Abstract
This review describes recent advances in biosensors of potential clinical applications. Biosensors are becoming increasingly important and practical tools in pathogen detection, molecular diagnostics, environmental monitoring, food safety control as well as in homeland defense. Electrochemical biosensors are particularly promising toward these goals arising due to several combined advantages including low-cost, operation convenience, and miniaturized devices. We review the clinical applications of electrochemical biosensors based on a few selected examples, including enzyme-based biosensors, immunological biosensors and DNA biosensors.
Introduction
Biosensors are sensors that transduce bio-recognition processes into measurable signals via a physicochemical transducer, with electronic and optical techniques as two major transducers (CitationFan et al 2005). The development of biosensors meets the rapidly increasing need for clinical diagnostics in these days. The use of biosensors brings about a combination of advantages. First, biosensors are highly sensitive. This is because biomolecules often possess high affinity toward their targets, for example, antibodies captures antigens with a dissociation constant at the nanomolar scale, and DNA DNA interactions are even stronger than antigen-antibody. Second, biological recognition is usually very selective. An example is that enzyme and substrate are just like lock and key. Such high selectivity often leads to selective biosensors. Third, arising due to the development of modern electronic industry, it has been relatively easy to develop inexpensive, integrated and ready-to-use biosensor devices. These biological sensors certainly improve the ability to detect pathogens or perform genetic analysis in hospitals; more importantly, they are particularly useful for small clinics and even for point-of care analysis.
A variety of new strategies have been developed toward biosensors with clinical applications. In principle, biosensors are analytical devices composed of a biological recognition element and an optical/electronic transducer. The biological element is in charge of capturing analytes in solution and the transducer converts the binding event to a measurable signal variation. The type of biosensors can be categorized by the nature of recognition, that is, enzyme-based biosensors, immunological biosensors, and DNA biosensors. Alternatively, based upon the type of transducers, there are electronic biosensors (electrical or electrochemical) (CitationFan et al 2003; CitationPark et al 2002), optical biosensors (fluorescent, surface plasmon resonance, or Raman) (CitationTaton et al 2000; CitationGaylord et al 2002; CitationXu et al 2005), and piezoelectric biosensors (quartz crystal microbalance) (CitationCooper et al 2001; CitationHook et al 2001). In this review, we will focus on electrochemical biosensors and introduce a few selected examples of enzyme, immunological and DNA biosensors as well as their potential clinical applications.
Electrochemical sensing
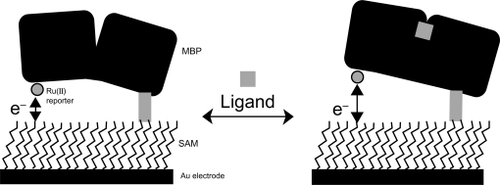
Electrochemical techniques are particularly useful for biological sensing, where electrodes serve as either electron donors or electron acceptors. Extensive electrochemical studies have provided evidence that heterogeneous electron transfer between electrodes and surface-confined redox molecules, analogous to donor-acceptor pairs in homogeneous solutions, also abides by Marcus electron-transfer theory (CitationHeeger 2000; CitationWosnick and Swager 2000; CitationAdams et al 2003). This means that small distance changes of surface-confined redox molecules might induce large variations in heterogeneous electron-transfer rates that should translate into measurable changes of electrochemical signals. For example, Hellinga and co-workers proposed an electrochemical sensing strategy that exploits ligand-mediated hinge-bending motions in proteins. In their approach, a gold electrode was first coated with self-assembled monolayer (SAM), which provides a versatile platform for site-specific immobilization of proteins (CitationBenson et al 2001). The maltose-binding protein (MBP) was then tethered to the gold electrode surface with a specific orientation such that the ruthenium (Ru(II)) redox reporter group is fixed at a certain distance above the electrode. When the ligand maltose binds to the active site, it induces a hinge-bending motion of MBP that moves the Ru(II) reporter away from the electrode (). This maltose-binding induced distance change induces concentration-dependent decrease of electrochemical signals, thus providing a way to electronically sensing maltose. They also demonstrated the use of this highly generalized sensing approach to detect diverse analytes with a family of proteins or enzymes that undergo ligand-binding induced conformational changes.
