). The double strand NF-κB consensus oligonucleotide probe (5′-AGTTGAGGGGACTTTCCCAGGC-3′) was end-labeled with [32P]-ATP (3,000 Ci/mmol at 10 mCi/ml) using T4 polynucleotide kinase. Binding reactions (10 μl) containing 35 fmol/ml of oligonucleotide probe, 5 mg of nuclear extract and 5 × binding buffer (10 mM Tris-HCl, 1 mM MgCl2, 50 mM NaCl, 0.5 mM EDTA, 0.5 mM DTT, 4% vol/vol glycerol, 0.5 mg poly dI-dC) were incubated for 20 min at room temperature. Following the binding procedure, 10-fold loading buffer (250 mM Tris-HCl, 0.2% bromophenol blue, 40% glycerol, 0.2% xylane cyanol) was added to stop the reaction. The reactions were subjected to nondenaturing 6% polyacrylamide gel electrophoresis (NOVEX, San Diego, CA) in 0.5 × TBE buffer (45 mM Tris-boric acid, 1 mM EDTA-2H2O) at 65 V for 85 min. Gels were vacuum dried for 45 min and exposed to X-ray film (BMR; Kodak, Rochester, NY) at −70 °C. The relative intensity of each band was quantitated using scanning laser densitometry and Scion Image Software (Scion Corporation, Frederick, MD).
Supershift assays were performed as previously described (CitationMalley et al 2003). Nuclear proteins (0.3 mg/ml) were incubated with 3.5 nM [32P]ATP-labeled NF-κB consensus oligonucleotide and an antibody against one subunit of NF-κB (0.2 mg/ml) for 30 min prior to electrophoresis. Polyclonal antibodies against RelA (sc-7151) and p50 (sc-1192) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Protein-oligonucleotide complexes were separated from protein-free oligonucleotides by polyacrylamide gel electrophoresis and detected by autoradiography. Independent experiments with proteins collected from the lungs of different mice yielded consistent results.
Statistics
Data are presented as the mean ± SEM. One-way analysis of variance and Scheffe's test were used. Differences were considered statistically significant when p < 0.05.
Results
Neutrophil emigration in alveolar spaces
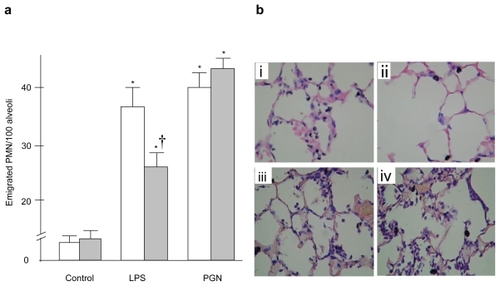
To evaluate the effect of TLR4 gene mutation on neutrophil emigration induced by bacterial products, emigrated neutrophils were quantified morphologically in histologic sections. The number of neutrophils per 100 cross-sectioned alveolar spaces is shown in . In C3H/HeN, or wild type mice, intratracheal LPS induced marked neutrophil emigration within the lungs, whereas a much smaller extent of neutrophil emigration occurred after LPS in C3H/HeJ or TLR4-mutant mice. The number of emigrated neutrophils was significantly less in C3H/HeJ than in C3H/HeN mice (p < 0.05). PGN induced significant neutrophil emigration within the lungs, and there was no difference in the emigrated neutrophils between the two strains examined. The doses of LPS and PGN that we selected caused similar degrees of neutrophil emigration. Representative microscopic findings are shown in .
Figure 1 (a) Neutrophil emigration in the lung. Although LPS-induced neutrophil emigration was significantly attenuated in C3H/HeJ mice, PGN-induced neutrophil accumulation did not differ between the two strains. (b) Representative example of lung pathology. Shown are images of lung sections obtained from C3H/HeN (i and iii) and C3H/HeJ (ii and iv) mice 6 hours after intratracheal instillation of LPS (i and ii) or PGN (iii and iv). Significant attenuation of neutrophil emigration was observed in the C3H/HeJ mice followed by LPS instillation.
