Abstract
Age-related macular degeneration (AMD) is the leading cause of blindness in developed countries. The etiology of AMD remains poorly understood and no treatment is currently available for the atrophic form of AMD. Atrophic AMD has been proposed to involve abnormalities of the retinal pigment epithelium (RPE), which lies beneath the photoreceptor cells and normally provides critical metabolic support to these light-sensing cells. Cumulative oxidative stress and local inflammation are thought to represent pathological processes involved in the etiology of atrophic AMD. Studies of tissue culture and animal models reveal that oxidative stress-induced injury to the RPE results in a chronic inflammatory response, drusen formation, and RPE atrophy. RPE degeneration in turn causes a progressive degeneration of photoreceptors, leading to the irreversible loss of vision. This review describes some of the potential major molecular and cellular events contributing to RPE death and inflammatory responses. In addition, potential target areas for therapeutic intervention will be discussed and new experimental therapeutic strategies for atrophic AMD will be presented.
Introduction
Age-related macular degeneration (AMD) is an idiopathic retinal degenerative disease and the cause of irreversible, profound vision loss in people over the age of 60 years (CitationEvans and Wormald 1996). Due to a rapidly aging population, the number of people with AMD in the US is estimated to increase from 1.75 million in 2000 to 2.95 million in 2020 (CitationFriedman et al 2004). Similar proportional increases in the worldwide prevalence of AMD can be expected placing an increasing burden on health services. The market for AMD therapies is projected to increase from $638 million in 2005 to $2722 million in 2015 (CitationAugustin and Offermann 2006).
AMD occurs in two major forms: atrophic (dry) AMD and exudative (wet) AMD. Atrophic AMD, characterized by retinal pigment epithelial (RPE) cell atrophy and subjacent photoreceptor degeneration, accounts for approximately 25% of cases with severe central vision loss (CitationKlein et al 1997). In contrast, exudative AMD is characterized by choroidal neovascularization (CNV) through the outer layers of Bruch’s membrane under the basement membrane of the RPE or through the RPE into the subretinal space with subsequent retinal hemorrhage, detachment, and scarring, and accounts for approximately 75% of cases with severe central vision loss (CitationKlein et al 1997). These two forms of AMD are both part of the same disease process and share similar risk factors for their development. In fact, patients may have CNV in one eye and geographic atrophy, the advanced form of atrophic AMD, in the second eye. In some cases, the dry form of AMD may progress to wet AMD (CitationSunness et al 1999). The specific biological processes that determine which form of advanced AMD occurs and why and how some of dry AMD later progress to wet AMD are still elusive. AMD is an aging-related syndrome caused by multiple factors including environmental, nutritional, and behavioral (CitationSnodderly 1995; CitationSeddon et al 1996) as well as different susceptibilities to these factors based upon an individual’s genetic background (CitationHeiba et al 1994; CitationSeddon et al 1997). The early pathological changes of atrophic AMD begin with extracellular lipid-containing deposits in the macular area (CitationBressler et al 1990). Patients with drusen greater than 63 μm in diameter are diagnosed as having early AMD (CitationBird et al 1995; CitationCurcio and Millican 1999). Large drusen can merge, become confluent and eventually lead to geographic atrophy of the central retina. Although the vision loss results from photoreceptor damage in the central retina and abnormalities of the photoreceptor cells or the choroid might contribute to the development of AMD, the initial pathogenesis of atrophic AMD has been proposed to involve the degeneration of RPE cells (CitationSpraul et al 1996; CitationZarbin 1998). The specific genetic and biochemical mechanisms responsible for RPE degeneration in AMD have not been well determined. Recent progress in our understanding of atrophic AMD suggests that inflammation initiated by RPE cell death and RPE cell inflammatory responses plays an important role in drusen biogenesis and maybe central to the etiology of this disease (CitationHageman et al 2001; CitationRodrigues 2007).
The RPE is a monolayer of cuboidal cells located subjacent to the neural retina (CitationStrauss 2005). The basal membrane of the RPE is in contact with Bruch’s membrane, a multilayered matrix that separates the RPE from the underlying choroidal vasculature. The apical membrane of the RPE is intimately associated with the outer segments of retinal photoreceptor cells. The major function of RPE cells is to support the survival and normal functioning of photoreceptors by acting as part of the outer blood-retinal barrier to control exchange of nutrients, waste products, ions, and gases between the underlying choroidal blood vessels and overlying photoreceptors (CitationMcBee et al 2001). Additionally, the RPE is involved in the shuttling of retinoids required for visual pigment synthesis to photoreceptors (CitationThompson and Gal 2003), and in phagocytizing shed photoreceptor outer segment (POS) membrane discs for photoreceptor renewal (CitationNguyen-Legros and Hicks 2000). RPE cells also produce trophic factors necessary for photoreceptor survival, and immunological factors necessary for establishing immune privilege of the eye (CitationTanihara et al 1997; CitationStreilein et al 2002). Injury to RPE cells causing failure of these functions may contribute to the retinal degeneration observed in AMD. Thus, there is considerable interest in elucidating the mechanisms responsible for RPE injury that would provide the basis for designing new strategies to treat or prevent AMD. In this review, the molecular and cellular events involved in RPE injury and inflammation, and potential therapeutic strategies are discussed.
Factors causing RPE cell injury
Oxidative stress
Histological staining with biomarkers of oxidative damage has demonstrated widespread oxidative damage in the retinas of patients with atrophic AMD and to a lesser extent also in the retinas of patients with wet AMD (CitationShen et al 2007). Moreover, intravitreal injection of oxidants results in lipid peroxidation and retinal degeneration in animal models (CitationCingolani et al 2006). RPE cells suffer from cumulative oxidative stress due to normal physiological activity as well as environmental factors (). RPE cells generate reactive oxygen species (ROS) during phagocytosis (CitationMiceli et al 1994). RPE phagocytosis takes up the tips of the shed POS that contain high concentrations of radicals, photo-damaged proteins and lipids. Some of these oxidative-damaged proteins and lipids are not completely digested by RPE lysosomes and accumulate within RPE cells as a major source of the photosensitizer, lipofuscin during RPE aging (CitationKennedy et al 1995). Once irradiated by light, lipofuscin produces ROS that damage surrounding tissues (CitationBoulton et al 1993). Thus, the unique phagocytic function of RPE places an additional oxidative burden on the RPE. This is particularly true of RPE cells in the macula, where the predominant photoreceptors are cones that have a higher energy demand than rods (CitationPerkins et al 2003). Additionally, the ratio of photoreceptors to RPE cells in the macula is significantly higher than in the para-macular region or the periphery (CitationDorey et al 1989).
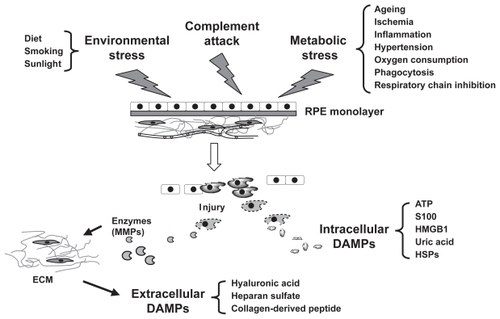
Figure 1 Conditions that trigger retinal pigment epithelial (RPE) cell injury and subsequent release of damage-associated molecule pattern (DAMP) molecules. RPE cell injury is triggered by environmental stress, metabolic stress, and complement attack. Injured or dying RPE cells then produce danger signals that are defined as DAMP molecules. Some of these danger signals are intracellular molecules and others are the cleaved products of extracellular components such as extracellular matrix (ECM).
Abbreviations: HMGB1, high mobility group box 1; HSPs, heat-shock proteins; MMPs, matrix metalloproteinases.
In addition to the physiological oxidative stress, aging and pathological conditions can also contribute to oxidative damage to macromolecules via a reduction in defense mechanisms or an increase in ROS production or both (CitationAmes et al 1993). Environmental factors such as cigarette smoke (CitationChan 1998), light exposure (CitationCruickshanks et al 1993), diet, and cataract surgery also enhance ROS generation and can increase the risk for AMD. Heavy cigarette smokers have significantly higher risks of AMD (CitationThornton et al 2005; CitationCong et al 2008). Furthermore, smoking can also cause a reduction in retinal and choroidal blood flow (CitationTamaki et al 1999; CitationWimpissinger et al 2004, Citation2005). The relative contribution of these two processes to AMD remains to be determined. Cataract surgery to remove the lens increases the amount of harmful ultraviolet and blue light reaching the retina. Epidemiological studies have revealed an increased risk of late AMD in patients with cataract surgery (CitationKlein et al 2002; CitationHo et al 2008). Data analysis of participants of the Rotterdam study shows that a history of cataract surgery is linked to an increased incidence of atrophic AMD, but no correlation with wet AMD was observed (CitationHo et al 2008).
Recent evidence suggests that genetic factors may also contribute to the accumulation of oxidative stress. Polymorphisms in the age-related maculopathy susceptibility 2 gene (ARMS2, also known as LOC387715) were shown to be strongly associated with increased risk of AMD. The A69S polymorphism shows a strong association with risk of AMD (odds ratio [OR] 2.66 for GT heterozygotes and 7.05 for TT homozygotes) (CitationKanda et al 2007). A deletion-insertion polymorphism (del443ins54) in ARMS2 also causes an increased risk in individuals carrying single copy of this allele (OR, 2.9) or those that are homozygous (OR, 8.1) (CitationFritsche et al 2008). The ARMS2 gene codes for a hypothetical protein of approximately 12 ~ 13.5 kDa expressed in RPE and photoreceptor cells that is associated with the mitochondrial outer membrane, leading to the hypothesis that ARMS2 dysfunction could alter mitochondrial energy metabolism and homeostasis, thereby generating ROS (CitationKanda et al 2007; CitationFritsche et al 2008). Animal studies have also provided genetic evidence for a role of oxidative stress in AMD. Deletion of the superoxide dismutase 1 (SOD1) gene, the product of which is responsible for scavenging superoxide, results in mice that develop many of the hallmark features of AMD including drusen, RPE atrophy, and CNV (CitationImamura et al 2006; CitationHashizume et al 2008).
The factors described above suggest that oxidative stress is particularly significant in the RPE. Indeed, it has been demonstrated that a variety of these oxidative stressors can cause RPE cell injury and death in cultured RPE cell lines (CitationLu et al 2006; CitationQin 2007). Once the protection afforded by the antioxidant system has been overwhelmed, dysfunction and death of RPE cells can occur. Thus, boosting the antioxidant capacity of these cells has the potential to prevent or delay AMD progression (CitationSnodderly 1995; CitationWinkler et al 1999).
