Abstract
Hepatic encephalopathy (HE) is a complex neuropsychiatric syndrome present in patients with liver disease that includes impaired intellectual function. To develop therapeutic treatments to restore cognitive function, it is important to understand the molecular mechanisms that impair cognitive function in HE. This review summarizes data showing that: (a) cognitive function and learning are impaired in patients with liver disease and in animal models of chronic liver failure or hyperammonemia; (b) the glutamate–NO–cGMP pathway modulates some forms of learning; and (c) the function of this pathway is impaired in brain in vivo in rats with chronic hyperammonemia or liver failure and from patients who died from HE. Learning ability of hyperammonemic rats was restored by increasing cGMP by: (1) continuous intracerebral administration of zaprinast, an inhibitor of the cGMP-degrading phosphodiesterase; (2) chronic oral administration of sildenafil, an inhibitor of the phosphodiesterase that crosses the blood–brain barrier; and (3) continuous intracerebral administration of cGMP. The data summarized indicate that impairment of learning ability in rats with chronic liver failure or hyperammonemia is due to impairment of the glutamate–NO–cGMP pathway. Moreover, increasing extracellular cGMP by pharmacological means may be a new therapeutic approach to improve cognitive function in patients with HE.
Hepatic encephalopathy
Hepatic encephalopathy (HE) is a complex neuropsychiatric syndrome present in patients with chronic or acute liver disease. HE covers a wide range of neuropsychiatric disturbances ranging from minimal changes in personality or altered circadian rhythms (sleep–waking cycle) to alterations in the intellectual function, personality, conscience, and neuromuscular coordination. HE is usually reversible, but in the worse cases can lead to coma and death.
The neurological alterations in HE are the result of a previous failure of liver function. Liver failure leads to impaired detoxification of ammonia and other toxic substances that can reach the brain and alter its function. Many studies have been carried out to identify factors responsible for the neurological alterations in HE. Clinical experience and basic research indicate that ammonia is the main factor responsible for HE. Ammonia is a product of degradation of proteins and other nitrogenated compounds but at high concentrations ammonia is toxic, leading to alteration of cerebral function which can lead to coma and death.
Hyperammonemia is therefore considered the main factor contributing to the neurological alterations found in HE both in acute and chronic liver disease (CitationFerenci et al 1984; CitationLockwood et al 1991; CitationFelipo and Butterworth 2002; CitationWang and Saab 2003). Classical clinical treatment of HE is mainly directed to reducing ammonia concentration by lowering ammonia production by the intestinal bacteria and by reducing ammonia transport from intestine to the blood flow by acidification of the intestinal lumen. Overt HE is usually elicited by a precipitating factor (high protein ingestion, gastrointestinal constipation, bleeding, diuretics) usually associated with increased ammonia levels. Cognitive, motor (extrapiramidal and cerebellar signs), and sleep alterations (impairment of sleep–wake cycle) are commonly observed in patients with HE and their intensities vary with the grade of HE. Patients with HE show alterations in cognition, consciousness, attention, memory, and learning.
Motor alterations include increase in muscular tone, reduced speed of rapid alternating movement, ataxia, an increase in deep tendon reflexes, abnormal movements such as tremors, and, particularly, asterixis. Also hypomimia, dysarthria, bradykinesia, and hypokinesia could be detected on careful neurological examination.
Alterations in the regulation of biological rhythms such as sleep, appetite, melatonin production, and in sexuality are common in patients with liver disease (CitationIguchi et al 1982; CitationSteindl et al 1995; CitationGarfinkel and Zisapel 1996; CitationCordoba et al 1998).
Patients with liver cirrhosis with normal neurological and mental status examination may present minimal forms of HE, showing intellectual function impairment that cannot be detected through general clinical examination but can be unveiled using specific neuropsychological and neurophysiologic examination (CitationFerenci et al 2002; CitationAmodio et al 2004).