Notes: Open columns: C3H/HeN mice, Gray columns, C3H/HeJ mice; Data are presented as the mean ± SEM (n = 6); *p < 0.05 versus control; †p < 0.05 versus C3H/HeN mice. Magnification × 400.
Abbreviations: LPS, lipopolysaccharide; PGN, peptidoglycan.
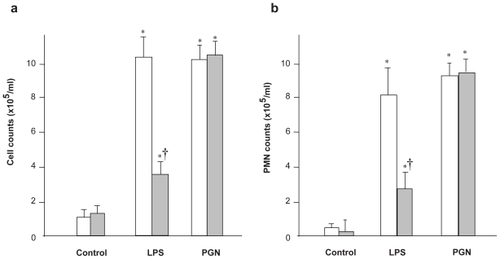
Neutrophil emigration in alveolar spaces following intratracheal instillation of bacterial product was also evaluated using differential cell count in BAL fluid (). In C3H/HeN mice, LPS induced remarkable increases in the counts of both total cells and neutrophils, compared with the PBS control group of the same strain (p < 0.001). The recruitment of inflammatory cells after LPS was significantly decreased in C3H/HeJ mice, compared with C3H/HeN mice (p < 0.05). PGN challenge also induced significant increases in the counts of both total cells and neutrophils, which did not differ between the two strains examined. These findings suggested that TLR4 is responsible for LPS-induced, but not PGN-induced, recruitment of inflammatory cell within the lungs.
Figure 2 Cellularity of bronchoalveolar lavage (BAL) fluid. The total cell (a) and neutrophil (b) counts in BAL fluid after LPS challenge were significantly decreased in C3H/HeJ mice, compared with C3H/HeN mice (p < 0.01). PGN challenge also induced significant increases in the counts of both total cells and neutrophils, which did not differ between the two strains examined.
Notes: Open columns, C3H/HeN mice; Gray columns, C3H/HeJ mice; Data are presented as the mean ± SEM (n = 6); *p < 0.05 versus control; †p < 0.05 versus C3H/HeN mice.
Abbreviations: LPS, lipopolysaccharide; PGN, peptidoglycan; SEM, standard error of mean.
Circulating WBC and neutrophil counts
Circulating WBC and neutrophil counts in blood samples obtained from the inferior vena cava are shown in . Neither WBC nor neutrophil counts was significantly changed by intratracheal LPS administration. Although there was no significant difference between C3H/HeJ and C3H/HeN mice, leukocyte and neutrophil counts were increased by PGN instillation (p < 0.05). The absence of TLR4 signaling made no significant difference in either circulating WBC or neutrophil counts.
Table 1 Circulating leukocyte and neutrophil counts
Cytokine and chemokine production
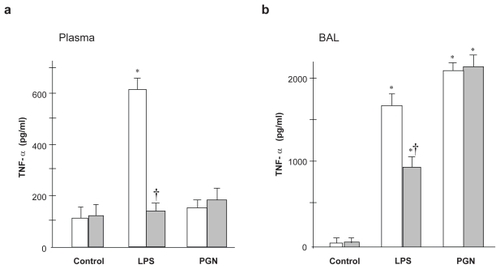
To validate the effect of TLR4 gene mutation on LPS-induced upregulation of inflammatory mediators, the levels of inflammatory cytokines (TNF-α and IFN-γ) and chemokines (KC and CXCL10) were measured in plasma and lung lavage fluid with ELISA, and the results are shown in –. In C3H/HeN mice, the plasma TNF-α level was significantly increased after LPS instillation, whereas LPS did not increase the plasma TNF-α level compared with the control group in C3H/HeJ mice (). The TNF-α level in the lavage fluid sampled after LPS instillation was decreased in C3H/HeJ mice, compared with C3H/HeN mice (). Intratracheal PGN caused no significant change in plasma TNF-α levels regardless of the strain. In contrast, PGN induced significant upregulation of TNF-α in the lung lavage fluid, which did not differ between the two strains.
