Abstract
Pulmonary arterial hypertension (PAH) may occur as an idiopathic process or as a component of a variety of diseases, including connective tissue diseases, congenital heart disease, and exposure to appetite suppressants or infectious agents such as HIV. Untreated, it is a potentially devastating disease; however, diagnosis can be difficult due to the non-specific nature of symptoms during the early stages, and the fact that patients often present to a range of different medical specialties. The past decade has seen remarkable improvements in our understanding of the pathology associated with the condition and the development of PAH-specific therapies with the ability to alter the natural history of the disease. This article reviews the evidence for screening and diagnosis of susceptible patient groups and discusses treatment selection and recommendations based on data available from randomized controlled trials. In addition, due to the complexity of the diagnostic evaluation required and the treatment options available, this review mandates for a multidisciplinary approach to the management of PAH. We discuss the roles and organizational structure of a specialized PAH center in Perth, Western Australia to highlight these issues.
Introduction
Pulmonary arterial hypertension (PAH) is a group of diseases which form a subset of those with pulmonary hypertension (PHT). PAH occurs as an idiopathic process (iPAH) or as a component of a variety of disease processes, including connective tissue diseases (CTD), congenital heart disease, and exposure to exogenous factors including appetite suppressants or infectious agents such as human immunodeficiency virus (HIV). All of these conditions share a common pattern of vascular remodeling of the small pulmonary arteries. An increase in pulmonary vascular resistance (PVR) results in progressive dyspnea, profound functional limitation, and if untreated, progression to right ventricular failure and death.Citation1
PAH is often perceived as a disease with a uniformly poor outcome. However, the past decade has witnessed significant advances in PAH-specific therapies with the ability to change the course of the disease. For example, untreated PAH has an estimated median survival of 2.8 years, with 1-year, 3-year, and 5-year survival rates of 68%, 48% and 34%, respectively.Citation2 In contrast, a prospective study of observed survival in 139 patients with iPAH treated with bosentan and 346 similar patients treated with epoprostenol, reported survival estimates after 1 and 2 years of 97% and 91%, respectively, in the bosentan cohort and 91% and 84% in the epoprostenol cohort.Citation3 In patients with CTD-related PAH, earlier treatment is associated with better outcomes – the risk of death increasing by 11% for every 10 mmHg increase in mean pulmonary arterial pressure (mPAP).Citation4 Unfortunately, given the range of comorbidities associated with the disease, and the fact that various specialities can be confronted with PAH, translating the importance of early diagnosis and treatment into clinical practice represents an ongoing challenge.
In the initial stages, the most common symptoms of PAH include breathlessness, fatigue and near syncope.Citation5 Since these symptoms are non-specific, PAH is often overlooked or under-recognized until its later, more advanced stages (such as the onset of right heart failure). This pattern of presentation may also be responsible for underestimating the true prevalence of the disease and explain, in part, why PAH is sometimes managed by cardiologists and sometimes by respiratory physicians. The aim of this article is twofold. Firstly, to provide an overview of PAH and appropriate diagnostic and treatment approaches, and secondly, to discuss the rationale and importance of a multidisciplinary approach to the management of this disease. The process of integrated PAH care provided by the Royal Perth Hospital, Western Australia is used as the basis for this discussion.
Classification and pathophysiology of PAH
Classification
PHT is defined by a mPAP >25 mmHg at rest or >30 mmHg with exercise. In 2003, the World Health Organization (WHO) revised the classification of PHT into five categories based in part on etiology: PAH, pulmonary venous hypertension, PHT associated with lung diseases and/or hypoxemia, PHT resulting from chronic thrombotic or embolic disease, and miscellaneous ().Citation6 The classification system aims to frame whether PHT is a manifestation of an underlying disease and provides an understanding of the contexts in which PHT occurs. PAH is a sub-category of PHT (the two terms are not synonymous) and is defined as a mPAP > 25 mmHg at rest or >30 mmHg during exercise with a normal pulmonary artery wedge pressure (PAWP) which excludes pulmonary venous hypertension.Citation7–Citation9 Patients with PAH can also be classified according to their ability to function and symptom severity. The WHO classification of functional capacity, an adaptation of the New York Heart Association (NYHA) system, has been useful in this regard ().Citation10
Table 1 Revised clinical classification of pulmonary hypertension (Venice 2003)Citation6
Table 2 Modified New York Heart Association classification of functional status in patients with pulmonary hypertension (PHT)
Regardless of the cause, the common end result of processes leading to PAH is elevation of PVR, with resultant elevation of PAP and progression to right-sided heart failure.