Figure 1 A ruthenium-labeled maltose-binding protein (MBP) is site specifically attached to a self-assembled monolayer (SAM) coated gold electrode. The protein-ligand binding process mediates dependent changes between the Ru(II) redox reporter group and the surface-modified gold electrode, which thereby alters current flow between the two components. Reprinted with permission from CitationBenson DE, Conrad DW, de Lorimier RM, et al. 2001. Design of bioelectronic interfaces by exploiting hinge-bending motions in proteins. Science, 293:1641–4. Copyright 2001 © AAAS.
Enzyme-based biosensors
Glucose oxidase (GOD)-based biosensors were the first biosensors ever reported, which was developed by Clark and Lyons in 1962 (CitationClark and Lyons 1962). Diabetes mellitus is characteristic of hyperglycemia, a chronically raised concentration of blood glucose. As a result, it is critical for diabetic patients to frequently monitor their glucose concentration in blood. This biosensor, and its more recent versions, takes the advantage of electrochemistry coupled with enzyme catalysis (CitationTurner et al 1987). Clark’s biosensor was an electrode immobilized with GOD. In the presence of glucose, the oxidized form of GOD reacts with glucose and produces gluconic acid and reduced GOD, involving two electrons and two protons. This oxidation of glucose also consumes oxygen in solution since dissolved oxygen reacts with reduce GOD, thus forms hydrogen peroxide and oxidized GOD, and lowers oxygen pressure. As a result, the electrode can sense the glucose by electrochemically sensing oxygen with a Clark oxygen electrode. This kind of sensor is called “first-generation” biosensor. This first-generation biosensor was commercialized in the 1970s by the Yellow Springs Instrument Company (Ohio, USA).
The “second-generation” biosensor replaces the naturally existing substrate, oxygen, with artificial small redox molecules, which serve as the redox mediator and exchange electrons between electrodes and enzymes. A variety of soluble redox molecules, such as ferrocene, thionine, methylene blue, methyl viologen, were employed to improve the sensor performance, that is, sensitivity and signal-to-noise ratio. Initially these mediators were dissolved in solution. They obtain electrons from the electrodes and then these electrons are shuttled to the redox center of enzymes, or vice versa. As a step further, immobilized mediators were proposed in order to develop reagentless biosensors. For example, Ruan et al reported a reagentless, solid-state sensor for hydrogen peroxide (CitationRuan et al 1998). They first modified gold electrodes with L-cysteine, and then multilayers of horseradish peroxidase (HRP) was linked to the amine group of cysteine by using glutaraldehyde, thionine was further linked to the enzyme by the same linking chemistry. As a result, both the enzyme and the mediator were immobilized at the gold electrode, which could sensitively detect hydrogen peroxide in the test solution without further addition of reagents. An obvious advantage of this biosensor configuration is that mediator is fixed at the electrode surface, thus obviating the problem arising due to the diffusion. Heller reported an alternative approach that involved the use of redox polymers (CitationGao et al 2002; CitationRajagopalan and Heller 1997). They first prepared a polymer doped with Os2+ complex. This kind of polymer serves as a “molecular wire” and exchanges electrons between the electrode and the enzyme. Therefore, they co-immobilized the Os-polymer and glucose oxidase on carbon electrode which produces sensitive response to the presence of glucose. By using these redox polymers, they were able to make nearly 100% immobilized enzyme molecules electroactive, which led to a glucose detection method with very high sensitivity.