Figure 3 TNF-α concentration in plasma (a) and BAL fluid (b) LPS-induced increase in plasma TNF-α concentration was significantly attenuated in C3H/HeJ mice, compared with C3H/HeN mice. PGN-induced upregulation of TNF-α did not differ between the two strains.
Notes: Open columns, C3H/HeN mice; Gray columns, C3H/HeJ mice; Data are presented as the mean ± SEM (n = 6); *p < 0.05 versus control; †p < 0.05 versus C3H/HeN mice.
Abbreviations: BAL, bronchoalveolar lavage; LPS, lipopolysaccharide; PGN, peptidoglycan; SEM, standard error of mean.
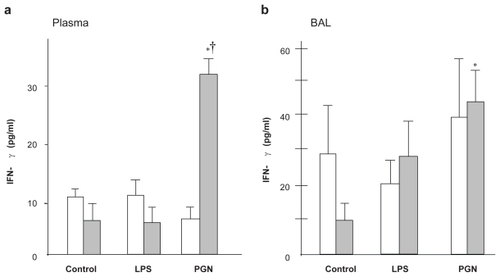
PGN instillation significantly increased the INF-γ levels in both plasma and BAL in C3H/HeJ mice (p < 0.05) but not in C3H/HeN mice (). Although the plasma IFN-γ level was not increased by LPS challenge, the IFN-γ level in BAL fluid was significantly increased after intratracheal PGN only in C3H/HeJ mice ().
Figure 4 IFN-γ concentration in plasma and BAL fluid. (a) PGN increased plasma IFN-γ only in C3H/HeJ mice, whereas LPS did not change the plasma IFN-γ level. (b) In BAL fluid, the IFN-γ level after LPS was greater in C3H/HeJ than in C3H/HeN mice, while PGN-induced upregulation of IFN-γ did not differ between the strains.
Notes: Open columns, C3H/HeN mice; Gray columns, C3H/HeJ mice; Data are presented as the mean ± SEM (n = 6); *p < 0.05 versus control; †p < 0.05 versus C3H/HeN mice.
Abbreviations: BAL, bronchoalveolar lavage; IFN, interferon; LPS, lipopolysaccharide; PGN, peptidoglycan; SEM, standard error of mean.
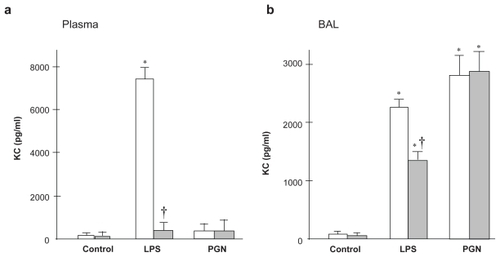
The plasma KC level was markedly increased after LPS challenge in C3H/HeN mice, which was completely suppressed in TLR4 mutant mice (p < 0.01; ). Intratracheal LPS upregulated the KC level also in BAL fluid, which was significantly greater in C3H/HeN than in C3H/HeJ mice (p < 0.05; ). PGN caused significant increase in KC levels in lung BAL fluid compared with the control group, whereas there was no difference in plasma KC levels between the control and PGN groups.
Figure 5 KC concentration in plasma and BAL fluid. (a) Plasma KC was increased after LPS challenge only in C3H/HeN mice (p < 0.01). (b) In BAL fluid, LPS-induced increase in KC was attenuated by the absence of TLR4 (p < 0.05), whereas PGN-induced upregulation of KC did not differ between the strains.
Notes: Open columns, C3H/HeN mice; Gray columns, C3H/HeJ mice; Data are presented as the mean ± SEM (n = 6); *p < 0.05 versus control. †p<0.05 versus C3H/HeN mice.
Abbreviations: BAL, bronchoalveolar lavage; LPS, lipopolysaccharide; PGN, peptidoglycan; SEM, standard error of mean; TLR4, Toll-like receptor 4.