Pathophysiology
Under normal conditions, the pulmonary circuit is a low pressures system (mPAP 12–16 mmHg). Healthy individuals can accommodate up to a 4-fold rise from the resting cardiac output with little increase in PAP, due to distensibility of the thin-walled pulmonary vasculature and to recruitment of vessels that are normally closed when at rest.Citation11 The excess capacity is such that approximately 70% of the vascular bed must be lost before there is an increase in resting PAP.Citation11
Most forms of PAH share a common pathophysiology which includes pulmonary vasoconstriction, vascular wall hypertrophy and thrombosis in situ.Citation12–Citation14 The primary mechanism is believed to involve endothelial dysfunction, resulting in over expression of endothelin (ET)-1, a 21-amino acid peptide which, at normal levels contributes to the maintenance of vascular tone, mediates cardiac, renal and endocrine function, and plays a role in mitogenesis.Citation15 Endothelial dysfunction also reduces the synthesis of nitric oxide and prostacyclin (PgI2), impairing vasodilatory responses and exacerbating the dysfunctional vasoresponses caused by elevated ET levels.Citation14,Citation16 Other important pathways in the process of pulmonary vascular remodeling include changes in potassium channel expression, activation of vascular elastases, and increased expression of inflammatory chemokines.Citation17 Various growth factors, including platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), insulin growth factor-1 (IGF-1), and epidermal growth factor (EGF) have also been postulated to be involved.Citation18
A mutation of the bone morphogenetic protein receptor type II (BMPR2) gene has been identified in approximately 50% patients with familial and iPAH.Citation19 However, the relationship of this gene mutation to the broad range of associated causes of PAH remains unknown, and not all individuals carrying the BMPR2 mutation develop PAH. A subject who possesses the mutation has a 10% to 20% lifetime risk of acquiring PAH, while an individual without the mutation has a lifetime risk of PAH no different to the general population.Citation19
Despite the multitude of perturbations that have been demonstrated in clinical PAH as well as in animal models of pulmonary hypertension, it remains unclear which are “causes” versus consequences of this disorder. However, the end result includes increased vasoconstriction, smooth muscle cell proliferation, decreased vasodilation, and fibrotic changes in medium- to small-sized pulmonary arteries.Citation12–Citation14
Both vasoconstrictive and hypertrophic changes lead to increased PVR, increasing the workload of the right ventricle. Initially, the right ventricle compensates to maintain adequate pulmonary flow, but as the increased workload causes the right ventricle to dilate, efficiency eventually falls. Symptoms such as dyspnea and fatigue appear, initially on exertion. Eventually and often suddenly, the right ventricle decompensates and right heart failure ensues.Citation20 Death occurs as a result of end-stage right heart failure or arrhythmia.Citation21
As a result of the interconnection between pulmonary and cardiac pathologies, the diagnosis of PAH remains clinically challenging, with an umbrella of potential presentations. Furthermore, various specialities can be confronted with patients with PAH (eg, cardiologists, respiratory physicians and rheumatologists). It is this diverse and often non-specific presentation of patients that provides justification for a multidisciplinary approach to the detection and management of PAH.
Epidemiology
The incidence of idiopathic and familial PAH in the general population is almost certainly higher than the historical estimate of 1 to 2 per million population.Citation10 More recent estimates vary from 5 to 52 cases per million population.Citation22,Citation23 iPAH may manifest in both genders and all ages. During childhood, the condition affects both genders equally.Citation24 After puberty however, detection is more frequent in females (approximately 2:1 ratio).Citation25,Citation26
For other causes, PAH is associated with scleroderma in 4.9% to 38.6% of patients,Citation27–Citation31 a condition which itself has a point prevalence of between 30.8 and 286 cases per million population.Citation21 Taken together, these estimates indicate that the prevalence of PAH in the community may be much higher than previously proposed. Recently, a retrospective database review of all patients undergoing Doppler echocardiographic measures in a major regional cardiology center identified a significant number of patients with PAH, subsequently confirmed by right-heart catheterization (RHC). The estimated minimum prevalence of PAH in this community was 75.2 cases per million population (47.9 case per million for iPAH and 27.4 cases per million for scleroderma-related PAH).Citation32
Diagnosis and screening
Clinical presentation
Despite the gravity of the disease, detection of PAH remains elusive until the later stages of the disease, owing to the often subtle and non-specific nature of symptoms (). Symptoms are often difficult to dissociate from those caused by a known underlying pulmonary or cardiac disorder and frequently, there is a delay of up to 3 years between first symptoms and diagnosisCitation33 – an interval that has remained stable over the last 20 years. The most common symptoms – exertional dyspnea and fatigue – reflect an inability to increase cardiac output during activity. Typical angina may also occur despite normal coronary arteries. The mechanism is unclear, but anginal chest pain may be due to pulmonary artery stretching or right ventricular ischemia.Citation34 Over time, increasing fatigue, palpitations, chest pain, and increasing exercise intolerance develop, while hypoxemia, syncope and right-sided heart failure are markers of end-stage disease.
Table 3 Symptoms and signs of pulmonary hypertensionCitation34
Since PHT may be associated with a variety of comorbid conditions, past medical history and symptomatic evidence of a related illness should also be considered. Potential exposure to toxic agents should be explored. This includes previous use of appetite suppressants and chemotherapy agents (including mitomycin-C, carmustine, etoposide, cyclophosphamide and bleomycin). Known exposure to HIV infection should also be considered together with a history of pulmonary embolism or deep vein thrombosis.Citation19 Orthopnea and paroxysmal nocturnal dyspnea suggest elevated pulmonary venous pressure and pulmonary congestion due to left-sided cardiac disease. Raynaud phenomenon, arthralgias, or swollen hands and other symptoms of CTD in the setting of dyspnoea should raise the possibility of PAH related to CTD. A history of snoring or apnea provided by the patient’s partner warrants evaluation for sleep-disordered breathing as a potential causative or contributory factor.Citation19
Diagnosis
The diagnosis of PAH is, in part, through the exclusion of other diseases that cause PHT. Formal guidelines have been published by the European Society of Cardiology (ESC)Citation7 and the American College of Chest Physicians (ACCP),Citation8,Citation19,Citation35 while systemic sclerosis groups have published proposed assessment pathways for the detection of PAH in this cohort.Citation20 Assessment follows a logical sequence of determining whether there is a risk of PAH being present, whether PAH is likely to be present based on initial, non-invasive evaluation, clarifying the underlying etiology of PAH, and delineating the specific hemodynamic profile, including the acute response to vasodilator testing.Citation19
Physical examination
Clinical suspicion should arise in any patient presenting with breathlessness without overt signs of specific heart or pulmonary disease, particularly in diseases which may be associated with PAH.Citation33 On physical examination, the likelihood of PAH increases when certain findings are present. For example, a left parasternal lift produced by the impulse of the hypertrophied right ventricle, an accentuated pulmonary component of the second heart sound (S2), pansystolic murmur of tricuspid regurgitation, diastolic murmur of pulmonary insufficiency, right ventricular S3, jugular vein distention, hepatomegaly, peripheral edema, ascites and cool extremities.Citation19,Citation35 However, the absence of these findings does not exclude PAH.