Complement-mediated bystander lysis
The activation of complement (McGeer and McGeer 2004) is a sophisticated attack system designed to destroy invaders, stimulate inflammation and assist in the phagocytosis of waste materials. A role for the complement pathway in AMD was first recognized through the biochemical characterization of the composition of drusen. By histochemical analysis and immunocytochemical studies drusen were found to contain numerous components of the complement pathway including complement activators, inhibitors, activation-specific complement fragments as well terminal pathway components (CitationHageman et al 2001; CitationAnderson et al 2002). Complement cascades consist of the classical, alternative, and lectin pathways (CitationWalport 2001). Antibodies are considered to be the chief activators of the classical complement pathway while molecules, such as lipopolysaccharides, activate the alternative complement pathway. Once the complement system is fully activated, it proceeds to assemble the terminal components (C5b, C6, C7, C8, C9) into the lytic macromolecule C5b-9, known as the membrane attack complex (MAC) that mediates the destruction of the foreign bacteria and viruses. However, if host cells are inadequately protected, they may also suffer a MAC attack in a process termed bystander lysis. Under normal physiological conditions, RPE cells protect themselves from complement-mediated bystander lysis, by expressing high levels of complement pathway regulators, such as complement factor H (CFH) (CitationChen et al 2007; CitationWu et al 2007) and CD46 (CitationMcLaughlin et al 2003; CitationVogt et al 2006) that inhibit activation of alternative pathway. However, acquisition of functional mutations or dys-regulation of these regulators during aging can lead to activation of alternate pathway. The Y402H polymorphism in CFH in particular has been proposed to be the major risk factor for acquiring AMD (CitationEdwards et al 2005; CitationHaines et al 2005; CitationKlein et al 2005). In addition, CFH-related proteins (CFHR) including CFHR1, CFHR2, CFHR3, CFHR4, and CFHR5, which lack the complement regulatory activity of CFH can indirectly modulate activation of the complement pathway, likely via competing with CFH (CitationJozsi and Zipfel 2008). Deletion of CFHR1 and CFHR3 has been reported to be associated with decreased risk of AMD. Meta-analysis reveals that the ORs are 0.35 and 0.63 for a Caucasian population of 173 cases and 170 controls in the UK (CitationHughes et al 2006) and a Caucasian population of 780 cases and 265 controls in the US (CitationSpencer et al 2008). Similarly, the R32G polymorphism in complement factor B (CFB), an activator of alternative pathway, is significantly associated with a reduced incidence AMD. In a Caucasian population of 698 cases and 282 controls evaluated at Medical Centers at Vanderbilt and Duke Universities, the R32Q variance was strongly associated with reduced risk of AMD (OR, 0.21) after controlling for age, the Y402H polymorphism, and smoking (CitationSpencer et al 2007). A similar result is obtained in the combined Columbia and Iowa cohorts with 551 cases and 269 controls (OR 0.32) (CitationGold et al 2006). In contrast, genotyping demonstrated that a single nucleotide polymorphism in Complement factor 3 (C3) is significantly correlated with an increased risk of AMD (CitationMaller et al 2007; CitationYates et al 2007; CitationSpencer et al 2008). In combined English and Scottish groups with a population of 847 cases and 701 controls, the ORs for S/F heterozygotes and F/F homozygotes are 1.7 and 2.6, respectively (CitationYates et al 2007).
Together, these observations demonstrate a role for complement, particularly the alternative pathway, in the development of AMD. Consistent with this notion, it has been shown that in vitro, RPE cell synthesis of CFH is downregulated by direct hydrogen peroxide challenge (CitationWu et al 2007) or exposure to oxidized POS (CitationChen et al 2007). Furthermore, light exposure activates complement pathways in A2E-loaded RPE cells (CitationZhou et al 2006). Thus, it is reasonable to speculate that with aging, increased numbers of RPE cells could be subjected to bystander lysis due to oxidative stress-induced activation of the complement system. While the alternative pathway seems to play the major role in AMD, it was recently reported that mice immunized with mouse serum albumin (MSA) adducted with carboxylpyrrole, an oxidation fragment of docosahexaenoic acid (DHA) develop phenotypes mimicking atrophic AMD such as RPE cell lysis and drusen formation (CitationHollyfield et al 2008), suggesting that the classical pathway may also be involved in the development of atrophic AMD.
RPE cell inflammatory responses
Drusen biogenesis, in particular soft drusen, is a biological marker of atrophic AMD. Protein components of drusen include immunoglobulins, complement components, acute-phase inflammatory response molecules, and major histocompatibility complex class II antigens (CitationJohnson et al 2000; CitationMullins et al 2000). Cellular components of drusen include RPE debris, lipofuscin, and dendritic cells (DCs) (CitationHageman et al 2001). The mechanisms by which drusen is formed are poorly understood. However, compelling evidence from a number of laboratories suggest that inflammatory responses of injured RPE cells serve as the initial events in drusen biogenesis (CitationHageman et al 2001; CitationAnderson et al 2002). Potential triggers of RPE cell inflammatory responses discussed here include molecules derived from necrotic RPE cells, complement attack, and oxidized adducts.
Pro-inflammatory danger molecules: DAMPs
Tissue damage arising from nutrient, oxidative, genomic, or mechanical stress is a principal stimulus of inflammation. Since this inflammation occurs without infection, it is defined as sterile inflammation to distinguish it from pathogen-induced inflammation. Although the mechanisms that stimulate the inflammatory response to injured cells remain poorly defined, progress has been made recently in our understanding of the process. In a fashion similar to defenses against invading microorganisms, the innate immune system alerts neighboring cells to the imminent danger by reacting to damage signals generated from dying cells called damage-associated molecular patterns (DAMPs). These signals lead to an inflammatory response by releasing mediators that dilate local vasculature and recruit leukocytes from the blood into the tissue. These DAMP molecules can be in general divided into two categories, intracellular and extracellular DAMPs, based on their origin and mechanism of action. Intracellular DAMPs are bioactive mediators of cellular origin that directly stimulate cells of the innate immune system. Extracellular DAMPs are the cleaved fragments of extracellular components generated by enzymes released from dead cells.
As shown in , intracellular DAMPs include the high mobility group box 1 protein (HMGB1) (CitationKlune et al 2008), heat-shock proteins (HSPs) (CitationChen et al 1999; CitationAsea et al 2000), S100 proteins (CitationHofmann et al 1999; CitationFoell et al 2007), nucleosomes (CitationMuruve et al 2008), ATP (CitationIdzko et al 2007), and uric acid (CitationMartinon et al 2006). These molecules are expressed in all cell types. Most intracellular DAMPs exhibit a double life. As intracellular molecules, they play a role in cell homeostasis, for example as calcium binding proteins, chaperones, or chromatin-stabilizing molecules. Once released into the extracellular compartment as a result of cell injury, they become ‘danger’ signals which serve to warn other cells and tissues of imminent danger by activating innate and pro-inflammatory immune responses. For example, S100B, a member of the S100 family of calcium-binding proteins is a ligand for the receptor for advanced glycation endproducts (RAGE) and can stimulate expression of vascular endothelial growth factor (VEGF), transforming-growth factor-β1 and fibronectin in RPE cells (CitationKim et al 2007; CitationMa et al 2007). Induction of a variety of cytokines by intracellular DAMPs in RPE cells could contribute to disease progression and potentially the conversion of atrophic to wet AMD.
Extracellular DAMPs are generated through the cleavage of extracellular matrix molecules such as collagen, hyaluronic acid, and heparan sulfate into pro-inflammatory fragments (CitationWrenshall et al 1999; CitationJohnson et al 2002; CitationTaylor et al 2004; CitationScheibner et al 2006; CitationWeathington et al 2006). Collagen, heparan sulfate, and hyaluronan are major components of RPE extracellular matrix that have been shown to be expressed in cultured RPE cells (CitationGross-Jendroska et al 1992; CitationdeS et al 2001). Degradation of extracellular matrix proteins is regulated by matrix metalloproteinases (MMPs), a family of zinc-binding, calcium-dependent enzymes (CitationAlexander et al 1990). Release of MMPs from necrotic RPE cells would be expected to disrupt extracellular matrix, generating extracellular DAMPs. However, the extent to which these cellular triggers and the extracellular DAMPs that they generate contribute to the inflammatory response in AMD has not yet been addressed.
The detection of danger signals is made by antigen-presenting cells of the innate immune systems through receptors that recognize DAMP molecules. The known DAMP receptors include CD14, the NACHT-, LRR-, and Pyrin-domain-containing protein-3 (NALP3), P2 receptor, RAGE, and toll-like receptors (TLR) (). CD14, NALP3, and P2 receptors detect HSPs (CitationChen et al 1999; CitationAsea et al 2000), monosodium urate (CitationMartinon et al 2006), and extracellular ATP (CitationIdzko et al 2007), respectively. RAGE (discussed below) is a multi-ligand receptor for advanced glycation end (AGE) product, HMGB1, and S100 proteins (CitationBarile and Schmidt 2007). TLRs recognize heparin suflate, HMGB1, HSP, and hyaluronate (CitationMiyake 2007).
Figure 2 Receptors that sense danger signals released from neighboring injured retinal pigment epithelial (RPE) cells. Toll-like receptors (TLRs) and the receptor for AGE (RAGE) can detect danger signals released from or generated by dying RPE cells. Once stimulated, TLRs and RAGE activate the transcription factor nuclear factor kappa B (NFkB), thereby turning on the transcription of many pro-inflammatory genes and consequently inducing an inflammatory response. The DAMP molecules ATP and monosodium urate stimulate a pathway that works through Nod-like receptor NACHT, LRR, and PYD containing protein-3 (NALP3) and leads to the production of the proinflammatory cytokine interleukin-1β (IL-1β) and IL-18. Those pathways surrounded by dotted lines indicate a current lack of data supporting a functional pathway in RPE cells.
Abbreviations: AGE, advanced glycation endproducts; OxLDL, oxidized low density proteins.
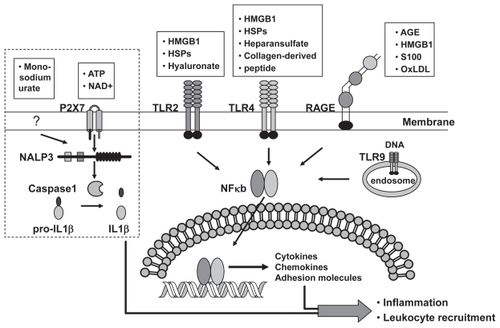
RPE cells make up the first line of defense against pathogens by expressing TLRs, which recognize microbial motifs. The ligation of TLRs leads to a cascade of events that culminates in the production of chemokines, and to the production of cytokines that can induce DC maturation. RPE cells express almost all TLR iso-forms except TLR-8 (CitationKumar et al 2004). While, activation of TLR-2, 4, and 9 have been demonstrated to result in cytokine production when cells are challenged with DAMP molecules in other systems (CitationScheibner et al 2006; CitationMiyake 2007) it remains to be addressed whether TLRs have similar roles in mediating RPE cell danger responses.