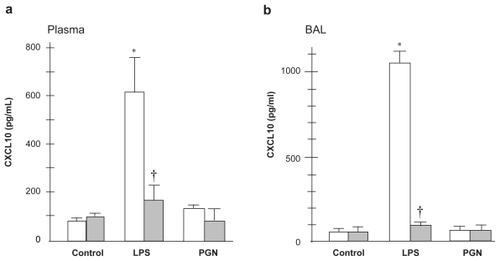
The plasma CXCL10 level was significantly increased after intratracheal LPS only in C3H/HeN mice (). There was a significant difference in the CXCL10 levels after LPS challenge between the two strains examined (p < 0.01). The CXCL10 level in BAL fluid was significantly increased by LPS only in wild type mice (). PGN instillation induced no upregulation of CXCL10 regardless of the mouse strain.
Figure 6 CXCL10 concentration in plasma and BAL fluid. LPS-induced increase in CXCL10 in plasma and BAL fluid was attenuated by the absence of TLR4 (p < 0.01). PGN did not upregulate CXCL10 in plasma or BAL fluid.
Notes: Open columns, C3H/HeN mice; Gray columns, C3H/HeJ mice; Data are presented as the mean ± SEM (n = 6); *p < 0.05 versus control; †p < 0.05 versus C3H/HeN mice.
Abbreviations: BAL, bronchoalveolar lavage; LPS, lipopolysaccharide; PGN, peptidoglycan; SEM, standard error of mean; TLR4, Toll-like receptor 4.
NF-κB translocation
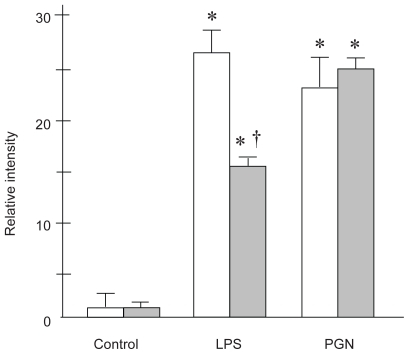
NF-κB translocation after instillation of PBS, LPS, or PGN was estimated using EMSA of whole lung nuclear extracts at 3 h (). In C3H/HeN, or wild type mice, LPS caused significant NF-κB translocation in the lungs (p < 0.001), which was attenuated in those with TLR4 gene mutation (p < 0.05). In animals that received PGN instillation, significant NF-κB translocation was observed, compared with the control mice with PBS instillation (p < 0.01). There was no difference in lung NF-κB activation after PGN between the two strains examined. These results suggest that LPS-induced NF-κB translocation may be TLR4-dependent, whereas NF-κB activation after PGN may not be TLR4- dependent.
Figure 7 NF-κB translocation in the lungs 3 hours after LPS or PGN instillation. (a) Representative example of the gel appearance. Significant attenuation of NF-κB translocation was observed in the C3H/HeJ mice followed by LPS instillation. (b) LPS-induced translocation of NF-κB was significantly attenuated by the absence of TLR4 (p < 0.05), whereas PGN-induced NF-κB translocation did not differ between the strains.
Notes: Open columns, C3H/HeN mice; Gray columns, C3H/HeJ mice; Data are presented as the mean ± SEM (n = 6); * p < 0.05 was considered to be significantly different from the corresponding value of the control of the same strain; † p < 0.05 was considered to be significantly different from the corresponding value of the C3H/HeN mice with the same treatment.
Abbreviations: BAL, bronchoalveolar lavage; LPS, lipopolysaccharide; NF-κB, nuclear factor-κB; PGN, peptidoglycan; SEM, standard error of mean; TLR4, Toll-like receptor 4.