Electrocardiogram
The electrocardiogram (ECG) lacks sufficient sensitivity to serve as an effective screening tool for PAH. However, it does provide important prognostic information on anatomical and arrhythmic problems. ECG changes associated with PAH include right ventricular hypertrophy and right axis deviation in 87% and 79% of patients, respectively.Citation19,Citation35
Chest X-ray
In the majority of patients with mild PAH, a chest X-ray (CXR) is normal. In more advanced disease, signs suggestive of PAH include enlarged main and hilar pulmonary arterial shadows with concomitant attenuation of peripheral pulmonary vascular markings (‘pruning’). Right ventricular enlargement is often detected by impingement of the anteriorly situated right ventricle silhouette into the retrosternal clear space on the lateral CXR.Citation19,Citation35
Pulmonary function tests
In the diagnosis of PAH, pulmonary function tests (PFTs) are used to exclude significant lung disease. Although lung function abnormalities have been described in association with PAH they are generally mild and unlikely to be the primary cause of symptoms. The exception is a marked impairment in the diffusion capacity for carbon monoxide (DLCO) associated with systemic sclerosis. The DLCO of 20% of patients with limited systemic sclerosis is below normal; however, a DLCO of <55% of predicted increases the likelihood of the presence or future development of PAH.Citation19,Citation35
Ventilation-perfusion (V/Q) lung scanning
V/Q lung scans are an important component of assessment and should be performed to rule out chronic thrombolic pulmonary hypertension (CTEPH), a potentially curable cause of PHT.Citation19,Citation35 Patients with PHT who have normal V/Q scans are unlikely to have chronic pulmonary embolism and more likely to have iPAH. In three studies, V/Q scanning showed sensitivity of 90% to 100% with a specificity of 94% to 100% for distinguishing between iPAH and CTEPH. A positive V/Q scan in patients with CTEPH generally shows one or more segmental-sized or larger mismatched perfusion defects and warrants pulmonary angiography for definitive diagnosis.Citation19,Citation35
Assessment of exercise capacity
Assessment of exercise capacity, using the 6-minute walk test (6MWT) or cardiopulmonary exercise testing is an important part of the evaluation of PAH. The goals of exercise testing include, but are not limited to: determining maximal exercise tolerance; identifying functional capacity; obtaining prognostic data; uncovering PHT silent at rest; establishing a baseline measure of exercise capacity and following the response to therapy. Furthermore, the assessment of exercise capacity may provide evidence of contributory reasons for symptoms (eg, myocardial ischemia).Citation19,Citation35
Doppler echocardiogram
When PAH is suspected Doppler echocardiography should be employed to assess pulmonary artery systolic pressure (PASP), right ventricular enlargement (RVE), right atrial enlargement (RAE) and RV dysfunction.Citation19,Citation35 An adequate Doppler signal from the tricuspid regurgitation jet is required to estimate PASP and this may be absent in approximately 25% of patients.Citation36 Furthermore, the parameters for estimating PASP and abnormal thresholds for defining PAH have not been standardized. Peak velocity of the tricuspid regurgitant jet (TRV), a range of tricuspid gradients, and RVSP have all been used.Citation20 To exclude PHT due to left-heart disease, left ventricular systolic and diastolic function and valve morphology and function should also be assessed. Finally, echocardiography with contrast should be used to identify or rule out PAH due to congenital heart disease (eg, abnormal morphology; shunt).
Right-heart catheterization
RHC is required to confirm the presence of PAH, establish the specific diagnosis including exclusion of pulmonary venous hypertension, determine the severity of hemodynamic impairment, test the vasoreactivity of the pulmonary circulation, and to guide subsequent therapy.Citation19,Citation35 A response to a vasodilator challenge indicates that a patient may be suitable for a trial of high dose calcium-channel blockers (CCBs). A PCWP > 15 mmHg may indicate left heart disease and requires careful evaluation as PAH specific therapies may be contraindicated.
Screening
High risk conditions and the likelihood of having PAH are shown in . The ACCP Consensus Statement recommends that patients with scleroderma should undergo periodic Doppler echocardiography as part of a screening program because of the relatively high detection rates of PAH and disease severity when it develops.Citation19 Other potential causes of PAH do not warrant routine screening (eg, previous use of appetite suppressant, HIV infection, other CTDs) because of the low likelihood of discovering PAH. The question of when to begin screening in patients with the potential for having familial PAH remains open. Patients with more than one family member with PAH related to a mutation in the BMPBR2 gene might be considered for genetic testing, since a negative test would imply that there is no higher than normal risk of developing PAH. However, any test should be preceded by extensive family and genetic counseling.Citation37
Table 4 Patients at risk of developing pulmonary arterial hypertensionCitation37
The availability of new therapies that have been shown to slow or prevent progression of PAH has caused a growing interest among physicians to diagnose PAH at an early stage. Since the range of diagnostic tests required is broad, and often specialized in itself, multidisciplinary clinics, consisting of a rheumatologist, cardiologist, respiratory physician and a specialist nurse together with allied health services (eg, sonography, physiotherapy and radiology) appear warranted. Given the complex nature of PAH, there can be little doubt, that such clinics would facilitate early diagnosis and facilitate access to available therapies according to best practice guidelines.Citation38
Treatment approaches and priorities
The treatment of PAH has witnessed a dramatic change in the past decade. Management guidelines for PAH have been published by the ESCCitation7 and the ACCP.Citation39–Citation41 Both of these guidelines are in the process of being updated with new publications due in 2009. Furthermore, the National Pulmonary Hypertension Centres of the United Kingdom and Ireland recently released an updated consensus statement on the management of PAH in clinical practice.Citation33 Treatment is often complex and patients benefit from referral to a center that specializes in the treatment of this disorder.Citation34 Here we will consider ‘conventional’ therapy, defined as that which has been used for patients with PAH in the years prior to the development of disease-specific, targeted therapy, and PAH-specific therapy separately. An evidence-based treatment algorithm is also proposed. The intention is to provide a guide to the selective use of each form of therapy.