The commercialization of the second-generation enzyme-based biosensor was very successful. In 1987, MediSense was founded and they launched the pen-sized Exactech™ glucose sensors. This success has led to a revolution for health care of diabetic patients. They have been able to monitor their blood glucose concentration at home instead of going to the clinics. The MediSense and other later amperometric biosensor system consists of disposable, screen-printed carbon electrodes coated with GOD and mediators (test strips). Upon applying a droplet of blood on the test strip, the sensor begins to work and records amperometric response, which is converted to a digit displayed on LCD, representative of glucose concentration.
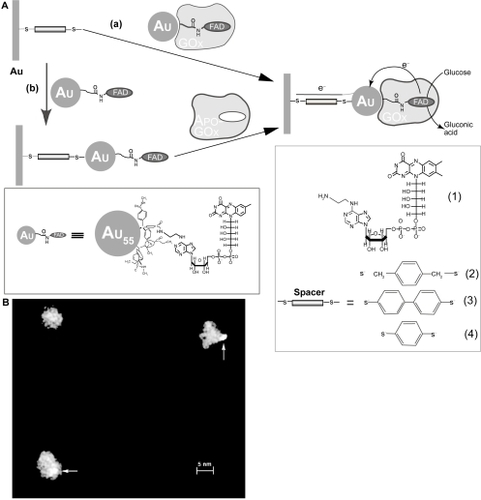
More recently, CitationXiao et al (2003)reported a new generation of glucose biosensor by designing a reconstructed GOD enzyme. They first prepared apo-GOD that was free of the flavin adenine dinucleotide (FAD) cofactor, then they functionalized a 1.4-nm gold nanoparticle with FAD and insert it into the apo-GOD by reconstruction. Such a reconstructed enzyme was linked to gold electrodes by using a dithiol monolayer (). They showed that the electron transfer turnover of this artificial enzyme is as high as 5000 s−1, approximately 8-fold higher than the natural enzyme (700 s−1). In this system, gold nanoparticle acts as an electron relay for electrical wiring of the redox center of the enzyme. The glucose biosensor developed by Xiao et al represents a new direction in this area, which is free of any mediator and is highly sensitive. More recently, Willner’s group reported a modified version of this sensor by using single wall carbon nanotubes (SWNTs) instead of gold nanoparticles and realized similarly high efficiency (CitationPatolsky et al 2004). While the commercialization of this technology is still not available, it is anticipated that it may further improve the state-of-the-art biosensors.
Figure 2 (A) Assembly of Au-NPreconstituted GOx electrode by (a) the adsorption of Au-NPreconstituted GOx to a dithiol monolayer associated with a Au electrode and (b) the adsorption of Au-NPs functionalized with FAD on the dithiol-modified Au electrode followed by the reconstitution of apo-GOx on the functional NPs (16). (B) A STEM image of GOx reconstituted with the Au-FAD hybrid NP. Arrows show Au clusters. Reprinted with permission from CitationXiao Y, Patolsky F, Katz E, et al. 2003. Plugging into enzymes: nanowiring of redox enzymes by a gold nanoparticle. Science, 299:1877–81. Copyright 2003 © AAAS.
Immunological biosensor
Immunological biosensors rely on highly specific immunological system, ie, antibody and antigen, to detect environmentally or clinically relevant targets. Immunological biosensors are actually a new version of enzyme-linked immunosorbent assay (ELISA), with reduced cost, improved speed and operation convenience, and comparable or even higher sensitivity. Electrochemical immunological biosensors are among the most popular ones. There are two types of immunological biosensor. First, a capture antibody is immobilized at the electrode, which captures specific target antigen. Signal transduction is realized via a secondary antibody tagged with redox molecules or enzymes. Second, an antigen is immobilized at the electrode, which detects specific antibody.
Ju and co-workers developed an amperometric immunological biosensor for carcinoembryonic antigen (CEA) (CitationDai et al 2004). They co-immobilized thionine and HRP-labeled CEA antibody on a glassy carbon electrode, which were crosslinked with glutaraldehyde. HRP catalytically reduced hydrogen peroxide in solution, which was coupled to the electrode reaction of thionine, leading to a catalyzed signal. Capturing CEA partially blocked the redox center of HRP, thus leading to attenuation of amperometric signals.