Oxidized adducts
RPE cells are challenged with oxidative stress due to light irradiation, high oxygen consumption, and phagocytosis of photo-bleached POS containing high levels of oxidized unsaturated fatty acids and free radicals. Many oxidized molecules, which are toxic to RPE cells, accumulate within and surround RPE cells with aging. Here we will focus our discussion on the relevance of AGE products, oxidized low-density proteins (oxLDL) and carboxyethyl pyrole (CEP) adducts to the etiology of AMD.
AGEs are an heterogeneous group of products derived from nonenzymatic glycation and oxidation of amino groups of proteins, lipids, and nucleic acids. AGEs accumulate during normal aging with their formation being accelerated in a setting of oxidative stress and inflammation (CitationSchleicher et al 1997). There is little or no AGE products detected in normal retina, but expression of AGE products increases concomitantly with drusen formation and development of early atrophic AMD (CitationHowes et al 2004). AGE products are also present in RPE lipofuscin, an enzymatically nondegradable heterogeneous mixture of numerous biomolecules (CitationSchutt et al 2003). With the accumulation of AGE products during aging, RAGE, receptors for AGEs, are simultaneously induced (CitationYamada et al 2006). RAGE belongs to the immunoglobulin super-family of cell surface molecules that are constitutively expressed at very low levels in numerous cells, including Muller cells, photoreceptor cells, RPE cells, and vascular endothelial cells (CitationHowes et al 2004; CitationMcFarlane et al 2005; CitationBarile and Schmidt 2007). Cellular expression of RAGE increases upon ligand binding thus further amplifying cellular activation. In vivo, the levels of RAGE are correlated with drusen formation and early development of AMD (CitationHowes et al 2004). Additionally, induction of AGE formation in vivo leads to the increased transcription of inflammatory genes in the RPE/choroid (CitationTian et al 2005). In cultured human RPE cells, activation of AGE-RAGE pathway stimulates expression of VEGF (CitationMa et al 2007), platelet derived growth factor-b (CitationHanda et al 1998), and production of interlukin-8 (IL-8) and monocyte chemotactic protein-1 (MCP-1) (also known as CCL2) (CitationBian et al 2001). Moreover in vitro, ligand-dependent activation of RAGE can trigger RPE cell death (CitationHowes et al 2004). Similarly, exposure of RPE cells to AGE-modified matrix but not normal matrix triggers RPE cell death (CitationHonda et al 2001). These observations suggest that activation of the RAGE axis is involved in disease progression and potentially in the conversion of atrophic to wet AMD. Consistent with this notion, transgenic expression of RAGE augmented blood-retinal barrier breakdown and leukostasis, accompanied by increased expression of VEGF and ICAM-1 in the retina in a murine diabetic model (CitationKaji et al 2007). Moreover, these changes were significantly inhibited by systemic administration of a soluble form of RAGE.
Oxidation of LDL generates malondialdehyde lysine protein adducts or 4-hydroxynoneal cysteine adducts that are the major modifications observed in RPE lipofuscin (CitationSchutt et al 2003). OxLDLs have been shown to accumulate in Bruch’s membrane in AMD patients (CitationYamada et al 2008). Significantly, treatment of RPE cells in culture with oxLDL alters phagosome maturation (CitationHoppe et al 2004) and inhibits phagocytosis of POS (CitationHoppe et al 2001), an important RPE cell function essential for outer segment renewal and survival of photoreceptors. Moreover, exposure of RPE cells to oxLDL induces transcriptional alterations in genes related to lipid metabolism, oxidative stress, inflammation and apoptosis (CitationYamada et al 2008). Consistent with this, ingestion of oxidized POS stimulates production of IL-8 and MCP-1 in cultured RPE cells (CitationHiggins et al 2003). Additionally, as oxLDLs are ligands for scavenger receptors expressed on macrophages oxLDL accumulation leads to the recruitment of macrophages (CitationKamei et al 2007).
Proteomic analysis of drusen revealed the presence of a number of oxidized proteins (CitationCrabb et al 2002). Most notable of these is CEP adducts. CEP adducts are uniquely generated by the oxidation of docosahexaenoate-containing lipids. Interestingly, docosahexaenoate is abundant in photoreceptors and is the most oxidizable fatty acid in humans (CitationFliesler and Anderson 1983). Western analysis and immunocytochemistry confirmed that CEP adducts are more abundant in photoreceptors in AMD than normal tissue (CitationCrabb et al 2002; CitationGu et al 2003). Moreover, CEP immunoreactivity as well as autoantibodies to CEP was higher in plasma from AMD patients (CitationGu et al 2003). As previously noted, mice immunized with MSA adducted with CEP develop an atrophic AMD-like phenotype (CitationHollyfield et al 2008). Macrophages are observed in the interphotoreceptor matrix in these mice. Additionally, immunized mice fix C3 in Bruch’s membrane (CitationHollyfield et al 2008). Interestingly, CEP modified human serum albumin was shown to stimulate angiogenesis in ex-vivo models and a subretinal injection of MSA-CEP exacerbated laser-induced CNV in mice (CitationEbrahem et al 2006). Together, these observations implicate oxidized molecules in the perturbation of RPE cell function and inflammatory responses observed in AMD.
Complement-driven inflammation
Activated complement will injure host cells and induce inflammatory responses under the conditions where loss of function mutations or downregulation of inhibitory regulators of the pathway occurs. In the retina, host cells are protected from complement attack by complement factors such as CFH, a negative regulator of the complement alternative pathway. The RPE is the major local source of CFH at the retina/choroid interface. Thus, mutations or downregulation of CFH may increase the chance of RPE cells being attacked by activated complement systems. During the process of complement cascade activation, the small fragments of cleaved complement components C3a, C4a, and C5a, known as anaphylotoxins, stimulate inflammation. C3a and in particular C5a, are potent inflammatory factors, recruiting leucocytes from blood and stimulating secretion of cytokines that lead to amplification of inflammatory responses (CitationEmber et al 1994; CitationDaffern et al 1995). In cultured RPE cells, treatment with C5a stimulates production of IL-8 (CitationFukuoka and Medof 2001; CitationFukuoka et al 2003) and MCP-1 (CitationAmbati et al 2003). Furthermore, C3a and C5a have been shown to be present in drusen (CitationJohnson et al 2001; CitationAmbati et al 2003), supporting the idea that RPE cells are constantly stimulated by the presence of C3a and C5a, which induce the release of inflammatory mediators that subsequently cause further damage to retina in vivo. In addition, C3a and C5a are generated early in the course of laser-induced CNV and activation of C3aR or C5aR has been shown to be required for CNV formation (CitationNozaki et al 2006). Moreover, the alternative pathway has been suggested to play a central role in the CNV development in laser-induced murine model (CitationBora et al 2006). Taken together, these results suggest that complement-driven inflammation is involved in the etiology of both dry and wet AMD as well as progression of dry to wet AMD.
Chronic infection
RPE cells can be infected in vitro with viruses and bacteria such as cytomegalovirus (CitationBodaghi et al 1999) and Bacillus cereus (CitationMoyer et al 2008). The presence of viruses in RPE cells has also been demonstrated in patients (CitationHenderly et al 1988) and in animal models (CitationVann and Atherton 1991). Additionally, pathogen replication and toxin production can cause RPE death (CitationVann and Atherton 1991; CitationMoyer et al 2008). Evidence is accumulating that pathogen-induced chronic infection may be a novel risk factor in the etiology of AMD. An increase in the prevalence of higher-titer antibody to Chlamydia pneumoniae was found more often in wet AMD than in dry AMD (CitationMiller et al 2004). Subjects with high antibody titers had a threefold greater risk of progression than those with low antibody titers (CitationRobman et al 2005). Pathogen infection may stimulate macrophage activation and entrapment between the choroid and RPE layer. Local release of cytokines from these macrophages in concert with cytokine release from overlying RPE cells might be involved in the manifestation of certain AMD phenotypes. This is supported by the observation that infection of macrophages and RPE cells with C. pneumoniae in vitro induces production of cytokines such as VEGF, IL-8, and MCP-1 (CitationKalayoglu et al 2005). Moreover, the risk of AMD progression was increased by about 12-fold when, in addition to having the CFH C-risk allele, subjects also presented with high antibodies titers to the bacterial pathogen C. pneumoniae (CitationBaird et al 2008). These observations suggest that infection-induced complement activation may participate in AMD progression, although a causal and functional tie between infection and AMD remains to be established.
Recruitment of inflammatory cells
Injury to RPE cells caused by oxidative stress induced by a number of processes including ischemia, photooxidative damage, phagocytosis, and lipofuscin toxicity, serves as the critical seeding event in the initiation of drusen formation by secreting soluble cytokines that initiate recruitment of macrophages and DCs (CitationHageman et al 2001; CitationHoltkamp et al 2001). Activated macrophages and DCs have been found to accumulate in the subretinal space of patients with AMD (CitationGupta et al 2003). However, it is unclear whether endogenous macrophages serve protective or destructive functions in these patients. Recruited macrophages have been shown to be required for clearance of drusen and other debris (CitationDuvall and Tso 1985). While activated macrophages and DCs phagocytize RPE debris they also produce mediators that amplify local inflammation (Citationvan der Schaft et al 1993; CitationHageman et al 2001; CitationPenfold et al 2001). In support of a protective role for these cells, impairment in recruitment of macrophages leads to manifestations of atrophic AMD in CCL2 (MCP-1) knockout mice (CitationAmbati et al 2003). Consistent with this, an age-dependent increase in the expression of CCL2 (MCP-1) in the RPE and in macrophage infiltration is observed in the choroids of wild-type mice (CitationAmbati et al 2003). Additionally, IL-10−/− mice have significantly reduced laser-induced CNV formation with increased macrophage infiltrates compared to wild type mice. Moreover, direct injection of macrophages into the eye of these IL-10−/− mice inhibits CNV (CitationApte et al 2006). However, depletion of macrophages is also reported to inhibit experimental CNV (CitationEspinosa-Heidmann et al 2003; CitationSakurai et al 2003). These findings suggest that macrophages can serve in both pro- and anti-inflammatory capacities, and that dysregulation of clearance functions may fuel disease progression.