This neurophysiological examination includes the use of EEG as a tool to monitor the severity of HE and the changes due to treatment. Alterations of EEG patterns in cirrhosis are roughly related to the alterations of mental state (CitationParsons-Smith et al 1957). Both qualitative and semi-quantitative scales for grading of the EEG alterations due to HE have been proposed (eg, CitationParsons-Smith et al 1957; CitationAmodio, Marchetti, et al 1999). Spectral analysis of EEG tracing to quantify and classify the alterations due to HE is also being used (eg, CitationVan der Rijt et al 1992; CitationAmodio et al 1996, Citation2001). This technique provides a quantitative estimation of the EEG based on the relative power of frequency bands and the mean dominant frequency. These EEG techniques may detect minimal HE.
The prevalence of minimal EH ranges from 30% to 84% depending on the kind and number of tests used and the population (etiology and severity of the liver disease) investigated. Cirrhotic patients with minimal HE are “clinically normal” but present cognitive alterations which can be unveiled by a detailed analysis of the patients’ history and by neurological and neuropsychiatric assessement of consciousness and of sensory, cognitive, and motor function.
Even minimal HE has been associated with reduced quality of life and ability to work and to drive (CitationSchomerus et al 1981; CitationSrivastava et al 1994; CitationGroeneweg et al 1998; CitationMarchesini et al 2001; CitationSchomerus and Hamster 2001). Moreover, patients suffering minimal HE have increased probability of suffering later overt HE (CitationAmodio, Del Piccolo, et al 1999; CitationHartmann et al 2000; CitationRomero-Gomez et al 2001).
Intellectual function is impaired in HE
Early manifestations of intellectual dysfunction in HE include psychomotor slowing and impaired ability to perform tasks that require sustained attention (CitationMcCrea et al 1996; CitationSchomerus and Hamster 1998; CitationAmodio, Del Piccolo, et al 1999). As encephalopathy worsens, impairment in speech and inability to copy simple drawings (eg, a star) appear. In grade II HE, temporal and spatial disorientation and reduced vigilance state or delirium have been observed. Grade IV HE is characterized by the appearance of stupor and coma.
Patients with minimal HE show impaired ability to perform memory tasks, mainly because of deficits in attention and visual perception (CitationTarter et al 1987; CitationWeissenborn et al 2003). These patients also perform worse than healthy controls in motor function, visual perception, visual orientation, visuo-constructive abilities, and attention (CitationRehnström et al 1977; CitationRikkers et al 1978; CitationGilberstadt et al 1980; CitationTarter et al 1984). Performance in recognition and free recall tasks is also impaired in these patients (CitationWeissenborn et al 2003).
The psychomotor slowing present in patients with minimal HE is due to cognitive rather than motor deficits, as indicated by the fact that delays in choice reaction times are more strongly impaired than the delays in simple reaction times (CitationRikkers et al 1978; CitationSchomerus et al 1981).
Sustained attention is also impaired in cirrhotic patients even when memory, language, constructive, or pure motor alterations are absent (CitationMcCrea et al 1996; CitationWeissenborn et al 2001, Citation2003). Patients with minimal HE have a tendency to be easily distracted.
Animal models of chronic HE show neurological alterations similar to those in patients with HE
Hyperammonemia is considered the main contributor to the pathogenesis of HE, but the mechanisms by which liver failure and hyperammonemia lead to the associated neurological alterations are not well understood.
To try to study these mechanisms a few animal models are being used. The most usual model to study the neurological alterations in HE is the rat with chronic liver failure induced surgically by portacaval anastomosis. In this model a surgical shunt is constructed between the portal and the inferior cava veins. Thus blood does not pass through the liver, and ammonia and other substances are not properly detoxified, can reach the brain, and lead to neurological alterations.
This animal model reproduces some of the neurological alterations found in patients with HE. Rats with portacaval anastomosis show impaired circadian rhythms of locomotor activity and food intake (CitationZee et al 1991; CitationSteindl et al 1996; CitationCordoba et al 1997; CitationLozeva et al 2000, Citation2002; CitationLopez et al 2002).
Reduced motor activity is also observed in rats with portacaval anastomosis, which is similar to the motor slowing, hypokinesia and bradykinesia present in patients with HE (CitationBengtsson et al 1986, Citation1989; CitationMartin et al 1986; CitationApelqvist et al 1998, Citation1999; CitationLozeva et al 2000). Reduced motor activity has been also observed in rats with bile-duct ligation, another model of HE (CitationChan et al 2004).