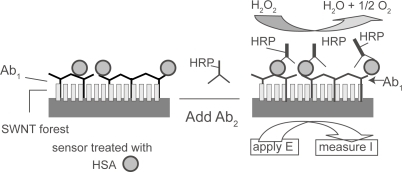
Recently, Rusling and co-workers took advantage of SWNTs to improve the performance of immunological biosensors (CitationYu et al 2005) (). They prepared vertically aligned arrays of SWNTs (SWNT forest) at pyrolytic graphite electrodes by using metal mediated self-assembly. Anti-HSA was then covalently linked to the carboxylated ends of SWNT forest by using EDC/NHS. After capturing HSA target, the electrode was further incubated with a secondary anti-HSA antibody labeled with HRP. Based on the catalytic signal of HRP for hydrogen peroxide, one can detect the HSA target in the test solution. Notably, the use of SWNT forests significantly improved the detection sensitivity, which was approximately 1 nM. This was possibly because of the enhanced electron transfer reactivity of HRP encapsulated in SWNT forests.
Figure 3 Schematic diagram of HSA sandwich assay procedure. Reprinted with permission from CitationYu X, Kim SN, Papadimitrakopoulos F, et al. 2005. Protein immunosensor using single-wall carbon nanotube forests with electrochemical detection of enzyme labels. Mol Biosyst, 1:70–8. Copyright 2003 © Royal Society of Chemistry.
Immunological assays have become one of the most important clinical tools. Nevertheless, current assay methods, eg, ELISA, require large and expensive instrument as well as well-trained technicians. Electrochemical methods are well suited for the development of inexpensive, miniaturized and portable devices. As a result, it is highly attractive to develop electrochemical immunological biosensors in order to meet field and point-of-care analysis. It is worthwhile to note that, similar to glucose biosensors, the use of disposable screen-printed electrodes should be critical toward this goal. Also of note, it is important to develop antibody microarrays based on electrochemistry in order to perform high-throughput (HTS) assays.
DNA biosensors
Detection of DNA hybridization events has been of significant scientific and technological interest. This significance has been particularly manifested by rapidly growing interest of chip-based clinical diagnosis (CitationGao et al 2002) Consequently, a variety of techniques have been developed over the years, including optical (CitationTaton et al 2000; CitationGaylord et al 2002; CitationXu et al 2005), acoustic (CitationCooper et al 2001; CitationHook et al 2001), and electronic approaches (CitationBoon et al 2000a; CitationPatolsky et al 2001; CitationPark et al 2002). Among them, the fluorescent detection has dominated state of the art genosensors in past decades (CitationBowtell 1999; CitationWinzeler et al 1999; CitationGao et al 2002). However, electrochemical methods, which have proven successful in simple chemical species especially metal ions, have attracted rapidly increasing attention in applications of sensing biologically related species (CitationFritz et al 2002; CitationKuhr 2000; CitationWillner 2002). Advantages of electronic detection include: 1) electrochemical detection is usually inexpensive while enables highly sensitive and rapid screening (CitationBard and Faulkner 2001); 2) unlike fluorophores that often have “photo-bleaching” problems, many electroactive labels, eg, metallocenes, are stable and environmentally insensitive; 3) “multi-color” labeling has been possible by suitable molecular design and synthesis that produce a spectrum of derivatives, each having a unique redox potential (CitationBrazill et al 2001); 4) the highly developed silicon industry has paved the road to mass-production of integrated circuits which renders electronic detection especially suitable and compatible with microarray-based technologies; 5) the rapidly developing interfacial science and technology has been unraveling mysteries in precisely controlling surface properties which acts as one of the major barricades in bioelectronic applications (CitationMrksich and Whitesides 1996; CitationYu et al 2001; CitationWhitesides and Grzybowski 2002).