DCs are powerful antigen-presenting cells that participate in the induction of immunity. Histochemical studies have revealed that DCs are present in drusen and are thus proposed to play a role in drusen biogenesis (CitationHageman et al 2001). According to this hypothesis, injured RPE cells recruit and activate DCs which sustain and amplify the local inflammation by producing inflammatory mediators, activating complement system, and degrading extracellular matrix. The CX3C chemokine receptor 1 (CX3CR1) is expressed in retinal microglia cells (macrophages and DCs) and mediates migration and adhesion of these cells in response to its ligand. Interestingly, mice deficient in CX3CR1 develop some cardinal features of AMD with accumulation of microglia cells in the subretinal space at the sites of retinal degeneration and CNV (CitationCombadiere et al 2007). Manifestations of AMD-like retinal lesions are exacerbated by further knockdown of the chemokine, CCL2 (MCP-1). AMD lesions are found in 6-week-old CX3CR1/CCL2 double-deficient mice while none of the single knockout mice develop retinal lesions at this age (CitationTuo et al 2007). This may be because of the retention of CX3CR1-deficient microglia cells at the sites of cell and/or tissue injury leading to sustained inflammation. In the laser-induced CNV model, DCs infiltrate transiently into the lesion sites (CitationEter et al 2008; CitationNakai et al 2008) and intravenous DC transplantation augments CNV (CitationNakai et al 2008), supporting a contribution of DCs to the development of AMD.
Recent evidence suggests that lymphoid cells may also be involved in the development of experimental atrophic AMD. When RAG-deficient mice lacking mature lymphocytes were challenged with MSA adducted with CEP, unlike wild-type mice they failed to produce antigen-specific antibodies and fix C3 in Bruch’s membrane. Nor did these animals develop an AMD-like phenotype (CitationHollyfield et al 2008).
Potential therapeutic approaches for atrophic AMD
Damage caused by oxidative stress and inflammation lead to progressive loss of cell function and thus contributes to the development of atrophic AMD. Protecting RPE cells by neutralizing ROS, promoting cell survival, and inhibiting inflammatory responses could potentially delay the onset or progression of this disease. Discussed below are two treatment strategies: cytoprotection and anti-inflammation therapy.
Cytoprotection
RPE cell death is believed to constitute the primary events in atrophic AMD and subsequent dysfunction and degeneration of photoreceptor cells lead to the loss of cone-mediated high resolution central and color vision. Thus, preservation of RPE and photoreceptor cells could help prevent progression of AMD.
ROS scavengers
Antioxidants have the capacity to alleviate or reduce the detrimental effects of ROS in response to a variety of stress stimuli. Extensive laboratory investigations have demonstrated the role of oxidative stress and the inhibitory effects of antioxidants on RPE cellular function (CitationChoudhary et al 2005; CitationFernandez-Robredo et al 2005; CitationVoloboueva et al 2005). Clinically, antioxidant supplements may delay the onset of AMD and progression to advanced AMD. The Age-related Eye Disease Study (AREDS) was comprised of 3640 participants who were randomized into cohorts receiving daily high-dose antioxidants (vitamin C 500 mg, vitamin E 400 IU, β-carotene 15 mg), or zinc (80 mg zinc oxide, 2 mg copric oxide), or antioxidants plus zinc or placebo (CitationAREDS. 2001). With an average 6.5-year follow-up, comparison with placebo showed a statistically significant reduction in the development of advanced AMD in those patients receiving antioxidants plus zinc (CitationAREDS. 2001). At 5 years in the absence of supplementation of antioxidants plus zinc, the estimated probability of progression of participants with advanced AMD or vision loss due to nonadvanced AMD to advanced AMD in the second eye was 43%. The 5-year estimated probability of progression to advanced AMD in either eye with extensive intermediate drusen, large drusen, or noncentral geographic atrophy is 18% (CitationAREDS 2001). Thus, people at risk of developing advanced AMD lowered this risk by about 25% when treated according to the AREDS recommendations. It has been estimated that if the 8 million people in the USA at risk of advanced AMD receive AREDS-recommended supplements, 329, 000 of them would avoid developing advanced AMD during the next 5 years (CitationBressler et al 2003). In addition, dietary lutein/zeaxanthin intake has been shown to be inversely associated with the occurrence of geographic atrophy and large or extensive intermediate drusen (CitationSanGiovanni et al 2007). Taken together these data demonstrate that dietary supplementation with antioxidants reduce the risk of progression of AMD. OT-551, a catalytic antioxidant with anti-inflammatory activity has been shown to functionally and morphologically protect photoreceptor cells against acute light-induced damage in an animal model (CitationTanito et al 2007) and is currently being assessed in clinical trials in AMD patients with geographic atrophy ().
Table 1 Developing therapies for atrophic age-related macular degeneration
Induction of phase 2 antioxidant enzymes including SOD, catalase, glutamate cysteine ligase, and heme oxygenase-1 by phase 2 enzyme inducers provides an alternative way to protect cells against ocular oxidative stress. High amounts of catalase (CitationTate et al 1995), SOD (CitationOliver and Newsome 1992), and glutathione (CitationNewsome et al 1994; CitationBeatty et al 2000) can efficiently detoxify and remove ROS once they are generated in RPE cells. GSH synthesis can be induced in cultured RPE cells by a number of naturally occurring compounds that induce phase 2 detoxification enzymes, including the rate-limiting enzyme for GSH synthesis, glutamate cysteine ligase (CitationNelson et al 1999; CitationVoloboueva et al 2005; CitationHa et al 2006; CitationQin et al 2006). RPE cells can be protected from peroxide-induced damage by pre-treatment with a number of agents that induce GSH synthesis including dimethylfumarate (CitationNelson et al 1999), 15-deoxy-Δ12,14-prostaglandin J2 (CitationQin et al 2006), (R)-α-lipoic acid (CitationVoloboueva et al 2005), or zinc (CitationHa et al 2006). Sulforaphane, a potent inducer of phase 2 antioxidant enzymes, has been demonstrated to protect RPE cells from photooxidative damage in vitro (CitationGao and Talalay 2004; CitationTanito et al 2005; CitationZhou et al 2006) and in vivo (CitationTanito et al 2005). A single dose of oltipraze, another potent phase 2 enzyme inducer, significantly stimulated GSH level in lymphocytes isolated from human blood (CitationGupta et al 1995), and induced a four- to fivefold increase in mRNA transcripts for glutamate cysteine ligase and quinone reductase in colon mucosa (CitationO’Dwyer et al 1996). These findings provide a strong rationale to test whether phase 2 enzyme inducers, such as sulforaphane and oltipraz, can elevate the antioxidant enzymes in the retina and whether they can prevent oxidative damage in humans at risk for AMD.
Antiapoptotic approaches
In addition to the maintenance of antioxidant capacity, RPE cells and photoreceptors can be persevered by stimulating cell survival signaling and/or inhibiting death pathways.
RPE cell survival
Cell viability is precisely regulated by the balanced expression of pro- and antiapoptotic factors such as the Bcl-2 family of proteins. As expected, ectopic expression of antiapoptotic proteins Bcl-2 renders RPE cells resistant to A2E-mediated toxicity (CitationSparrow and Cai 2001) whereas knockdown of Bcl-xL expression leads to RPE cell death (CitationZhang et al 2007). Neuroprotectin D1 (NPD1), a metabolite of DHA that is rich in the retina, has been shown to protect against RPE cell apoptosis induced by A2E, a byproduct of phototransduction (CitationMukherjee et al 2007), as well as hydrogen peroxide and tumor necrosis factor-α (CitationMukherjee et al 2004). NPD1 and DHA protect RPE cells via upregulation of antiapoptotic proteins and downregulation of pro-apoptotic proteins of Bcl-2 family (CitationMukherjee et al 2004, Citation2007). Pigment epithelium-derived factor (PEDF), secreted by RPE cells, is a trophic factor required for RPE cell function and survival under stress conditions. PEDF can protect RPE cells from oxidant-mediated barrier dysfunction (CitationTsao et al 2006) and death (CitationHo et al 2006). ERK activation and NPD1 production stimulated by PEDF appear to be involved in mediating PEDF’s cyto-protective effects (CitationTsao et al 2006; CitationMukherjee et al 2007). Interestingly, PEDF levels in RPE cells are significantly reduced in AMD patients (CitationBhutto et al 2006). Thus, increases in PEDF levels could potentially delay the onset of RPE abnormalities and progression to RPE cell atrophy.
Photoreceptor cell survival
RPE cell death initiates the development of atrophic AMD. However, loss of vision is caused by the death of rod and cone photoreceptors. Thus, the preservation of macular photoreceptors is a valid anti-AMD strategy. Many trophic factors can support photoreceptor survival (CitationLaVail et al 1992; CitationSteinberg 1994). Among them, PEDF and ciliary neurotrophic factor (CNTF) have demonstrated consistent photoreceptor protection. PEDF protects cultured retinal neurons from hydrogen peroxide injury (CitationCao et al 1999) and inhibits macrophage-driven inflammation (CitationZamiri et al 2006). In vivo, intravitreal injection or intraocular gene transfer of PEDF rescues photoreceptors from light-induced damage (CitationCao et al 2001; CitationImai et al 2005). Moreover, a PEDF (82–121) peptide is as effective as full-length PEDF in protecting retina from ischemia and longer protection can be achieved if delivered in poly(lactide-co-glycolide) (PLGA) nanospheres (CitationLi et al 2006). Taken together with the RPE protective effects, these observations suggest that PEDF is a promising therapeutic treatment for AMD.
CNTF is another trophic factor that has reached clinical development for retinal degeneration (CitationSieving et al 2006). Injection or gene transfer of CNTF protects photoreceptor degeneration in animal models (CitationLaVail et al 1998; CitationHuang et al 2004). Delivery of CNTF by encapsulated cell-based technology reduces photoreceptor degeneration in animal model of retinitis pigmentosa (CitationTao et al 2002). This delivery system, which can deliver CNTF to the vitreous for at least 1 year (CitationThanos et al 2004), is currently being evaluated clinically.