Learning ability is reduced in animal models of HE
Cognitive function is also altered in animal models of chronic liver disease. When normal rats are moved from the cage where they usually live and are placed in a novel cage, they start to explore it, resulting in increased motor activity. If rats are placed in the same cage during consecutive days, the exploratory behaviour decreases since the animal remembers the novel cage and the interest for it lowers. In rats with portacaval anastomosis this long-term habituation is impaired. This has been interpreted as consequence of a possibly impaired learning–memory capacity in these rats (CitationApelqvist et al 1999).
A more direct demonstration of impairment of learning ability in rats with portacaval anastomosis has been reported recently by CitationErceg et al (2005a), who showed that the ability of these rats to learn a conditional discrimination task in a Y maze is lower than that of control rats.
Rats with biliar obstruction (another model of HE) show impaired learning ability in a working memory test consisting of an object recognition task, ie, show decreased ability to discriminate between novel objects and previously known objects (CitationGarcia-Moreno et al 2005). Working memory is also altered in patients with HE (CitationElithorn et al 1975).
As mentioned above, hyperammonemia is considered the main factor contributing to the pathogenesis of HE. However, liver failure induces, in addition to hyperammonemia, other alterations (such as decreased muscle mass, altered metabolism of other compounds). To discern the contribution of hyperammonemia to the neurological alterations in HE, we developed an animal model of chronic hyperammonemia without liver failure:rats fed an ammonium-containing diet (CitationAzorin et al 1989). These rats present a level of hyperammonemia similar to that of patients with liver cirrhosis or of rats with portacaval anastomosis, but do not present other alterations associated with liver failure and may be considered therefore as a model of “pure” hyperammonemia.
Learning of a conditional discrimination task in a Y maze is impaired in these rats with chronic hyperammonemia without liver failure (CitationAguilar et al 2000) as well as in rats with chronic liver failure due to portacaval anastomosis (CitationErceg et al 2005a).
In patients with liver disease, increase in hyperammonemia worsens (and reduction of hyperammonemia improves) neuropsychiatric and neurological scores in patients with HE.
In summary the above studies show that animal models of HE reproduce some of the neurological alterations found in patients with HE and are therefore adequate to study the molecular mechanisms by which liver failure leads to these neurological alterations. Moreover, the above studies also show that chronic hyperammonemia is responsible for at least some of the cognitive alterations observed in HE.
Molecular mechanisms modulating learning
The studies summarized above show that cognitive function and learning ability are impaired both in patients with liver disease and HE (either minimal, non clinical HE) and in animal models of hyperammonemia and liver failure.
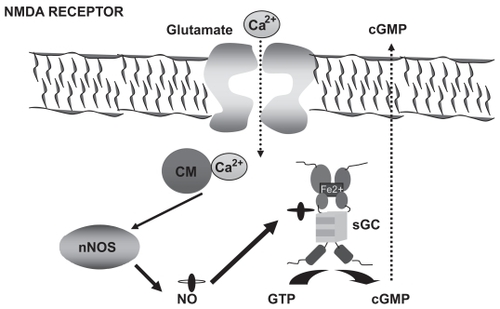
To try to develop appropriate therapeutic treatments to restore cognitive function it is important to understand the molecular mechanisms that modulate learning ability as well as the mechanisms by which these processes are altered in hyperammonemia and HE. The molecular bases for different types of learning are not well known. It has been proposed that N-methyl-D-aspartic acid (NMDA) receptors play a crucial role in some types of learning. However, the molecular mechanisms by which activation of NMDA receptors modulate learning remain unclear. Activation of NMDA receptors leads to increased calcium in the post-synaptic neuron. Calcium binds to calmodulin and activates neuronal nitric oxide synthase, increasing nitric oxide (NO), which activates soluble guanylate cyclase, increasing guanosine 3′,5′-(cyclic)phosphate (cGMP). Part of this cGMP is released to the extracellular space (). Several reports suggest that activation of this glutamate–NO–cGMP pathway is involved in some forms of learning (CitationDanysz et al 1995; CitationChen et al 1997; CitationMeyer et al 1998).