DNA itself is electrochemically silent at moderate applied voltages while severe interferences are expected at high voltages that enable oxidation/reduction of DNA bases (CitationPalecek and Jelen 2002). Millan was the first that proposed sequence-selective DNA target detections based on electroactive hybridization indicators, which provide electronic signals as well as discrimination between double and single stranded DNA (CitationMillan and Mikkelsen 1993). In an attempt to reduce high background deriving from the minor binding of hybridization indicators to ssDNA, “sandwich” type detections has been proposed (CitationIhara et al 1996; CitationUmek et al 2001; CitationYu et al 2001). Besides an immobilized DNA probe, a DNA strand possessing an electroactive label has been introduced to act as the signaling molecule. Similarly, CitationPark et al (2002) have developed an array-based electrical DNA detection with nanoparticle probes which demonstrates high sensitivity and selectivity. CitationThorp (2003) developed a technology based on the relatively high oxidation activity of guanine and its facilitation by exogenous redox catalysts. The ds/ss discrimination is achieved by the fact that guanine in duplexes, due to the steric effect, has relatively low electron transfer reactivity. This method is highly sensitive in detection of PCR products however relatively poor in discrimination of hybridization events. Moreover, this method is only possible at ITO surfaces till now the high oxidation potential still excludes the use of gold.
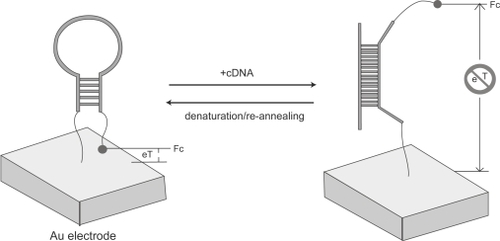
In spite of the progress, it is still highly important to develop an all-in-one (ie, reagentless) sensor that directly signals upon target capturing (ie, obviating further treatment with either hybridization indicators or signal molecules). DNA or RNA aptamers provide a viable means to this end. Aptamers are well-structured DNA or RNA which, as well as natural enzymes, possess high affinity and selectivity to specific targets whereas demonstrate superior robustness to fragile enzymes (CitationChang and Varani 1997; CitationBurgstaller et al 2002). They have emerged as a very promising therapeutic and diagnostic tool (CitationTurner et al 1987). In the meantime, the well developed in vitro selection has been able to produce aptamers for virtually any given target (CitationTurner et al 1987; CitationGriffiths and Tawfik 2000). Given these advantages, the oligonucleotide aptamers have been anticipated to be the next-generation biosensing elements (CitationRobertson and Ellington 1999; CitationSullivan 2002). CitationFan et al (2003) employed a simple structured, hairpin-like DNA with an electroactive label (electronic DNA hairpin) as the building block to detect hybridization events (). Hairpin-like DNA has been an extremely interesting aptamer that forms the basis of fluorescent “molecular beacons” for homogeneous hybridization detection. The DNA sequence have been designed such that this “beacon” is in the close state in the absence of targets while will be “turned on” when it meets its specific gene target. The existence of the stem-loop structure in the design provides an on/off switch as well as a stringency to discriminate single mismatch in DNA hybridization. In this electronic DNA hairpin, a thiolated terminus provides a sticky end to the gold surface while a ferrocene tag at the other end transducts electronic signals. The initial hairpin localizes the ferrocene proximal to the electrode surface, thus allowing interfacial electron transfer. After hybridization, the formation of the linear duplex structure disrupts the hairpin and forces apart the ferrocene and the electrode. This significant distance change (up to a few nm) effectively blocked the interfacial electron transfer and leads to the diminution of corresponding electrochemical current signals (Figure 5). This strategy offers the opportunity to identify 10 pM DNA targets. More importantly, such a design takes the advantage of integrating the capturing part (probe sequence) and the signaling part (electroactive species) within a single surface-confined hairpin structure. Therefore, in contrast to most previously proposed solid-state DNA sensors, this design is effectively reagentless, ie, aside from DNA targets, no exogenous reagent is necessary during the recognition process. This provides the basis to construct a portable, continuous DNA analyzer that might be useful in medical and military applications (CitationPalecek 2004; CitationThorp 2003).