Anti-inflammation therapy
Steroids
Clinical studies have indicated that corticosteroid, triamcinolone acetonide (TA), has the capacity to reduce the permeability of the outer blood–retinal barrier (CitationAntoszyk et al 1993), promote resorption of subretinal exudation (CitationPenfold et al 1995) and downregulate inflammatory stimuli (CitationTanner et al 1998). After 18 months of treatment with TA in patients with exudative AMD, the rate of development of severe visual loss in treated patients was 30%, compared with 55% for historical controls (CitationChalla et al 1998). Anecortave acetate is another angiostatic steroid that might be clinically efficacious for maintaining vision, preventing severe vision loss, and inhibiting subfoveal CNV lesion growth (CitationRussell et al 2007). Anecortave acetate was approved for use in wet AMD in Australia in 2005 with approval pending in the US. A third steroid, Posurdex (dexamethasone implant) is in phase III clinical trial for retinal vein occlusion and diabetic macular edema. Although no clinical data are available yet, the role of chronic inflammation in atrophic AMD pathogenesis (CitationHageman et al 2001; CitationAnderson et al 2002) has led to the consideration that atrophic AMD patients might be expected to benefit from anti-inflammatory therapy that attenuates the local inflammation, thereby preventing progression of the disease.
Nonsteroidal anti-inflammatory drug
Epidemiology and animal studies suggest that nonsteroidal anti-inflammatory drugs (NSAIDs) can delay the onset and progression of Alzheimer’s disease (AD) (CitationTownsend and Pratico 2005; CitationMcGeer and McGeer 2007). Amyloid-β (Aβ), a major trigger of AD pathogenesis, is present in drusen from retinas with AMD (CitationDentchev et al 2003). Owing to the compositional similarities between drusen and AD-associated plaques, atrophic AMD patients might be expected to benefit from NSAID therapy that may attenuate local inflammation (CitationMullins et al 2000; CitationLuibl et al 2006). Consistent with this, accumulation of Aβ causes RPE atrophy and basal deposit formation, characteristic features of human AMD (CitationYoshida et al 2005). Moreover, the addition of Aβ to cultured RPE cells upregulates expression of VEGF (CitationYoshida et al 2005; CitationMa et al 2007) and downregulates PEDF expression (CitationYoshida et al 2005). The anti-inflammatory compounds eicosapentaenoic acid and astaxanthin have been shown to inhibit cytokine production, macrophage activation, and CNV formation in mice (CitationKoto et al 2007; CitationIzumi-Nagai et al 2008). Consistent with these observations, the prevalence of AMD in rheumatoid arthritis patients with long term NSAID use is about 10-fold lower than in general populations of similar racial origin (CitationMcGeer and Sibley 2005). Thus, additional studies of the potential utility of NSAIDs for the treatment of AMD patients are warranted.
Complement pathway regulators
As discussed in the section III, certain CFH haplotypes are associated with increased frequency of AMD (CitationHageman et al 2005; CitationHughes et al 2006). In contrast, the L9H variant of CFB, which participates in the amplification of the complement alternative pathway, confers a significantly reduced risk for AMD (CitationGold et al 2006). Conceivably, delivery of a protective variant of the CFH or CFB through gene therapy or as a biologic could block activation of the alternative pathway and serve as an anti-AMD strategy. Alternatively, as CFB and CFD are both serine proteases small-molecule, inhibitors of these proteins could be used to inhibit activation of the alternate pathway.
Reduction of the CFH-associated AMD may also be achieved by inhibiting targets downstream the point of conversion for all three complement pathways. JPE-1375 and PMX53 are two small molecule peptidomimetic antagonists targeting the C5a receptor (CitationSchnatbaum et al 2006) and are in preclinical evaluation for AMD. POT-4 is a cyclic peptide capable of binding to human C3 and prevents its activation, resulting in broad and potent complement activation inhibition (CitationNilsson et al 1998). POT-4 is the first complement inhibitor that has entered into a Phase I clinical trial for AMD ().
Other novel therapeutic approaches
Besides antioxidant and anti-inflammation therapies, other novel therapeutic approaches are currently under development for the treatment of atrophic AMD. Lipofuscin in RPE cells is an autofluorescent lipid-retinoid aggregate derived from visual cycle retinoids in photoreceptor cells. The major component of lipofuscin is A2E, a product of condensation of two molecules of all-trans-retinal with one molecule of phosphatidylethanolamine (CitationSparrow et al 2003). Lipofuscin accumulation in RPE cells is the key feature of the aging retina (CitationSparrow and Boulton 2005). In vitro, A2E can inhibit lysosomal function, perturb cholesterol metabolism, cause light-induced oxidative stress to RPE cells as well as activate the complement pathway (CitationBergmann et al 2004; CitationZhou et al 2006; CitationLakkaraju et al 2007). Therefore, reducing A2E accumulation in RPE cells by inhibiting the visual cycle could lead to delay in visual loss in the patients with atrophic AMD. Fenretinide, a synthetic nontoxic retinoid, competes with all-trans-retinol binding to retinol binding protein, thereby reducing ocular all-trans-retinol uptake and inhibiting the visual cycle (CitationRadu et al 2005). Thus, administration of fenretinide effectively blocks A2E production and lipofuscin accumulation (CitationRadu et al 2005). Fenretinide is currently in clinical trials to evaluate its efficacy on AMD patients with geographic atrophy.
Issues and challenges
Etiology of atrophic AMD
Despite an increase in our understanding of the immunological factors involved in atrophic AMD, this knowledge has yet to be translated into new treatments. One possible reason for this is our lack of understanding of disease chronicity, and the causative rather than risk factors that contribute to disease. Do drusen materials in AMD represent an abnormal or normal accumulation of metabolic debris? Why do soft drusen appear in macular area? What specific factors lead to atrophy of the RPE and photoreceptors? The answers to these questions could one day be exploited to provide ideas for novel therapies. Another issue is the variability in disease characteristics at different stages of progression. As our understanding of the natural history of atrophic AMD grows, eligibility criteria for clinical trials might be tailored depending on the mechanism of action of a particular drug candidate.
Animal models
A variety of animal models have been developed to investigate the pathogenesis of atrophic AMD and test therapeutic compounds to ameliorate the pathology. Inflammatory models of AMD have been generated to elucidate the role of inflammation in the etiology of AMD. One such model can be induced by immunization of mice with MSA adducted with CEP, an oxidation fragment of DHA (CitationHollyfield et al 2008). Mice challenged with the CEP produce antigen-specific antibodies, develop drusen under the RPE and lesions mimicking atrophic AMD. Moreover, macrophages are observed in the interphotoreceptor matrix in these mice (CitationHollyfield et al 2008). As complement deposition is also observed below the RPE in this model the phenotype appears to be driven by activation of this pathway. Other inflammatory models include mice with knockouts of the chemokine receptor CX3CR1 with CCL2 (MCP-1) or CCR2 in combination with its ligand, CCL2 as well (CitationAmbati et al 2003; CitationCombadiere et al 2007; CitationTuo et al 2007). These knockout mice display many of the features of AMD including the accumulation of lipofuscin, A2E, drusen as well as photoreceptor atrophy and CNV. Additionally, in the case of the CX3CR1 knockouts, subretinal accumulation of microglial cells was observed (CitationCombadiere et al 2007; CitationTuo et al 2007).
There are also a number of noninflammatory models that have been developed. The sodium iodate model involves the selective induction of RPE necrosis followed by photoreceptor degeneration in a pattern resembling gyrate atrophy of the choroid and retina (CitationKiuchi et al 2002). The mcd/mcd mouse was generated using a dominant negative form of Cathepsin D which is required for RPE phagocytosis of shed POS membrane discs (CitationRakoczy et al 2002). This transgenic mouse model manifests features of AMD including lipofuscin accumulation and both photoreceptor and RPE atrophy. The SOD1-deficient mouse represents a novel model of ROS-mediated retinal degeneration, which similarly displays features of AMD such as drusen, RPE atrophy, and CNV (CitationImamura et al 2006; CitationHashizume et al 2008).
The extrapolation of experimental data obtained from animal studies to explain the in vivo pathological conditions must be done with caution. First, the mouse retina does not contain a specialized macula, thus, a murine model is not an exact replica of the human condition. Moreover, the components of drusen described in many animal models have not been molecularly characterized to confirm a similarity with human drusen. In this regard, some progress has been made to identify a nonhuman primate model of AMD. A cynomolgus monkey pedigree has been identified that develops early onset macular degeneration (CitationUmeda et al 2005). Interestingly, many components in the drusen of these animals are comparable to those found in human drusen (CitationUmeda et al 2005). Furthermore, the loss of ultrastructure and function as well as progression of the disease is similar to that observed in humans (CitationDawson et al 2008).
AMD is a multi-factor disease; however, the available animal models only evaluate the contributions of one or at most two risk factors. Thus this limits the utility of these animal models of AMD for drug testing. Frequently, in the case of genetically modified animals the penetrance of the observed phenotype is not complete. Moreover, the appearance of many of the AMD-like features occurs over a protracted period. Thus, a large number of animals and a prolonged treatment regimen would be required to test potential drug candidates. Efforts to accelerate the manifestation of the measurable features of these models are ongoing. Crossing various transgenic and knockout mice may accelerate the course of the disease. Alternatively, modification of environmental factors such as diet or the addition of other risk factors could similarly hasten disease progression. For example, the addition of paraquat to SOD1-defficient mice has been shown to result in changes in electroretinography measurements within two weeks (CitationDong et al 2006). These efforts have the potential to enhance the utility of these models for drug development significantly.
Conclusions and future studies
Currently, most treatments for AMD such as laser photocoagulation, photodynamic therapy, surgery, and anti-VEGF antibodies benefit patients in advanced stages of the disease. The progressive nature of atrophic AMD and the extensive cell damage in affected individuals at the time of diagnosis indicates that the future of atrophic AMD therapeutics is in the prevention of progression of the disease. Any drug for disease prevention needs to fulfill two crucial requirements. First, the drug should target a key mechanism responsible for the initiation or progression of the disease process. Second, the drug should have minimal side effects and be tolerated for extended periods. To date no such therapy is available for atrophic AMD. Thus, educating people about the risk factors associated with this disease such as smoking and diet is an important consideration at present. The development of pharmacological approaches to prevent onset or progression of AMD faces significant hurdles including a lack of understanding of the molecular mechanisms underlying the disease and the paucity of experimental models for testing potential drugs. Although a causal relationship based upon mechanistic studies of oxidative injury and inflammation has not been completely established, the cumulative evidence presented in this review suggests a strong correlation between oxidative stress, inflammation and AMD. This represents a significant step towards the goal of identifying more effective treatments for this disease.
Acknowledgments
The authors thank Dr Larry Wheeler for his critique of this manuscript.