Figure 1 The glutamate–NO–cGMP pathway. Activation of ionotropic (mainly NMDA) glutamate receptors leads to increased intracellular calcium (Ca2+) which, after binding to calmodulin (CM), activates neuronal nitric oxide synthase (nNOS) leading to increased production of NO, which in turn activates soluble guanylate cyclase (sGC), resulting in increased formation of cyclic 3′-5′-guanosine monophosphate (cGMP) from guanosine-5′-triphosphate (GTP). Part of the cGMP formed is released to the extracellular space.
NMDA receptors modulate some kinds of learning
There is a large amount of evidence that activation of NMDA receptors is involved in some forms of learning. We summarize below some of the data available.
The most frequent experimental approach to analyze the role of NMDA receptors in learning processes is the study of the effects of blocking NMDA receptors with selective antagonists on the ability of experimental animals to learn different kinds of tasks.
This kind of study has shown that both competitive and noncompetitive antagonists of NMDA receptors alter learning and memory processes in a Y-maze (CitationParada-Turska and Turski 1990; CitationMaurice et al 1994). Administration of NMDA receptor antagonists also alters learning of different kinds of tasks: passive avoidance (CitationRiekkinen et al 1996; CitationSmith et al 1997), active avoidance (CitationDelay 1996; CitationRedolat et al 1998), spacial tasks (CitationMurray and Ridley 1997), classical conditional tasks (CitationXu 1997), and 14-unit T-maze (CitationPatel et al 1998) and spatial learning in the Morris Water Maze (CitationMorris et al 1986; CitationPackard and Teacher 1997).
In addition to the studies using antagonists, there are genetic studies that support a role of NMDA receptors in certain types of learning (CitationMcHugh et al 1996; CitationTsien et al 1996; CitationHuerta et al 2000; CitationRampon and Tsien 2000). Mice that do not express the NMDA receptor subunit NR2A have decreased spatial learning ability (CitationSakimura et al 1995). On the other hand, overexpression of the NR2B subunit in transgenic mice increases NMDA receptor activation and improves learning and memory (CitationTang et al 1999).
It is therefore clear that NMDA receptors play a role in learning. The subsequent steps by which activation of NMDA receptors mediates learning processes are not so clear. Some reports suggest that activation of the glutamate–NO–cGMP pathway associated to NMDA receptors is involved in some forms of learning.
Role of NO, soluble guanylate cyclase, and cGMP in learning
Activation of NMDA receptors leads to activation of NO synthase and to increased formation of NO. NO seems to mediate at least part of the role of NMDA receptors in learning. This is supported by studies showing that inhibition of NO synthase reduces learning of some spatial tasks: 14-unit T-maze (CitationIngram, Spangler, Kametani, et al 1998; CitationIngram, Spangler, Meyer, et al 1998) or radial maze (CitationZou et al 1998). It also impairs memory consolidation in objects recognition tasks (CitationPrickaerts et al 1997) and learning of passive avoidance tasks (CitationMyslivecek 1997).
The role of NO in learning may be mediated by its activation of soluble guanylate cyclase and the increase in cGMP. Some reports indicate that soluble guanylate cyclase and cGMP are important in learning and memory. Bernabeu et al showed that, in rats, passive avoidance learning was associated with a time-dependent, learning-specific increase in cGMP (CitationBernabeu et al 1996) and in cGMP-dependent protein kinase activity in the hippocampus (CitationBernabeu et al 1997).
The same group also showed that administration of a membrane permeable analog of cGMP facilitated memory consolidation (CitationBernabeu et al 1996), while bilateral intrahip-pocampal administration of an inhibitor of soluble guanylate cyclase caused full amnesia for inhibitory avoidance learning when given immediately after training (CitationBernabeu et al 1997). These results support a role for soluble guanylate cyclase and cGMP in learning and memory.
CitationSmith et al (2000) tested later whether activation of soluble guanylate cyclase and increased cGMP formation in the brain would improve learning in cognitively impaired animals. They showed that a nitrate ester that activates soluble guanylate cyclase improved learning in scopolamine-pretreated animals in a time- and dose-dependent manner. The authors suggested that stimulation of cerebral soluble guanylate cyclase might be an effective strategy to improve learning and memory performance in individuals in whom cognitive abilities are impaired.