Figure 4 A stem loop oligonucleotide possessing terminal thiol and a ferrocene group is immobilized at a gold electrode through self-assembly. In the absence of target, the stem loop structure holds the ferrocene tag into close proximity with the electrode surface, thus ensuring rapid electron transfer and efficient redox of the ferrocene label. On hybridization with the target sequence, a large change in redox currents is observed, presumably because the ferrocene label is separated from the electrode surface. Reprinted with permission from CitationFan C, Plaxco KW, Heeger AJ. 2003. Electrochemical interrogation of conformational changes as a reagentless method for the sequence-specific detection of picomolar DNA. Proc Natl Acad Sci U S A, 100:9134–7. Copyright 2003 © The National Academy of Sciences of the United States of America.
DNA double helix has been suggested to be a medium for long-range electron transfer (ET) via its base stacking (CitationKelley and Barton 1999; CitationSchuster 2000). Although this issue has been of long-time debate, Barton and coworkers have electrochemically proven that well-oriented DNA films at gold electrode allows long-range electron transfer and that such ET is extremely sensitive to base stacking pertubations such as mismatches (CitationBoon et al 2003; CitationKelley et al 1999). They observed that electroactive intercalators such as methylene blue (MB) could be efficiently reduced at an electrode modified with fully matched DNA duplex. However, the presence of just a single mismatch converts the wire-like ET medium to an insulator, which completely disrupts ET between the MB and the electrode. Such a difference can be readily read out via cyclic voltammetric or coulometric assays, which forms the basis of a rapid DNA mutation screening sensor (CitationBoon et al 2000b; CitationDrummond et al 2003). Barton and coworkers further showed that the sensitivity of this approach could be improved by electrocatalysis. Addition of ferricyanide in solution repetitively pulls electrons from electrochemically reduced MB which amplifies electron flow through the DNA double helix (CitationBoon et al 2000b). This allows detection of ~108 DNA molecules with a 30-μm electrode. Parallel to DNA detection, they also constructed DNA-based sensor to detect DNA binding proteins (CitationBoon et al 2002). Certain DNA-binding proteins or enzymes are known to interfere with DNA base pair stacking, thus converting DNA double helix from efficient ET wires to insulators. Based on the similar sensing strategy, they developed a sensitive way to electrically assay a variety of DNA-binding proteins. Importantly, these sensors effectively discriminate against proteins that bind to DNA albeit do not perturb base stacking (CitationBoon et al 2002). This undoubtedly confirms that the signal cut-off upon protein binding is due to the alteration of base stacking-related ET medium.
The future of clinical biosensors
Despite the rapid progress in biosensor development, clinical applications of biosensors are still rare, with glucose monitor as an exception. This is in sharp contrast to the urgent need in small clinics and point-of-care tests. We believe the following requirements are necessary. First, high sensitivity: Sensitivity improvement is an ever-lasting goal in biosensor development. It is true that the requirement for sensitivity varies from case to case. For example, one does not need a very high sensitivity for glucose detection since glucose concentration is high in blood. This is actually part of reason for the success of glucose monitors. However, in many cases it is very important to develop highly sensitive biosensors, optimally single-molecule detection, in order to meet the requirement of molecular diagnostics and pathogen detection. Second, high selectivity: This might a major barricade in the application of biosensors. Most biosensors reported in the literature work very well in laboratories, however may meet series problems in test real samples. As a result, it is essential to develop novel surface modification approaches in order to avoid non-specific adsorption at surfaces. Third, multiplexing is critical for saving assay time, which is especially important for assays performed in laboratories or clinics. Thus it is important to develop high-density electrode arrays as well as electrochemical instrument that can simultaneously perform a large number of assays. Forth, it is important to develop miniaturized biosensors in order to increase portability, thus meet the requirement of field and point-of-care test. Fifth, an ideal biosensor should be integrated and highly automated. Current lab-on-a-chip technologies (microfluidics) offer a solution toward this goal. We can expect that successful biosensors in the future may incorporate all these features, and can conveniently detect minute targets within a short period.