Conventional treatment
The aim of therapy in patients with PAH is to improve survival, disease-related symptoms and quality of life (QoL). Conventional treatment options include oxygen therapy in cases of hypoxemia, anticoagulants such as warfarin, digoxin and diuretics in cases of right-sided heart failure. If, during RHC, the pulmonary vasoreactive tests are positive, the treatment of choice is high-dose CCBs, as improved survival with long-term use has been demonstrated in this cohort of patients.Citation42,Citation43 Vasoreactivity is considered to be present when the mPAP has decreased by at least 10 mmHg to 40 mmHg or less with normal or high cardiac output after intervention with pulmonary vasodilators, such as 100% oxygen, nitric oxide inhalation or intravenous prostacyclin.Citation35 However, less than 10% of patients with iPAH respond long term to CCB therapy and even less in other associated conditions, such as PAH related to CTD: in many cases the test is omitted because the response is so infrequent.Citation35 For well over 90% of patients, PAH specific therapies are indicated first line.
Specific treatment
Classes of drugs available for the treatment of PAH include prostanoids (epoprostenol and iloprost), endothelin receptor antagonists (ERAs, bosentan, sitaxsentan and ambrisentan) and a phosphodiesterase-5 (PDE-5) inhibitor (sildenafil). These drugs offer improved symptom control, exercise capacity, QoL and cardiopulmonary hemodynamics, as well as the prospect of extended survival. However, due to the short-term nature of the randomized controlled trials (12–16 weeks), only epoprostenol has demonstrated a survival benefit compared to patients treated with conventional therapy.Citation44 presents a proposed algorithm for the therapeutic management of patients with PAH.Citation45 This algorithm is based on evidence from clinical trials performed to date and is focused on patients in NYHA functional class II to IV. Furthermore, the algorithm is based on therapies that have been evaluated in iPAH, and in PAH associated with scleroderma or due to anorexigens. Extrapolation of trial results to other PAH subgroups should be done with caution.
Figure 1 Proposed algorithm for the medical management of pulmonary arterial hypertension (PAH).Citation41 A positive acute response to vasodilators is defined as a fall in mPAP of at least 10 mmHg to 40 mmHg, with an increased or unchanged cardiac output during acute challenge with inhaled nitric oxide, iv epoprostenol, or iv adenosine. Sustained response to calcium channel blockers is defined as patients being in functional class I or II with near-normal hemodynamics after several months of treatment. Most experts recommend that patients in functional class IV in unstable condition be treated with iv epoprostenol. In patients in functional class III, first-line therapy may include oral endothelin-receptor antagonists, phosphodiesterase inhibitors, long-term iv epoprostenol, or prostanoid analogues.
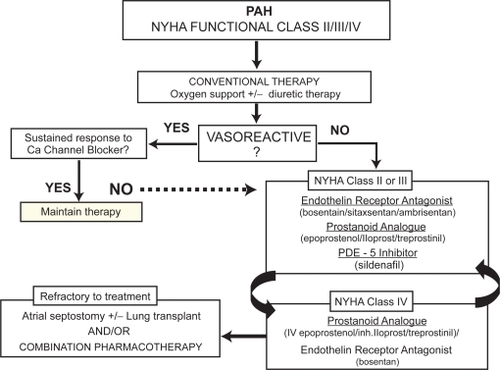
For patients classified as NYHA class II, III or IV, there are a number of therapeutic options. Bosentan is an oral dual endothelin-A (ETA) and endothelin-B (ETB)-receptor antagonist. It is the first molecule of this class of drugs to be synthesized. Activation of ETA and ETB-receptors on smooth muscle cells mediate the vasoconstrictive and mitogenic effects of ET-1, and the prominent role of ET-1 in the pathogenesis of PAH has been well documented.Citation46,Citation47 The efficacy of bosentan has been extensively studied in five randomized placebo-controlled trialsCitation48–Citation52 and five open-label studies.Citation53–Citation57 In these trials, bosentan demonstrated improvements in exercise capacity, functional class, hemodynamics, echocardiographic and Doppler variables, and time to clinical worsening. Additional ERAs have since been developed (eg, sitaxsentan and ambrisentan) that target the ETA receptor only, with similar efficacy results. Slightly less long-term evidence is available for these agents.Citation58–Citation61
The prostacyclin analogue, epoprostenol has been approved for the treatment of patients with iPAH in NYHA class III and IV. In several randomized controlled trials, the beneficial effects of epoprostenol were demonstrated on survival, exercise tolerance, functional class and pulmonary vascular hemodynamics.Citation44,Citation62–Citation64 Despite its benefits, the complexity of drug administration limit its use. Epoprostenol is recommended first line for those patients presenting with functional class IV symptoms. Several additional prostacylin analogues, namely iloprost, treprostinil and beraprost, have also been developed with less utility than epoprostenol.
More recently, sildenafil, an oral PDE-5 inhibitor, has been shown to be effective in patients with iPAH.Citation65 PDE-5 exerts its pharmacological effect by increasing the intracellular concentration of cGMP. The increase of this nucleotide induces relaxation and antiproliferative effects on vascular smooth muscle cells.Citation7
For patients in functional class II–III, therapeutic efficacy of the PAH-specific agents is evaluated after 3 months and every 3 to 6 months thereafter and more frequently in functional class IV. If treatment with initial therapy fails, the next treatment option, usually an agent with a different mode of action is administered, or continuous intravenous epoprostenol considered.