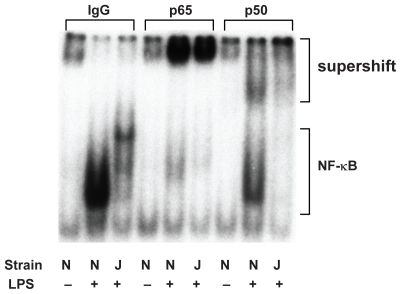
The identities of NF-κB proteins translocating to the nucleus in response to LPS were investigated with supershift analyses, using antibodies to the p65 (RelA) and p50 subunits (). Whereas the levels of NF-κB proteins in nuclear fractions were decreased in C3H/HeJ mice, the bands that were supershifted by Ab against RelA showed comparable intensity regardless of the strain. In contrast, the bands supershifted by the antibody against p50 were lighter in C3H/HeJ than in C3H/HeN mice, indicating that, in TLR4 mutant mice, the decreased translocation of p50 may be responsible for the attenuated NF-κB activation following intratracheal LPS. These results suggest that TLR4 gene mutation might inhibit nuclear binding of LPS-induced p65/p50 heterodimer. Another possibility is that the components of NF-κB may be different between the two strains.
Figure 8 Supershift analysis revealed a decrease in NF-κB in nuclear fractions in C3H/HeJ mice. The bands that were supershifted by p65 showed similar intensity regardless of the strain. In contrast, the bands supershifted by Ab against p50 was lesser in C3H/HeJ than in C3H/HeN mice.
Abbreviation: NF-κB, nuclear factor-κB.
Discussion
TLR4 is a key pattern recognition receptor that is essential for recognition of bacterial LPS, but its role in the inflammatory response to locally administered stimuli has been less clear. In this series of experiments, we evaluated the role of TLR4 in the development of acute lung inflammation after intratracheal challenge of bacterial cell constituents. In TLR4 mutant mice, LPS-induced neutrophil emigration within the lungs was attenuated, whereas PGN-induced emigration was not. LPS-induced expression of proinflammatory cytokine (TNF-α) and chemokines (KC and CXCL10) in plasma and BAL fluid was significantly attenuated in TLR4 mutant mice. In addition, NF-κB translocation after LPS was significantly attenuated in TLR4 mutant mice, whereas TLR4 gene mutation made no significant difference in PGN-induced NF-κB activation. We believe that these results suggest that TLR4 might play an important role in the development of acute lung inflammation after intratracheal instillation of LPS, a component of gram-negative bacteria.
Although there have been several reports about the importance of TLR4 in the inflammatory response to LPS, most of them described in vitro studies or an animal model of systemic administration of LPS (CitationSchwartz 2001; CitationFan et al 2003; CitationZhou et al 2005). The effect of TLR4 deficiency on lung inflammatory change following locally administered stimuli has not been evaluated quantitatively.
In C3H/HeJ mice, the missense mutation in the intracytoplasmic domain of TLR4 acts as a dominant negative molecule, possibly by sequestrating the downstream adaptor and signaling molecules (eg, MyD88 and IRAK1), and prevents LPS-induced signaling and NF-κB activation (CitationPoltorak et al 1998; CitationVogel et al 1999). Lack of TLR4-associated signaling results in the impaired transcription of various inflammatory mediators and adhesion molecules (CitationFaure et al 2000; CitationLi and Cherayil 2004). Although, in C3H/HeJ mice, attenuated response to systemic inflammation such as sepsis and impaired bacterial clearance in the lung have been reported, there have been few reports comparing the pulmonary inflammatory responses induced by LPS and PGN between TLR4 mutant and wild type mice (CitationWang et al 2002; CitationLee et al 2005).
In the present study, we observed that PGN from Staphylococcus aureus caused similar degrees of neutrophil accumulation in C3H/HeN and C3H/HeJ mice. Whereas S. aureus has been shown to be recognized by TLR2 (CitationTakeuchi et al 1999), the role of TLR4 in the inflammatory process initiated by Gram-positive organisms remains controversial (CitationMalley et al 2003; CitationBranger et al 2004). Further investigation will be needed to elucidate the role of TLRs in infection of gram-positive organisms.