References
- AlexanderJPBradleyJMGabourelJD1990Expression of matrix metalloproteinases and inhibitor by human retinal pigment epitheliumInvest Ophthalmol Vis Sci31252082176183
- AmbatiJAnandAFernandezS2003An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient miceNat Med91390714566334
- AmesBNShigenagaMKHagenTM1993Oxidants, antioxidants, and the degenerative diseases of agingProc Natl Acad Sci U S A907915228367443
- AndersonDHMullinsRFHagemanGS2002A role for local inflammation in the formation of drusen in the aging eyeAm J Ophthalmol1344113112208254
- AntoszykANGottliebJLMachemerR1993The effects of intravitreal triamcinolone acetonide on experimental pre-retinal neovascularizationGraefes Arch Clin Exp Ophthalmol23134408428678
- ApteRSRichterJHerndonJ2006Macrophages inhibit neovascularization in a murine model of age-related macular degenerationPLoS Med3e31016903779
- [AREDS] Age-Related Eye Disease Study2001A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no 8Arch Ophthalmol11914173611594942
- AseaAKraeftSKKurt-JonesEA2000HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokineNat Med64354210742151
- AugustinAJOffermannI2006Emerging drugs for age-related macular degenerationExpert Opin Emerg Drugs117254017064228
- BairdPNRobmanLDRichardsonAJ2008Gene-environment interaction in progression of AMD – the CFH gene, smoking and exposure to chronic infectionHum Mol Genet17129930518203751
- BarileGRSchmidtAM2007RAGE and its ligands in retinal diseaseCurr Mol Med77586518331234
- BeattySKohHPhilM2000The role of oxidative stress in the pathogenesis of age-related macular degenerationSurv Ophthalmol451153411033038
- BergmannMSchuttFHolzFG2004Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degenerationFASEB J18562414715704
- BhuttoIAMcLeodDSHasegawaT2006Pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in aged human choroid and eyes with age-related macular degenerationExp Eye Res829911016019000
- BianZMElnerVMYoshidaA2001Signaling pathways for glycated human serum albumin-induced IL-8 and MCP-1 secretion in human RPE cellsInvest Ophthalmol Vis Sci421660811381075
- BirdACBresslerNMBresslerSB1995An international classification and grading system for age-related maculopathy and age-related macular degeneration. The International ARM Epidemiological Study GroupSurv Ophthalmol39367747604360
- BodaghiBSlobbe-van DrunenMETopilkoA1999Entry of human cytomegalovirus into retinal pigment epithelial and endothelial cells by endocytosisInvest Ophthalmol Vis Sci40259860710509655
- BoraNSKaliappanSJhaP2006Complement activation via alternative pathway is critical in the development of laser-induced choroidal neovascularization: role of factor B and factor HJ Immunol1771872816849499
- BoultonMDontsovAJarvis-EvansJ1993Lipofuscin is a photoinducible free radical generatorJ Photochem Photobiol B1920148229462
- BresslerNMBresslerSBCongdonNG2003Potential public health impact of Age-Related Eye Disease Study results: AREDS report no 11Arch Ophthalmol1211621414609922
- BresslerSBMaguireMGBresslerNM1990Relationship of drusen and abnormalities of the retinal pigment epithelium to the prognosis of neovascular macular degeneration. The Macular Photocoagulation Study GroupArch Ophthalmol108144271699513
- CaoWTombran-TinkJChenW1999Pigment epithelium-derived factor protects cultured retinal neurons against hydrogen peroxide-induced cell deathJ Neurosci Res5778980010467250
- CaoWTombran-TinkJEliasR2001In vivo protection of photoreceptors from light damage by pigment epithelium-derived factorInvest Ophthalmol Vis Sci4216465211381073
- ChallaJKGilliesMCPenfoldPL1998Exudative macular degeneration and intravitreal triamcinolone: 18 month follow upAust N Z J Ophthalmol26277819843254
- ChanD1998Cigarette smoking and age-related macular degenerationOptom Vis Sci75476849703035
- ChenMForresterJVXuH2007Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segmentsExp Eye Res846354517292886
- ChenWSyldathUBellmannK1999Human 60-kDa heat-shock protein: a danger signal to the innate immune systemJ Immunol1623212910092772
- ChoudharySXiaoTSrivastavaS2005Toxicity and detoxification of lipid-derived aldehydes in cultured retinal pigmented epithelial cellsToxicol Appl Pharmacol2041223415808518
- CingolaniCRogersBLuL2006Retinal degeneration from oxidative damageFree Radic Biol Med40660916458197
- CombadiereCFeumiCRaoulW2007CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degenerationJ Clin Invest1172920817909628
- CongRZhouBSunQ2008Smoking and the risk of age-related macular degeneration: a meta-analysisAnn Epidemiol186475618652983
- CrabbJWMiyagiMGuX2002Drusen proteome analysis: an approach to the etiology of age-related macular degenerationProc Natl Acad Sci U S A9914682712391305
- CruickshanksKJKleinRKleinBE1993Sunlight and age-related macular degeneration. The Beaver Dam Eye StudyArch Ophthalmol11151488470986
- CurcioCAMillicanCL1999Basal linear deposit and large drusen are specific for early age-related maculopathyArch Ophthalmol1173293910088810
- DaffernPJPfeiferPHEmberJA1995C3a is a chemotaxin for human eosinophils but not for neutrophils. I. C3a stimulation of neutrophils is secondary to eosinophil activationJ Exp Med1812119277760001
- DawsonWWDawsonJCLakeKP2008Maculas, monkeys, models, AMD and agingVision Res48360517892891
- DentchevTMilamAHLeeVM2003Amyloid-beta is found in drusen from some age-related macular degeneration retinas, but not in drusen from normal retinasMol Vis91849012764254
- deSSPCalabroANishiyamaK2001Glycosaminoglycan synthesis and secretion by the retinal pigment epithelium: polarized delivery of hyaluronan from the apical surfaceJ Cell Sci11419920511112703
- DongAShenJKrauseM2006Superoxide dismutase 1 protects retinal cells from oxidative damageJ Cell Physiol2085162616741961
- DoreyCKWuGEbensteinD1989Cell loss in the aging retina. Relationship to lipofuscin accumulation and macular degenerationInvest Ophthalmol Vis Sci30169192759786
- DuvallJTsoMO1985Cellular mechanisms of resolution of drusen after laser coagulation. An experimental studyArch Ophthalmol1036947034039560
- EbrahemQRenganathanKSearsJ2006Carboxyethylpyrrole oxidative protein modifications stimulate neovascularization: Implications for age-related macular degenerationProc Natl Acad Sci U S A10313480416938854
- EdwardsAORitterR3rdAbelKJ2005Complement factor H polymorphism and age-related macular degenerationScience308421415761121
- EmberJASandersonSDHugliTE1994Induction of interleukin-8 synthesis from monocytes by human C5a anaphylatoxinAm J Pathol1443934037508686
- Espinosa-HeidmannDGSunerIJHernandezEP2003Macrophage depletion diminishes lesion size and severity in experimental choroidal neovascularizationInvest Ophthalmol Vis Sci4435869212882811
- EterNEngelDRMeyerL2008In vivo visualization of dendritic cells, macrophages, and microglial cells responding to laser-induced damage in the fundus of the eyeInvest Ophthalmol Vis Sci4936495818316698
- EvansJWormaldR1996Is the incidence of registrable age-related macular degeneration increasing?Br J Ophthalmol809148664242
- Fernandez-RobredoPMoyaDRodriguezJA2005Vitamins C and e reduce retinal oxidative stress and nitric oxide metabolites and prevent ultrastructural alterations in porcine hypercholesterolemiaInvest Ophthalmol Vis Sci461140615790871
- FlieslerSJAndersonRE1983Chemistry and metabolism of lipids in the vertebrate retinaProg Lipid Res22791316348799
- FoellDWittkowskiHRothJ2007Mechanisms of disease: a ‘DAMP’ view of inflammatory arthritisNat Clin Pract Rheumatol33829017599072
- FriedmanDSO’ColmainBJMunozB2004Prevalence of age-related macular degeneration in the United StatesArch Ophthalmol1225647215078675
- FritscheLGLoenhardtTJanssenA2008Age-related macular degeneration is associated with an unstable ARMS2 (LOC387715) mRNANat Genet40892618511946
- FukuokaYMedofEM2001C5a receptor-mediated production of IL-8 by the human retinal pigment epithelial cell line, ARPE-19Curr Eye Res23320511910520
- FukuokaYStrainicMMedofME2003Differential cytokine expression of human retinal pigment epithelial cells in response to stimulation by C5aClin Exp Immunol1312485312562384
- GaoXTalalayP2004Induction of phase 2 genes by sulforaphane protects retinal pigment epithelial cells against photooxidative damageProc Natl Acad Sci U S A101104465115229324
- GoldBMerriamJEZernantJ2006Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degenerationNat Genet384586216518403
- Gross-JendroskaMLuiGMSongMK1992Retinal pigment epithelium-stromal interactions modulate hyaluronic acid depositionInvest Ophthalmol Vis Sci33339491428712
- GuXMeerSGMiyagiM2003Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degenerationJ Biol Chem278420273512923198
- GuptaEOlopadeOIRatainMJ1995Pharmacokinetics and pharmacodynamics of oltipraz as a chemopreventive agentClin Cancer Res1113389815904
- GuptaNBrownKEMilamAH2003Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degenerationExp Eye Res764637112634111
- HaKNChenYCaiJ2006Increased glutathione synthesis through an ARE-Nrf2-dependent pathway by zinc in the RPE: implication for protection against oxidative stressInvest Ophthalmol Vis Sci4727091516723490
- HagemanGSAndersonDHJohnsonLV2005A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degenerationProc Natl Acad Sci U S A10272273215870199
- HagemanGSLuthertPJVictor ChongNH2001An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degenerationProg Retin Eye Res207053211587915
- HainesJLHauserMASchmidtS2005Complement factor H variant increases the risk of age-related macular degenerationScience3084192115761120
- HandaJTReiserKMMatsunagaH1998The advanced glycation endproduct pentosidine induces the expression of PDGF-B in human retinal pigment epithelial cellsExp Eye Res6641199593635
- HashizumeKHirasawaMImamuraY2008Retinal dysfunction and progressive retinal cell death in SOD1-deficient miceAm J Pathol17213253118372426
- HeibaIMElstonRCKleinBE1994Sibling correlations and segregation analysis of age-related maculopathy: the Beaver Dam Eye StudyGenet Epidemiol1151678013888