Combination therapy
The optimal management of patients who exhibit clinical deterioration despite targeted monotherapy remains a matter of debate.Citation33 Follow-up beyond the first 3 months of therapy with oral, inhaled or intravenous agents shows that a proportion of patients deteriorate. For example, the percentage of patients remaining stable on bosentan monotherapy at 1 year is 85%,Citation66 and 57% at 3 years.Citation67 Similarly, for sildenafil monotherapy, 85% of patients are stable at 1 year,Citation65 as are 85% on epoprostenolCitation64 and 42% to 77% on inhaled iloprost.Citation68,Citation69 With the development of several therapeutic agents with different mechanisms of action and potential synergy, considerable interest has developed in the possibility of combination therapy.
At the time of writing, concomitant combination therapy has not been adequately studied. The single small trial of epoprostenol initiation with add on bosentan 48 hours later showed only trends in improvement, without significant benefits.Citation37 In contrast, sequential add on therapy has achieved successful endpoints for a wide range of combinations (eg, bosentan add iloprost, epoprostenol add sildenafil, bosentan add sildenafil, beraprost or iloprost add bosentan, treprostinil add bosentan, and iloprost add sildenafil).Citation37 For example, in the PACES-1 trial, Simonneau et al randomized 267 patients with Class III or IV disease (iPAH, CTD-related PAH or CHD-PAH) receiving epoprostenol continuous iv infusion, to either added sildenafil or placebo.Citation70 At 16 weeks, sequential therapy significantly improved 6MWD compared to monotherapy (+26 m; p < 0.001), decreased mPAP (p < 0.001), and increased time to clinical worsening (p = 0.012).
Combination therapy remains an attractive option, and individuals who demonstrate an inadequate response to monotherapy should be considered for a combination of two or more disease-targeted therapies. However, given the limited amount of conclusive data pertaining to combination treatment, published guidelines have so far made no specific recommendation to guide clinicians on the next step to improve the status of their patients who continue to decline on monotherapy.Citation33
Transplant
A small proportion of patients with PAH are eligible for heart-lung or bilateral lung transplantation. This treatment option should be considered and early discussion with experienced transplant physicians is recommended. However, limited donors supply and a 5-year survival of approximately 50%, significantly restricts its utlility.Citation71 Consideration should be given to referring patients to centers that are experienced in atrial septostomy where available.Citation72
Patient management issues and non-drug interventions
PAH is a chronic, life-shortening disease and many patients suffer from limitations in their physical mobility, energy, emotional reactions and social isolation. It is therefore essential that the multidisciplinary team involved with the care of patients with PAH is highly skilled in managing the burden and impact of the disease, and its complex and often intrusive therapies at both physical and psychological levels. Understandably, many PAH patients are affected by a degree of anxiety and/or depression that can have a profound impact on their QoL. Assisting patients to adapt to the uncertainty associated with their illness is important, as is referral to psychologists or psychiatrists when needed. Support groups for patients, families and carers are useful in improving the understanding and the acceptance of the disease condition.
QoL has been shown to be low in patients with PAH using the 36-Item Short-Form Health Survey (SF-36),Citation73 the Nottingham Health Profile (NHP), the Congestive Heart Failure Questionnaire (CHQ) and the Hospital Anxiety and Depression Scale (HADS).Citation1 Improvements in QoL have been reported in trials of PAH-specific therapy and improvements over pre-treatment scores have been seen in association with increased exercise capacity.Citation44,Citation74–Citation79 However, exercise capacity and QoL in patients with PAH are not necessarily related. One study found an absence of any significant relationship in changes in 6MWT distance and changes in QoL in response to therapy,Citation80 concluding that these measures provide complementary data in terms of response to therapy. A new disease specific patient report outcome instrument, CAMPHOR has recently been developed and correlates well with current surrogate outcomes. An assessment such as CAMPHOR should be used in routine clinical practice to assess the QoL status of patients with PAH.Citation33
PAH is a contraindication to pregnancy. Patients with PAH require counseling regarding the very high risk of pregnancy-related mortality (approx. 30%) and the need for adequate contraception. Although the WHO advises discussing termination in the event of pregnancy, if a patient is fully informed and understands the risks of proceeding with pregnancy, treatment with some PAH-specific therapies may represent a realistic option and may improve the chances of maternal survival.Citation33
Finally, patients should be also be encouraged to be as active as their symptoms allow.Citation33 A recent study has demonstrated an improvement in exercise capacity in patients who took part in a training program.Citation81
Optimal structure and organization of multidisciplinary care
Effective diagnosis and treatment of PAH, where several clinical pathways and scenarios are possible, relies on the expertise of many clinical specialties with the knowledge and skill to meet the holistic needs of the patient and their carers.Citation33 These needs include information about the disease and its prognosis, education and support in managing complex drug therapies, psychological and social support, and access to local support in the community. It stands to reason that this care is best achieved when these specialists work together in multidisciplinary teams.
Multidisciplinary care involves a team approach to the provision of care by multiple medical and allied health disciplines. The liaison and cooperation of team members together with the patient ensures that all relevant treatment methods that have a place in the modern management of PAH are properly considered – from diagnosis through follow-up. The effectiveness of multidisciplinary care and coordinated specialty units has been well documented in other conditions, namely heart failure,Citation82 strokeCitation83,Citation84 and oncology.Citation85 To date, however, there is no comprehensive information describing how, or to what extent, institutions managing PAH in Australia have adopted a multidisciplinary approach. In Western Australia, the state referral center for PAH is located at the Royal Perth Hospital. The composition and roles of the multidisciplinary team are shown in . We conducted independent observations and interviews with members of the team to elicit further information about the organization, style, leadership and benefits of the multidisciplinary approach.