CitationTakeuchi and colleagues (1999) showed that S. aureus cell wall-mediated TNF-α production was fully dependent on TLR2, which coincides with our finding that TNF-α levels in plasma and BAL fluid after PGN was not different between the two strains examined. In this study, the plasma IFN-γ level after PGN instillation was significantly greater in TLR4 mutant mice, although in PGN-treated animals, none of the other mediators differ between the two strains. Since TNF-α plays a central role in the acute inflammatory response (CitationMizgerd et al 2004), the absence of a TLR4-mediated signaling pathway might have upregulated alternative pathways, leading to the enhanced IFN-γ production after PGN challenge. Further investigation will be needed to elucidate the role of TLR2 in the wall-mediated cytokine production, such as using the TLR2 mutant mice.
KC has been reported to be associated with neutrophil recruitment into the lung vascular compartment as well as neutrophil activation (CitationFrevert et al 1995). In this study, we observed a significant decrease in LPS-induced KC production in TLR4 mutant mice, a result which agrees with a previous report (CitationRamphal et al 2005). In PGN-treated animals, however, plasma KC levels remained as low as in the control mice, which suggest that KC might not contribute to the recruitment of circulating neutrophils in response to PGN.
CXCL10 plays important roles in the host innate immune response to bacterial infection (CitationNarumi and Hamilton 1991; CitationMichalec et al 2002; CitationRe and Strominger 2004). In the present study, we observed a marked decrease in LPS-induced CXCL10 production in TLR4-mutants, suggesting that TLR4-mediated signaling may be responsible for CXCL10 production. Although CXCL10 production is induced by IFN-γ, the upregulation of IFN-γ and CXCL10 was not parallel. In addition to the direct effect of LPS on CXCL10 production, we speculate that the induction of CXCL10 by IFN-γ might have not occurred 6 hours after the PGN challenge.
The intrapulmonary deposition of bacteria or their cell constituents induce the nuclear translocation of NF-κB complexes, including p65 (RelA) and p50 subunits (CitationDoerschuk et al 1999). It is still controversial as to which subunit is essential for effectively responding to bacterial stimuli in the lungs (CitationAlcamo et al 2001; CitationMizgerd et al 2002). In this study, we observed lesser p50 in the TLR4 mutant than in wild type mice, indicating that, in TLR4 mutants, the decreased translocation of p50 may contribute to the attenuated NF-κB activation following intratracheal LPS. Although our data can not elucidate whether the p50 activity observed was homo-dimer or with BCL-3, the p50 subunit may play a more significant role in LPS-induced neutrophil accumulation in the absence of the TLR4 signaling pathway.
Conclusion
TLR4 plays an important role in the development of LPS-induced lung inflammation through differential upregulation of TNF-α and chemokines, whereas the inflammatory response to intratracheally administered PGN was independent of TLR4. TLR4 contributes to the pathogenesis of pneumonia caused by Gram-negative organisms as well as systemic inflammation such as sepsis.
Acknowledgments
This work was supported by a grant-in-aid for Scientific Research (B), Ministry of Education, Science, Sports and Culture, 16390457. The authors report no conflicts of interest in this work.
References
- AkiraSUematsuSTakeuchiO2006Pathogen recognition and innate immunityCell12478380116497588
- AlcamoEMizgerdJPHorwitzBH2001Targeted mutation of TNF receptor I rescues the RelA-deficient mouse and reveals a critical role for NF-kappa B in leukocyte recruitmentJ Immunol167159260011466381
- BrangerJKnappSWeijerS2004Role of Toll-like receptor 4 in gram-positive and gram-negative pneumonia in miceInfect Immun727889414742522
- CiesielskiCJAndreakosEFoxwellBM2002TNFα-induced macrophage chemokine secretion is more dependent on NF-κB expression than lipopolysaccharides-induced macrophage chemokine secretionEur J Immunol3220374512115625