Acknowledgements
This work was supported by National Natural Science Foundation (60537030 and 20404016), Shanghai Municipal Commission for Science and Technology (0452nm068 and 03DZ14025), Shanghai Rising-Star Program and Chinese Academy of Sciences.
References
- AdamsDABrusLChidseyCED2003Charge transfer on the nanoscale: current statusJ Phys Chem B107666897
- BardAJFaulknerLR2001Electrochemical MethodsNew YorkJohn W Willey & Sons
- BensonDEConradDWde LorimierRM2001Design of bioelectronic interfaces by exploiting hinge-bending motions in proteinsScience2931641411533486
- BoonEMCeresDMDrummondTG2000aMutation detection by electrocatalysis at DNA-modified electrodesNature Biotech181096100
- BoonEMLivingstonALChmielNH2003DNA-mediated charge transport for DNA repairProc Natl Acad Sci U S A10012543714559969
- BoonEMSalasJEBartonJK2002An electrical probe of protein-DNA interactions on DNA-modified surfacesNat Biotechnol20282611875430
- BowtellDDL1999Options available – from start to finish – for obtaining expression data by microarrayNat Genet2125329915497
- BrazillSAKimPHKuhrWG2001Capillary gel electrophoresis with sinusoidal voltammetric detection: A strategy to allow four-“color” DNA sequencingAnal Chem7348829011681464
- BurgstallerPGirodABlindM2002Aptamers as tools for target prioritization and lead identificationDrug Discov Today71221812547005
- ChangKYVaraniG1997Nucleic acids structure and recognitionNature Struct Biol485489377158
- ClarkJLCLyonsC1962Electrode systems for continuous monitoring in cardiovascular surgeryAnn N Y Acad Sci102294514021529
- CooperMADultsevFNMinsonT2001Direct and sensitive detection of a human virus by rupture event scanningNature Biotechnol19833711533641
- DaiZYanFYuH2004Novel amperometric immunosensor for rapid separation-free immunoassay of carcinoembryonic antigenJ Immuno Methods2871320
- DrummondTGHillMGBartonJK2003Electrochemical DNA sensorsNat Biotechnol211192914520405
- FanCPlaxcoKWHeegerAJ2003Electrochemical interrogation of conformational changes as a reagentless method for the sequence-specific detection of picomolar DNAProc Natl Acad Sci U S A1009134712867594
- FanCPlaxcoKWHeegerAJ2005Biosensors based on binding-modulated donor-acceptor distancesTrends Biotechnol231869215780710
- FritzJCooperEBGaudetS2002Electronic detection of DNA by its intrinsic molecular chargeProc Natl Acad Sci U S A9914142612386345
- GaoZBinyaminGKimH-H2002Electrodeposition of redox polymers and co-electrodeposition of enzymes by coordinative crosslinkingAngew Chem Int Ed4181013
- GaylordBSHeegerAJBazanGC2002DNA detection using water-soluble conjugated polymers and peptide nucleic acid probesProc Nat Acad Sci U S A9910954
- GriffithsADTawfikDS2000Man-made enzymes – from design to in vitro compartmentalisationCurr Opin Biotech113385310975453
- HeegerAJ2000Nobel Lecture: Semiconducting and Metallic polymers: The fourth generation of polymeric materials [online]URL: http://wwwnobelse
- HookFRayANordenB2001Characterization of PNA and DNA immobilization and subsequent hybridization with DNA using acoustic-shear-wave attenuation measurementsLangmuir17830512
- IharaTMaruoYTakenakaS1996Ferrocene-oligonucleotide conjugates for electrochemical probing of DNANucleic Acids Res244273808932383