Table 5 Composition and roles of the Royal Perth Hospital PAH multidisciplinary team
At this center, patients suspected of having PAH are first seen by a specialist nurse coordinator. The mode of referral is variable. Some patients may be identified by hospital physicians or family doctors as outpatients. Others are referred from related initiatives such as the scleroderma screening program.Citation38 The nurse coordinator is responsible for organizing initial screening examinations including echocardiography, ECG and CXR. Once this information is collated, the patient is seen by a respiratory physician. When required, additional assessments are organized by the nurse coordinator, and may include consultation with a cardiologist. If the results of the examinations are indicative of PAH, RHC is carried out. After the results of all tests have become available, patients are discussed by the multidisciplinary team, appropriate treatment is discussed and a management plan developed. The management plan is then proposed to the patient during a follow up visit. All consenting patients with confirmed PAH are entered into a PAH registry.
Analysis of the observations and interviews with participants about their perceptions of the multidisciplinary structure revealed several common elements related to the success of a unit. These include, but are not limited to:
A team approach, involving core disciplines integral to the provision of PAH management, with input from other specialties as required (where ‘core’ disciplines are cardiology, rheumatology, respiratory, radiology, and supportive care).
Communication among team members regarding treatment planning via regular multidisciplinary meetings held at regular times, venues and with consistent formats to the delivery of information.
Equivalent access to care for all patients, regardless of geographical location.
Provision of care in accord with international guidelines.
Strong leadership was also raised by several members as a key element to the success of the multidisciplinary team: coordinating and chairing regular meetings, encouraging involvement of all participants in case discussions and decision-making and, at the conclusion of case discussions, summarizing the discussion and inviting any further input before moving to the next case. Motivational factors for attending meetings included, 1) perceived benefits of the meetings for both meeting participants and patients, 2) an opportunity to interact with other members of the multidisciplinary team in an inclusive atmosphere, 3) an opportunity for educational interaction and professional development, and 4) streamlining of referral pathways. Meetings may also serve as a resource of identifying patients suitable for clinical trial participation. An established center for managing disease also allows for the collection of registry data on patient presentations, patterns of care and treatment outcomes. This provides a comprehensive set of information available to guide future management, clinical research and treatment practices.
Clearly, the way in which multidisciplinary care is best structured and implemented will vary between sites and depend on local, political and financial resources. For example, geographical remoteness may require collaborations to use telemedicine for multidisciplinary case conferencing. In other situations, the appointment of a nurse coordinator as a focal point for patients being treated by several facilities within a collaborative may represent an avenue of streamlining patient care and ensuring that patients are managed according to agreed guidelines and receive adequate follow-up. However, regardless of the structure in place, the diverse nature and complexity of PAH is an area of medicine in need of multidisciplinary care. The next step, and an avenue for future research, will be to evaluate whether care provided by a multidisciplinary team can improve long-term outcomes in patients with PAH, as has been the case in patients suffering from heart failureCitation82 and stroke.Citation83,Citation84
Conclusion
The treatment of PAH has realized dramatic advances over the past decade, and it is clear that the PAH-specific therapies can significantly alter the natural history of the disease. Despite this, there remains a need to identify patients with PAH earlier, before the onset of extensive vascular remodeling. This can be achieved by performing a thorough assessment to rule out other diagnostic possibilities and to establish the type of PAH. Once identified, it is essential that patients are provided with adequate care and follow-up in line with evidence-based guidelines. Effective diagnosis and treatment of PAH is complex and challenging, relying on the expertise of a team of specialists including rheumatologists, respiratory physicians, cardiologists and specialized nurses. The collaboration of a well coordinated multidisciplinary team of healthcare professionals accounts for the most optimal diagnostic and therapeutic procedures and ensures that patients receive the best possible care.
Disclosures
EG has received research support from Actelion Pharmaceuticals Australia and CSL as well as honoraria for speaking and consulting engagements, and as a member of the Actelion, CSL and GSK Advisory Boards. The Heart and Lung Transplant Foundation of WA, of which EG is chair has received educational grants from Actelion, and CSL. EG has also received travel support from Bayer-Schering the manufacturers of iloprost, Encysive Pharmaceuticals, the manufacturers of sitaxentan and GSK, the distributors of ambrisentan in Australia.
GS is an employee of Actelion Pharmaceuticals Australia. BD has received consulting fees from Actelion.