- DingKShibuiAWangY2005Impaired recognition by Toll-like receptor 4 is responsible for exacerbated murine Pneumocystis pneumoniaMicrobes Infect719520315725383
- DoerschukCMMizgerdJPKuboH1999Adhesion molecules and cellular biomechanical changes in acute lung injury: Giles F. Filley LectureChest11637S43S10424587
- FanJFreyRSMalikAB2003TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidaseJ Clin Invest11212344314561708
- FaureEEquilsOSielingPA2000Bacterial lipopolysaccharide activates NF-κB through Toll-like receptor 4 (TLR-4) in cultured human dermal endothelial cellsJ Biol Chem275110586310753909
- FaureKSawaTAjayiT2004TLR4 signaling is essential for survival in acute lung injury induced by virulent Pseudomonas aeruginosa secreting type III secretory toxinsRespir Res511015040820
- FrevertCWHuangSDanaeeH1995Functional characterization of the rat chemokine KC and its importance in neutrophil recruitment in a rat model of pulmonary inflammationJ Immunol154335447995953
- HoggJCDoerschukCM1995Leukocyte traffic in the lungAnnu Rev Physiol57971147778886
- LeeJSFrevertCWMatute-BelloG2005TLR-4 pathway mediates the inflammatory response but not bacterial elimination in E. coli pneumoniaAm J Physiol Lung Cell Mol Physiol289L731816024722
- LiQCherayilBJ2004Toll-like receptor 4 mutation impairs the macrophage TNF-alpha response to peptidoglycanBiochem Biophys Res Commun32591615522205
- MalleyRHennekePMorseSC2003Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infectionProc Natl Acad Sci U S A10019667112569171
- MichalecLChoudhuryBKPostlethwaitE2002CCL7 and CXCL10 orchestrate oxidative stress-induced neutrophilic lung inflammationJ Immunol1688465211777981
- MizgerdJPScottMLSpiekerMR2002Function of IκB proteins in inflammatory responses to Eschericha coli LPS in mouse lungsAm J Respir Cell Mol Biol275758112397017
- MizgerdJPLupaMMHjobergJ2004Roles for early response cytokines during Escherichia coli pneumonia revealed by mice with combined deficiencies of all signaling receptors for TNF and IL-1Am J Physiol Lung Cell Mol Physiol286L13021014966082
- NarumiSHamiltonTA1991Inducible expression of murine IP-10 mRNA varies with the state of macrophage inflammatory activityJ Immunol1463038441901891
- PahlHL1999Activators and target genes of Rel/NF-κB transcription factorsOncogene1868536610602461
- PoltorakAHeXSmirnovaI1998Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutation in Tlr4 geneScience282208589851930
- RamphalRBalloyVHuerreM2005TLRs 2 and 4 are not involved in hypersusceptibility to acute Pseudomonas aeruginosa lung infectionsJ Immunol17539273416148139
- ReFStromingerJL2004IL-10 released by concomitant TLR2 stimulation blocks the induction of a subset of Th1 cytokines that are specifically induced by TLR4 or TLR3 in human dendritic cellsJ Immunol17375485515585882
- SchurrJRYoungEByrneP2005Central role of Toll-like receptor 4 signaling and host defense in experimental pneumonia caused by gram-negative bacteriaInfect Immun735324515618193
- SchwartzDA2001The role of TLR4 in endotoxin responsiveness in humansJ Endotoxin Res73899311753209
- StrieterRMBelperioJAKeaneMP2002Cytokines in innate host defense in the lungJ Clin Invest10969970511901175
- TakeuchiOHoshinoKKawaiT1999Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall componentsImmunity114435110549626
- TasakaSIshizakaAYamadaW2003Effect of CD14 blockade on endotoxin-induced acute lung injury in miceAm J Respir Cell Mol Biol29252812639839
- VogelSNJohnsonDPereraPY1999Functional characterization of the effect of the C3H/HeJ defect in mice that lack an Lpsn gene: in vivo evidence for a dominant negative mutationJ Immunol16256667010229796
- WangXMoserCLouboutinJP2002Toll-like receptor 4 mediates innate immune responses to Haemophilus influenzae infection in mouse lungJ Immunol168810511777976
- YamamotoMTakedaKAkiraS2004TIR domain-containing adaptors define the specificity of TLR signalingMol Immunol40861814698224
- ZhouXGaoXPFanJ2005LPS activation of Toll-like receptor 4 signals CD11b/CD18 expression in neutrophilsAm J Physiol Lung Cell Mol Physiol288L6556215563689