- KelleySOBartonJK1999Electron transfer between bases in double helical DNAScience283375819888851
- KelleySOJacksonNMHillMG1999Long-range electron transfer through DNA filmsAngew Chem Int Ed389415
- KuhrWG2000Electrochemical DNA analysis comes of ageNature Biotech1810423
- MillanKMMikkelsenSR1993Sequence-selective biosensor for DNA-based on electroactive hybridization indicatorsAnal Chem652317238238927
- MrksichMWhitesidesGM1996Using self-assembled monolayers to understand the interactions of man-made surfaces with proteins and cellsAnnu Rev Biophys Biochem255578
- PalecekE2004Surface-attached molecular beacons light the way for DNA sequencingTrends Biotechnol2255814757035
- PalecekEJelenF2002Electrochemistry of nucleic acids and development of DNA sensorsCrit Rev Anal Chem3226170
- ParkSJTatonTAMirkinCA2002Array-based electrical detection of DNA with nanoparticle probesScience2951503611859188
- PatolskyFLichtensteinAWillnerI2001Detection of single-base DNA mutations by enzyme-amplified electronic transductionNature Biotech192537
- PatolskyFWeizmannYWilnerI2004Long-range electrical contacting of redox enzymes by SWCNT connectorsAngew Chem Int Ed43211317
- RajagopalanRHellerA1997Eelectrical “wiring” of glucose oxidase in electron conducting hydrogels in molecular electronicsOxfordBlackwell Science
- RobertsonMPEllingtonA1999In vitro selection of an allosteric ribozyme that transduces analytes to ampliconsNature Biotech17626
- RuanCYangFLeiC1998Thionine covalently tethered to multilayer horseradish peroxidase in a self-assembled monolayer as an electron-transfer mediatorAnal Chem7017215
- SchusterGB2000Long-range charge transfer in DNA: transient structural distortions control the distance dependenceAcc Chem Res332536010775318
- SullivanCKO2002Aptasensors–the future of biosensing?Anal Bioanal Chem37244811939212
- TatonTAMirkinCALetsingerRL2000Scanometric DNA array detection with nanoparticle probesScience28917576010976070
- ThorpHH2003Reagentless detection of DNA sequences on chemically modified electrodesTrends Biotechnol21522414624859
- TurnerAPFKaubeIWilsonGS1987Biosensors fundamentals and applicationsOxfordOxford University Press, UK
- UmekRMLinSWVielmetterJ2001Electronic detection of nucleic acids–A versatile platform for molecular diagnosticsJ Mol Diag37484
- WhitesidesGMGrzybowskiB2002Self-assembly at all scalesScience29524182111923529
- WillnerI2002Biomaterials for sensors, fuel cells, and circuitryScience298240712493919
- WinzelerEASchenaMDavisRW1999Fluorescence-based expression monitoring using microarraysMethods Enzymol306310432445
- WosnickJHSwagerTM2000Molecular photonic and electronic circuitry for ultra-sensitive chemical sensorsCurr Opin Chem Biol47152011102879
- XiaoYPatolskyFKatzE2003Plugging into enzymes: nanowiring of redox enzymes by a gold nanoparticleScience29918778112649477
- XuHWuHHuangF2005Magnetically assisted DNA assays: High selectivity using conjugated polymers for amplified fluorescent transductionNucleic Acids Res33e8315905472
- YuCJWanYJYowantoH2001Electronic detection of single-base mismatches in DNA with ferrocene-modified probesJ Am Chem Soc123111556111697958
- YuXKimSNPapadimitrakopoulosF2005Protein immunosensor using single-wall carbon nanotube forests with electrochemical detection of enzyme labelsMol Biosyst170816880966