References
- ShafazandSGoldsteinMKDoyleRLHlatkyMAGouldMKHealth-related quality of life in patients with pulmonary arterial hypertensionChest20041261452145915539712
- D’AlonzoGEBarstRJAyresSMSurvival in patients with primary pulmonary hypertension. Results from a national prospective registryAnn Intern Med19911153433491863023
- McLaughlinVVSitbonOBadeschDBSurvival with first-line bosentan in patients with primary pulmonary hypertensionEur Respir J20052524424915684287
- WilliamsMHDasCHandlerCESystemic sclerosis associated pulmonary hypertension: improved survival in the current eraHeart20069292693216339813
- RichSDantzkerDRAyresSMPrimary pulmonary hypertension. A national prospective studyAnn Intern Med19871072162233605900
- SimonneauGGalieNRubinLJClinical classification of pulmonary hypertensionJ Am Coll Cardiol2004435S12S15194173
- GalieNTorbickiABarstRGuidelines on diagnosis and treatment of pulmonary arterial hypertension. The Task Force on Diagnosis and Treatment of Pulmonary Arterial Hypertension of the European Society of CardiologyEur Heart J2004252243227815589643
- RubinLJDiagnosis and management of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelinesChest20041267S10S15249491
- RunoJRLoydJEPrimary pulmonary hypertensionLancet20033611533154412737878
- RichSExecutive summary from the world symposium on primary pulmonary hypertension Available at http://www.who.int/ncd/cvd/pph.htm.1998
- MaglianoMIsenbergDAHillsonJPulmonary hypertension in autoimmune rheumatic diseases: where are we now?Arthritis Rheum2002461997200912209501
- FusterVSteelePMEdwardsWDGershBJMcGoonMDFryeRLPrimary pulmonary hypertension: natural history and the importance of thrombosisCirculation1984705805876148159
- HoeperMMGalieNSimonneauGRubinLJNew treatments for pulmonary arterial hypertensionAm J Respir Crit Care Med20021651209121611991870
- KimNHRubinLJEndothelin in health and disease: endothelin receptor antagonists in the management of pulmonary artery hypertensionJ Cardiovasc Pharmacol Ther2002791912000973
- RubinLJRouxSBosentan: a dual endothelin receptor antagonistExpert Opin Investig Drugs2002119911002
- RouxSBreuVErtelSIClozelMEndothelin antagonism with bosentan: a review of potential applicationsJ Mol Med19997736437610353441
- HumbertMMorrellNWArcherSLCellular and molecular pathobiology of pulmonary arterial hypertensionJ Am Coll Cardiol20044313S24S15194174
- BarstRJPDGF signaling in pulmonary arterial hypertensionJ Clin Invest20051152691269416200204
- McGoonMGuttermanDSteenVScreening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelinesChest200412614S34S15249493
- ProudmanSMStevensWMSahharJCelermajerDPulmonary arterial hypertension in systemic sclerosis: the need for early detection and treatmentIntern Med J20073748549417547726
- HachullaECoghlanJGA new era in the management of pulmonary arterial hypertension related to scleroderma: endothelin receptor antagonismAnn Rheum Dis2004631009101415308510
- HumbertMSitbonOChaouatAPulmonary arterial hypertension in France: results from a national registryAm J Respir Crit Care Med20061731023103016456139
- PeacockAJMurphyNFMcMurrayJJCaballeroLStewartSAn epidemiological study of pulmonary arterial hypertensionEur Respir J20073010410917360728
- RubinLJPrimary pulmonary hypertensionN Engl J Med19973361111178988890
- GaineSPRubinLJPrimary pulmonary hypertensionLancet19983527197259729004
- GalieNManesAUguccioniLPrimary pulmonary hypertension: insights into pathogenesis from epidemiologyChest1998114184S194S9741567
- BattleRWDavittMACooperSMPrevalence of pulmonary hypertension in limited and diffuse sclerodermaChest1996110151515198989070
- KohETLeePGladmanDDAbu-ShakraMPulmonary hypertension in systemic sclerosis: an analysis of 17 patientsBr J Rheumatol1996359899938883438
- MacGregorAJCanavanRKnightCPulmonary hypertension in systemic sclerosis: risk factors for progression and consequences for survivalRheumatology (Oxford)20014045345911312386
- PopeJELeePBaronMPrevalence of elevated pulmonary arterial pressures measured by echocardiography in a multicenter study of patients with systemic sclerosisJ Rheumatol2005321273127815996064
- SchachnaLWigleyFMChangBWhiteBWiseRAGelberACAge and risk of pulmonary arterial hypertension in sclerodermaChest20031242098210414665486
- GabbayEYeowWPlayfordDPulmonary arterial hypertension is an uncommon cause of pulmonary hypertension in an unselected population: The Armadale echocardiography studyProceedings of the American Thoracic Society Annual Meeting2007 May 18–23San Francisco, California
- Consensus statement on the management of pulmonary hypertension in clinical practice in the UK and IrelandHeart200894Suppl 1i14118276826
- NauserTDStitesSWDiagnosis and treatment of pulmonary hypertensionAm Fam Physician2001631789179811352291
- BarstRJMcGoonMTorbickiADiagnosis and differential assessment of pulmonary arterial hypertensionJ Am Coll Cardiol20044340S47S15194177
- BorgesonDDSewardJBMillerFAJrOhJKTajikAJFrequency of Doppler measurable pulmonary artery pressuresJ Am Soc Echocardiogr199698328378943443
- BarstRJPulmonary arterial hypertension: Evidence-based treatmentWiltshireWiley2008
- PhungSStrangeGChungLPPrevalence of pulmonary arterial hypertension in an Australian scleroderma population: Screening allows for earlier diagnosisIntern Med J2008
- BadeschDBAbmanSHAhearnGSMedical therapy for pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelinesChest200412635S62S15249494
- DoyleRLMcCroryDChannickRNSimonneauGConteJSurgical treatments/interventions for pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelinesChest200412663S71S15249495
- McLaughlinVVPresbergKWDoyleRLPrognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelinesChest200412678S92S15249497
- OgataMOheMShiratoKTakishimaTEffects of a combination therapy of anticoagulant and vasodilator on the long-term prognosis of primary pulmonary hypertensionJpn Circ J19935763698437343
- RichSKaufmannELevyPSThe effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertensionN Engl J Med199232776811603139
- BarstRJRubinLJLongWAA comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study GroupN Engl J Med19963342963028532025
- StewartSStrangeGA clinician’s guide to pulmonary arterial hypertensionSecond editionInforma Healthcare2008