- HenderlyDEAtallaLRFreemanWR1988Demonstration of cytomegalovirus retinitis by in situ DNA hybridizationRetina8177812852836
- HigginsGTWangJHDockeryP2003Induction of angiogenic cytokine expression in cultured RPE by ingestion of oxidized photoreceptor outer segmentsInvest Ophthalmol Vis Sci4417758212657621
- HoLBoekhoornSSLianaL2008Cataract surgery and the risk of aging macula disorder: The Rotterdam StudyInvest Ophthalmol Vis Sci49479580018599571
- HoTCYangYCChengHC2006Pigment epithelium-derived factor protects retinal pigment epithelium from oxidant-mediated barrier dysfunctionBiochem Biophys Res Commun342372816483542
- HofmannMADrurySFuC1999RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptidesCell9788990110399917
- HollyfieldJGBonilhaVLRaybornME2008Oxidative damage-induced inflammation initiates age-related macular degenerationNat Med14194818223656
- HoltkampGMKijlstraAPeekR2001Retinal pigment epithelium-immune system interactions: cytokine production and cytokine-induced changesProg Retin Eye Res20294811070367
- HondaSFarboudBHjelmelandLM2001Induction of an aging mRNA retinal pigment epithelial cell phenotype by matrix-containing advanced glycation end products in vitroInvest Ophthalmol Vis Sci4224192511527959
- HoppeGMarmorsteinADPennockEA2001Oxidized low density lipoprotein-induced inhibition of processing of photoreceptor outer segments by RPEInvest Ophthalmol Vis Sci4227142011581220
- HoppeGO’NeilJHoffHF2004Accumulation of oxidized lipid-protein complexes alters phagosome maturation in retinal pigment epitheliumCell Mol Life Sci6116647415224189
- HowesKALiuYDunaiefJL2004Receptor for advanced glycation end products and age-related macular degenerationInvest Ophthalmol Vis Sci4537132015452081
- HuangSPLinPKLiuJH2004Intraocular gene transfer of ciliary neurotrophic factor rescues photoreceptor degeneration in RCS ratsJ Biomed Sci11374814730208
- HughesAEOrrNEsfandiaryH2006A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degenerationNat Genet381173716998489
- IdzkoMHammadHvan NimwegenM2007Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cellsNat Med13913917632526
- ImaiDYoneyaSGehlbachPL2005Intraocular gene transfer of pigment epithelium-derived factor rescues photoreceptors from light-induced cell deathJ Cell Physiol202570815316929
- ImamuraYNodaSHashizumeK2006Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: a model of age-related macular degenerationProc Natl Acad Sci U S A10311282716844785
- Izumi-NagaiKNagaiNOhgamiK2008Inhibition of choroidal neovascularization with an anti-inflammatory carotenoid astaxanthinInvest Ophthalmol Vis Sci4916798518385091
- JohnsonGBBrunnGJKodairaY2002Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4J Immunol1685233911994480
- JohnsonLVLeitnerWPStaplesMK2001Complement activation and inflammatory processes in Drusen formation and age related macular degenerationExp Eye Res738879611846519
- JohnsonLVOzakiSStaplesMK2000A potential role for immune complex pathogenesis in drusen formationExp Eye Res70441910865992
- JozsiMZipfelPF2008Factor H family proteins and human diseasesTrends Immunol29380718602340
- KajiYUsuiTIshidaS2007Inhibition of diabetic leukostasis and blood-retinal barrier breakdown with a soluble form of a receptor for advanced glycation end productsInvest Ophthalmol Vis Sci488586517251488
- KalayogluMVBulaDArroyoJ2005Identification of Chlamydia pneumoniae within human choroidal neovascular membranes secondary to age-related macular degenerationGraefes Arch Clin Exp Ophthalmol24310809015909160
- KameiMYonedaKKumeN2007Scavenger receptors for oxidized lipoprotein in age-related macular degenerationInvest Ophthalmol Vis Sci481801717389514
- KandaAChenWOthmanM2007A variant of mitochondrial protein LOC387715/ARMS2, not HTRA1, is strongly associated with age-related macular degenerationProc Natl Acad Sci U S A104162273217884985
- KennedyCJRakoczyPEConstableIJ1995Lipofuscin of the retinal pigment epithelium: a reviewEye9Pt 6763718849547
- KimYSJungDHKimNH2007Effect of magnolol on TGF-beta1 and fibronectin expression in human retinal pigment epithelial cells under diabetic conditionsEur J Pharmacol56212917321517
- KiuchiKYoshizawaKShikataN2002Morphologic characteristics of retinal degeneration induced by sodium iodate in miceCurr Eye Res25373912789545
- KleinRKleinBEJensenSC1997The five-year incidence and progression of age-related maculopathy: the Beaver Dam Eye StudyOphthalmology1047219022098
- KleinRKleinBEWongTY2002The association of cataract and cataract surgery with the long-term incidence of age-related maculopathy: the Beaver Dam eye studyArch Ophthalmol1201551812427071
- KleinRJZeissCChewEY2005Complement factor H polymorphism in age-related macular degenerationScience308385915761122
- KluneJRDhuparRCardinalJ2008HMGB1: endogenous danger signalingMol Med144768418431461
- KotoTNagaiNMochimaruH2007Eicosapentaenoic acid is anti-inflammatory in preventing choroidal neovascularization in miceInvest Ophthalmol Vis Sci4843283417724224
- KumarMVNagineniCNChinMS2004Innate immunity in the retina: Toll-like receptor (TLR) signaling in human retinal pigment epithelial cellsJ Neuroimmunol15371515265658
- LakkarajuAFinnemannSCRodriguez-BoulanE2007The lipofuscin fluorophore A2E perturbs cholesterol metabolism in retinal pigment epithelial cellsProc Natl Acad Sci U S A104110263117578916
- LaVailMMUnokiKYasumuraD1992Multiple growth factors, cytokines, and neurotrophins rescue photoreceptors from the damaging effects of constant lightProc Natl Acad Sci U S A8911249531454803
- LaVailMMYasumuraDMatthesMT1998Protection of mouse photoreceptors by survival factors in retinal degenerationsInvest Ophthalmol Vis Sci395926029501871
- LiHTranVVHuY2006A PEDF N-terminal peptide protects the retina from ischemic injury when delivered in PLGA nanospheresExp Eye Res838243316822505
- LuLHackettSFMinceyA2006Effects of different types of oxidative stress in RPE cellsJ Cell Physiol2061192515965958
- LuiblVIsasJMKayedR2006Drusen deposits associated with aging and age-related macular degeneration contain nonfibrillar amyloid oligomersJ Clin Invest1163788516453022
- MaWLeeSEGuoJ2007RAGE ligand upregulation of VEGF secretion in ARPE-19 cellsInvest Ophthalmol Vis Sci4813556117325184
- MallerJBFagernessJAReynoldsRC2007Variation in complement factor 3 is associated with risk of age-related macular degenerationNat Genet391200117767156
- MartinonFPetrilliVMayorA2006Gout-associated uric acid crystals activate the NALP3 inflammasomeNature4402374116407889
- McBeeJKPalczewskiKBaehrW2001Confronting complexity: the interlink of phototransduction and retinoid metabolism in the vertebrate retinaProg Retin Eye Res2046952911390257
- McFarlaneSGlennJVLichanskaAM2005Characterisation of the advanced glycation endproduct receptor complex in the retinal pigment epitheliumBr J Ophthalmol891071215615757
- McGeerPLMcGeerEG2007NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studiesNeurobiol Aging286394716697488
- McGeerPLSibleyJ2005Sparing of age-related macular degeneration in rheumatoid arthritisNeurobiol Aging26119920315917104
- McLaughlinBJFanWZhengJJ2003Novel role for a complement regulatory protein (CD46) in retinal pigment epithelial adhesionInvest Ophthalmol Vis Sci4436697412882822
- MiceliMVLilesMRNewsomeDA1994Evaluation of oxidative processes in human pigment epithelial cells associated with retinal outer segment phagocytosisExp Cell Res21424298082727
- MillerDMEspinosa-HeidmannDGLegraJ2004The association of prior cytomegalovirus infection with neovascular age-related macular degenerationAm J Ophthalmol138323815364212
- MiyakeK2007Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptorsSemin Immunol1931017275324
- MoyerALRamadanRTThurmanJ2008Bacillus cereus induces permeability of an in vitro blood-retina barrierInfect Immun7613586718268029
- MukherjeePKMarcheselliVLBarreiroS2007Neurotrophins enhance retinal pigment epithelial cell survival through neuroprotectin D1 signalingProc Natl Acad Sci U S A10413152717670936
- MukherjeePKMarcheselliVLSerhanCN2004Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stressProc Natl Acad Sci U S A1018491615152078
- MullinsRFRussellSRAndersonDH2000Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit diseaseFASEB J148354610783137
- MuruveDAPetrilliVZaissAK2008The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune responseNature452103718288107
- NakaiKFainaruOBazinetL2008Dendritic cells augment choroidal neovascularizationInvest Ophthalmol Vis Sci4936667018408184
- NelsonKCCarlsonJLNewmanML1999Effect of dietary inducer dimethylfumarate on glutathione in cultured human retinal pigment epithelial cellsInvest Ophthalmol Vis Sci4019273510440245
- NewsomeDAMiceliMVLilesM1994Antioxidants in the retinal pigment epitheliumProg Retin Eye Res1310123
- Nguyen-LegrosJHicksD2000Renewal of photoreceptor outer segments and their phagocytosis by the retinal pigment epitheliumInt Rev Cytol19624531310730217
- NilssonBLarssonRHongJ1998Compstatin inhibits complement and cellular activation in whole blood in two models of extracorporeal circulationBlood92166179716594
- NozakiMRaislerBJSakuraiE2006Drusen complement components C3a and C5a promote choroidal neovascularizationProc Natl Acad Sci U S A10323283316452172
- O’DwyerPJSzarkaCEYaoKS1996Modulation of gene expression in subjects at risk for colorectal cancer by the chemopreventive dithiolethione oltiprazJ Clin Invest98121078787684
- OliverPDNewsomeDA1992Mitochondrial superoxide dismutase in mature and developing human retinal pigment epitheliumInvest Ophthalmol Vis Sci331909181582797
- PenfoldPLGyoryJFHunyorAB1995Exudative macular degeneration and intravitreal triamcinolone. A pilot studyAust N Z J Ophthalmol23293811980075
- PenfoldPLMadiganMCGilliesMC2001Immunological and aetiological aspects of macular degenerationProg Retin Eye Res2038541411286898
- PerkinsGAEllismanMHFoxDA2003Three-dimensional analysis of mouse rod and cone mitochondrial cristae architecture: bioenergetic and functional implicationsMol Vis9607312632036