- GalieNManesABranziAThe endothelin system in pulmonary arterial hypertensionCardiovasc Res20046122723714736539
- StewartDJLevyRDCernacekPLanglebenDIncreased plasma endothelin-1 in pulmonary hypertension: marker or mediator of disease?Ann Intern Med19911144644691994793
- ChannickRNSimonneauGSitbonOEffects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled studyLancet20013581119112311597664
- JaisXD’ArminiAMJansaPBosentan for treatment of inoperable chronic thromboembolic pulmonary hypertension: BENEFiT (Bosentan Effects in iNopErable Forms of chronIc Thromboembolic pulmonary hypertension), a randomized, placebo-controlled trialJ Am Coll Cardiol2008522127213419095129
- GatzoulisMABeghettiMGalieNLonger-term bosentan therapy improves functional capacity in Eisenmenger syndrome: results of the BREATHE-5 open-label extension studyInt J Cardiol2008127273217658633
- GalieNRubinLHoeperMTreatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trialLancet20083712093210018572079
- RubinLJBadeschDBBarstRJBosentan therapy for pulmonary arterial hypertensionN Engl J Med200234689690311907289
- BarstRJIvyDDingemanseJPharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertensionClin Pharmacol Ther20037337238212709727
- SitbonOGressinVSpeichRBosentan for the treatment of human immunodeficiency virus-associated pulmonary arterial hypertensionAm J Respir Crit Care Med20041701212121715317666
- DentonCPPopeJEPeterHHLong-term effects of bosentan on quality of life, survival, safety and tolerability in pulmonary arterial hypertension related to connective tissue diseasesAnn Rheum Dis2008671222122818055477
- GalieNHinderliterALTorbickiAEffects of the oral endothelin-receptor antagonist bosentan on echocardiographic and doppler measures in patients with pulmonary arterial hypertensionJ Am Coll Cardiol2003411380138612706935
- HumbertMSegalESKielyDGCarlsenJSchwierinBHoeperMMResults of European post-marketing surveillance of bosentan in pulmonary hypertensionEur Respir J20073033834417504794
- BarstRJLanglebenDBadeschDTreatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentanJ Am Coll Cardiol2006472049205616697324
- BarstRJLanglebenDFrostASitaxsentan therapy for pulmonary arterial hypertensionAm J Respir Crit Care Med200416944144714630619
- BenzaRLBarstRJGalieNSitaxsentan for the treatment of pulmonary arterial hypertension: a 1-year, prospective, open-label observation of outcome and survivalChest200813477578218625676
- GalieNBadeschDOudizRAmbrisentan therapy for pulmonary arterial hypertensionJ Am Coll Cardiol20054652953516053970
- BarstRJRubinLJMcGoonMDCaldwellEJLongWALevyPSSurvival in primary pulmonary hypertension with long-term continuous intravenous prostacyclinAnn Intern Med19941214094158053614
- McLaughlinVVShillingtonARichSSurvival in primary pulmonary hypertension: the impact of epoprostenol therapyCirculation20021061477148212234951
- SitbonOHumbertMNunesHLong-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survivalJ Am Coll Cardiol20024078078812204511
- GalieNGhofraniHATorbickiASildenafil citrate therapy for pulmonary arterial hypertensionN Engl J Med20053532148215716291984
- McLaughlinVVGaineSPBarstRJEfficacy and safety of treprostinil: an epoprostenol analog for primary pulmonary hypertensionJ Cardiovasc Pharmacol20034129329912548091
- ProvencherSSitbonOSimonneauGTreatment of pulmonary arterial hypertension with bosentan: from pathophysiology to clinical evidenceExpert Opin Pharmacother200561337134816013984
- HoeperMMMarkevychISpiekerkoetterEWelteTNiedermeyerJGoal-oriented treatment and combination therapy for pulmonary arterial hypertensionEur Respir J20052685886316264047
- OpitzCFWenselRWinklerJClinical efficacy and survival with first-line inhaled iloprost therapy in patients with idiopathic pulmonary arterial hypertensionEur Heart J2005261895190215888496
- SimonneauGRubinLJGalieNAddition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trialAnn Intern Med200814952153018936500
- Recommendations on the management of pulmonary hypertension in clinical practiceHeart200186Suppl 1I11311473937
- SandovalJRothmanAPulidoTAtrial septostomy for pulmonary hypertensionClin Chest Med20012254756011590848
- TaichmanDBShinJHudLHealth-related quality of life in patients with pulmonary arterial hypertensionRespir Res200569216092961
- ArchibaldCJAugerWRFedulloPFLong-term outcome after pulmonary thromboendarterectomyAm J Respir Crit Care Med199916052352810430723
- MikhailGWPrasadSKLiWClinical and haemodynamic effects of sildenafil in pulmonary hypertension: acute and mid-term effectsEur Heart J20042543143615033256
- OlschewskiHSimonneauGGalieNInhaled iloprost for severe pulmonary hypertensionN Engl J Med200234732232912151469
- SastryBKNarasimhanCReddyNKRajuBSClinical efficacy of sildenafil in primary pulmonary hypertension: a randomized, placebo-controlled, double-blind, crossover studyJ Am Coll Cardiol2004431149115315063421
- SimonneauGBarstRJGalieNContinuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trialAm J Respir Crit Care Med200216580080411897647
- SouzaRJardimCMartinsBEffect of bosentan treatment on surrogate markers in pulmonary arterial hypertensionCurr Med Res Opin20052190791115969891
- StrangeGGabbayEWilliamsTWlodarczykJMcNeilKKeoghABosentan therapy in patients with pulmonary arterial hypertension: the relationship between improvements in six-minute walk test and quality of lifeRespirology20081367468218713089
- MerelesDEhlkenNKreuscherSExercise and respiratory training improve exercise capacity and quality of life in patients with severe chronic pulmonary hypertensionCirculation20061141482148916982941
- McDonaldKLedwidgeMCahillJHeart failure management: multidisciplinary care has intrinsic benefit above the optimization of medical careJ Card Fail2002814214812140806
- Organised inpatient (stroke unit) care for strokeStroke Unit Trialists’ CollaborationCochrane Database Syst Rev2000CD00019710796318
- HankeyGJWarlowCPTreatment and secondary prevention of stroke: evidence, costs, and effects on individuals and populationsLancet19993541457146310543686
- AugustDACarpenterLCHarnessJKBenefits of a multidisciplinary approach to breast careJ Surg Oncol1993531611678331938