- QinS2007Oxidative damage of retinal pigment epithelial cells and age-related macular degenerationDrug Dev Res6821325
- QinSMcLaughlinAPDe VriesGW2006Protection of RPE cells from oxidative injury by 15-deoxy-delta12,14-prostaglandin J2 by augmenting GSH and activating MAPKInvest Ophthalmol Vis Sci47509810517065531
- RaduRAHanYBuiTV2005Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseasesInvest Ophthalmol Vis Sci46439340116303925
- RakoczyPEZhangDRobertsonT2002Progressive age-related changes similar to age-related macular degeneration in a transgenic mouse modelAm J Pathol16115152412368224
- RobmanLMahdiOMcCartyC2005Exposure to Chlamydia pneumoniae infection and progression of age-related macular degenerationAm J Epidemiol1611013915901621
- RodriguesEB2007Inflammation in dry age-related macular degenerationOphthalmologica2211435217440275
- RussellSRHudsonHLJerdanJA2007Anecortave acetate for the treatment of exudative age-related macular degeneration – a review of clinical outcomesSurv Ophthalmol52Suppl 1S799017240260
- SakuraiEAnandAAmbatiBK2003Macrophage depletion inhibits experimental choroidal neovascularizationInvest Ophthalmol Vis Sci4435788512882810
- SanGiovanniJPChewEYClemonsTE2007The relationship of dietary carotenoid and vitamin A, E, and C intake with age-related macular degeneration in a case-control study: AREDS Report No. 22Arch Ophthalmol12512253217846363
- ScheibnerKALutzMABoodooS2006Hyaluronan fragments act as an endogenous danger signal by engaging TLR2J Immunol17712728116818787
- SchleicherEDWagnerENerlichAG1997Increased accumulation of the glycoxidation product N(epsilon)-(carboxymethyl)lysine in human tissues in diabetes and agingJ Clin Invest99457689022079
- SchnatbaumKLocardiEScharnD2006Peptidomimetic C5a receptor antagonists with hydrophobic substitutions at the C-terminus: increased receptor specificity and in vivo activityBioorg Med Chem Lett1650889216876401
- SchuttFBergmannMHolzFG2003Proteins modified by malondialdehyde, 4-hydroxynonenal, or advanced glycation end products in lipofuscin of human retinal pigment epitheliumInvest Ophthalmol Vis Sci443663812882821
- SeddonJMAjaniUAMitchellBD1997Familial aggregation of age-related maculopathyAm J Ophthalmol1231992069186125
- SeddonJMWillettWCSpeizerFE1996A prospective study of cigarette smoking and age-related macular degeneration in womenJAMA276114168827966
- ShenJKDongAHackettSF2007Oxidative damage in age-related macular degenerationHistol Histopathol221301817701910
- SievingPACarusoRCTaoW2006Ciliary neurotrophic factor (CNTF) for human retinal degeneration: phase I trial of CNTF delivered by encapsulated cell intraocular implantsProc Natl Acad Sci U S A103389690116505355
- SnodderlyDM1995Evidence for protection against age-related macular degeneration by carotenoids and antioxidant vitaminsAm J Clin Nutr621448S1461S7495246
- SparrowJRBoultonM2005RPE lipofuscin and its role in retinal pathobiologyExp Eye Res8059560615862166
- SparrowJRCaiB2001Blue light-induced apoptosis of A2E-containing RPE: involvement of caspase-3 and protection by Bcl-2Invest Ophthalmol Vis Sci4213566211328751
- SparrowJRFishkinNZhouJ2003A2E, a byproduct of the visual cycleVision Res4329839014611934
- SpencerKLHauserMAOlsonLM2007Protective effect of complement factor B and complement component 2 variants in age-related macular degenerationHum Mol Genet1619869217576744
- SpencerKLHauserMAOlsonLM2008Deletion of CFHR3 and CFHR1 genes in age-related macular degenerationHum Mol Genet17971718084039
- SpencerKLOlsonLMAndersonBM2008C3 R102G polymorphism increases risk of age-related macular degenerationHum Mol Genet171821418325906
- SpraulCWLangGEGrossniklausHE1996Morphometric analysis of the choroid, Bruch’s membrane, and retinal pigment epithelium in eyes with age-related macular degenerationInvest Ophthalmol Vis Sci372724358977488
- SteinbergRH1994Survival factors in retinal degenerationsCurr Opin Neurobiol4515247812140
- StraussO2005The retinal pigment epithelium in visual functionPhysiol Rev858458115987797
- StreileinJWMaNWenkelH2002Immunobiology and privilege of neuronal retina and pigment epithelium transplantsVision Res424879511853765
- SunnessJSGonzalez-BaronJBresslerNM1999The development of choroidal neovascularization in eyes with the geographic atrophy form of age-related macular degenerationOphthalmology106910910328389
- TamakiYAraieMNagaharaM1999Acute effects of cigarette smoking on tissue circulation in human optic nerve head and choroid-retinaOphthalmology106564910080215
- TaniharahInataniMHondaY1997Growth factors and their receptors in the retina and pigment epitheliumProg Retin Eye Res16271301
- TanitoMLiFElliottMH2007Protective effect of TEMPOL derivatives against light-induced retinal damage in ratsInvest Ophthalmol Vis Sci481900517389526
- TanitoMMasutaniHKimYC2005Sulforaphane induces thioredoxin through the antioxidant-responsive element and attenuates retinal light damage in miceInvest Ophthalmol Vis Sci469798715728556
- TannerVKanskiJJFrithPA1998Posterior sub-Tenon’s triamcinolone injections in the treatment of uveitisEye12Pt 4679859850264
- TaoWWenRGoddardMB2002Encapsulated cell-based delivery of CNTF reduces photoreceptor degeneration in animal models of retinitis pigmentosaInvest Ophthalmol Vis Sci433292812356837
- TateDJJrMiceliMVNewsomeDA1995Phagocytosis and H2O2 induce catalase and metallothionein gene expression in human retinal pigment epithelial cellsInvest Ophthalmol Vis Sci36127197775104
- TaylorKRTrowbridgeJMRudisillJA2004Hyaluronan fragments stimulate endothelial recognition of injury through TLR4J Biol Chem279170798414764599
- ThanosCGBellWJO’RourkeP2004Sustained secretion of ciliary neurotrophic factor to the vitreous, using the encapsulated cell therapy-based NT-501 intraocular deviceTissue Eng1016172215684670
- ThompsonDAGalA2003Vitamin A metabolism in the retinal pigment epithelium: genes, mutations, and diseasesProg Retin Eye Res2268370312892646
- ThorntonJEdwardsRMitchellP2005Smoking and age-related macular degeneration: a review of associationEye199354416151432
- TianJIshibashiKReiserK2005Advanced glycation endproduct-induced aging of the retinal pigment epithelium and choroid: a comprehensive transcriptional responseProc Natl Acad Sci U S A102118465116081535
- TownsendKPPraticoD2005Novel therapeutic opportunities for Alzheimer’s disease: focus on nonsteroidal anti-inflammatory drugsFASEB J19159260116195368
- TsaoYPHoTCChenSL2006Pigment epithelium-derived factor inhibits oxidative stress-induced cell death by activation of extracellular signal-regulated kinases in cultured retinal pigment epithelial cellsLife Sci795455016504212
- TuoJBojanowskiCMZhouM2007Murine ccl2/cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degenerationInvest Ophthalmol Vis Sci4838273617652758
- UmedaSAyyagariRAllikmetsR2005Early-onset macular degeneration with drusen in a cynomolgus monkey (Macaca fascicularis) pedigree: exclusion of 13 candidate genes and lociInvest Ophthalmol Vis Sci466839115671300
- UmedaSSuzukiMTOkamotoH2005Molecular composition of drusen and possible involvement of anti-retinal autoimmunity in two different forms of macular degeneration in cynomolgus monkey (Macaca fascicularis)FASEB J191683516099945
- van der SchaftTLMooyCMde BruijnWC1993Early stages of age-related macular degeneration: an immunofluorescence and electron microscopy studyBr J Ophthalmol77657618218037
- VannVRAthertonSS1991Neural spread of herpes simplex virus after anterior chamber inoculationInvest Ophthalmol Vis Sci322462721714427
- VogtSDBarnumSRCurcioCA2006Distribution of complement anaphylatoxin receptors and membrane-bound regulators in normal human retinaExp Eye Res838344016764856
- VolobouevaLALiuJSuhJH2005(R)-alpha-lipoic acid protects retinal pigment epithelial cells from oxidative damageInvest Ophthalmol Vis Sci4643021016249512
- WalportMJ2001Complement. First of two partsN Engl J Med34410586611287977
- WeathingtonNMvan HouwelingenAHNoeragerBD2006A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammationNat Med123172316474398
- WimpissingerBReschHBerishaF2004Response of choroidal blood flow to carbogen breathing in smokers and non-smokersBr J Ophthalmol887768115148211
- WimpissingerBReschHBerishaF2005Response of retinal blood flow to systemic hyperoxia in smokers and nonsmokersGraefes Arch Clin Exp Ophthalmol2436465215657773
- WinklerBSBoultonMEGottschJD1999Oxidative damage and age-related macular degenerationMol Vis53210562656
- WrenshallLEStevensRBCerraFB1999Modulation of macrophage and B cell function by glycosaminoglycansJ Leukoc Biol6639140010496308
- WuZLauerTWSickA2007Oxidative stress modulates complement factor H expression in retinal pigmented epithelial cells by acetylation of FOXO3J Biol Chem282224142517558024
- YamadaYIshibashiKBhuttoIA2006The expression of advanced glycation endproduct receptors in rpe cells associated with basal deposits in human maculasExp Eye Res82840816364296
- YamadaYTianJYangY2008Oxidized low density lipoproteins induce a pathologic response by retinal pigmented epithelial cellsJ Neurochem10511879718182060
- YatesJRSeppTMatharuBK2007Complement C3 variant and the risk of age-related macular degenerationN Engl J Med3575536117634448
- YoshidaTOhno-MatsuiKIchinoseS2005The potential role of amyloid beta in the pathogenesis of age-related macular degenerationJ Clin Invest115279380016167083
- ZamiriPMasliSStreileinJW2006Pigment epithelial growth factor suppresses inflammation by modulating macrophage activationInvest Ophthalmol Vis Sci473912816936104
- ZarbinMA1998Age-related macular degeneration: review of pathogenesisEur J Ophthalmol81992069891890
- ZhangNPeairsJJYangP2007The importance of Bcl-xL in the survival of human RPE cellsInvest Ophthalmol Vis Sci4838465317652760
- ZhouJGaoXCaiB2006Indirect antioxidant protection against photooxidative processes initiated in retinal pigment epithelial cells by a lipofuscin pigmentRejuvenation Res92566316706653
- ZhouJJangYPKimSR2006Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epitheliumProc Natl Acad Sci U S A10316182717060630