CitationYamada et al (1996) also showed that blocking NMDA receptors with dizocilpine or inhibiting NO synthase impaired spatial working memory in mice and suggested that the reduction in NO–cGMP production in the brain may be responsible for dizocilpine-induced impairment of learning.
On the other hand, administration of zaprinast, a selective inhibitor of the phosphodiestease that degrades cGMP, improves early stages of object recognition memory consolidation (CitationPrickaerts et al 1997). Microinjection of 8 Br-cGMP, a membrane permeable analog of cGMP, into the dorsal hippocampus also improves performance in object recognition tasks (CitationPrickaerts et al 2002).
These data indicate that the increase in cGMP produced by guanylate cyclase in response to NO has a crucial role in some types of learning.
Chronic hyperammonemia with or without liver failure impairs glutamate–NO–cGMP pathway in rat cerebellum in vivo
Chronic moderate hyperammonemia in rats, similar to that present in patients with liver cirrhosis, impairs the glutamate–NO–cGMP pathway in the cerebellum in vivo, as shown by brain microdialysis in freely moving rats by CitationHermenegildo et al (1998). Microdialysis probes were inserted in the cerebellum of control or hyperammonemic rats without liver failure. Administration of NMDA through the microdialysis probe activates the glutamate–NO–cGMP pathway and increases cGMP formation. Part of the cGMP formed is released to the extracellular fluid (), and the increase in extracellular cGMP is a good measure of the function of the glutamate–NO–cGMP pathway in the cerebellum in vivo. CitationHermenegildo et al (1998) showed that the NMDA-induced increase in extracellular cGMP in the cerebellum was significantly lower in hyperammonemic rats than in control rats, indicating that chronic hyperammonemia impairs the glutamate–NO–cGMP pathway in the rat cerebellum in vivo ().
Figure 2 Hyperammonemia impairs the function of the glutamate–NO–cGMP pathway in the cerebellum in vivo by reducing NO-induced activation of soluble guanylate cyclase. The effects of NMDA or SNAP on extracellular cGMP in the cerebellum of control, hyperammonemic rats without liver failure (2a and 2b) and with chronic liver failure (2c) were analyzed by in vivo microdialysis in freely moving rats. NMDA (0.1 and 0.3 mM) or SNAP (0.3 mM) was administered in the perfusion stream for 20 minutes at the times indicated by the horizontal bars. Perfusion was carried out at 3 μL/minute, samples were collected every 20 minutes, and cGMP was determined. Data are presented as a percentage of basal values. Data were analysed using two-way ANOVA.
a intragroup comparison values significantly different from baseline (p < 0.05 by Kruskal-Wallis test with post hoc Dunnett’s test; * values significantly different from control (p < 0.05).
Figure 2a and 2b from: CitationHermenegildo C, Montoliu C, Llansola M, et al. 1998. Chronic hyperammonemia impairs glutamatenitric oxide-cyclic GMP pathway in cerebellar neurons in culture and in the rat in vivo. Eur J Neurosci, 10:3201–9;
Figure 2c from: CitationMonfort P, Corbalán R, Martínez L, et al. 2001. Altered content and modulation of soluble guanylate cyclase in the cerebellum of rats with portacaval anastomosis. Neuroscience, 104:1119–25.
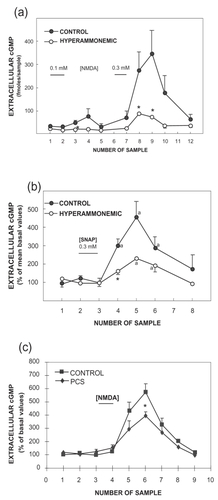
To assess whether the impairment occurs at the level of activation of soluble guanylate cyclase by NO, a NO-generating agent, S-nitroso-d,l-penicillamine (SNAP), was administered through the microdialysis probe to activate directly guanylate cyclase. The increase in extracellular cGMP induced by SNAP was also significantly reduced in hyperammonemic rats (). This indicates that chronic moderate hyperammonemia impairs activation of soluble guanylate cyclase by NO in the cerebellum in vivo, resulting in impairment of the glutamate–NO–cGMP pathway.
Chronic liver failure, induced by portacaval anastomosis, also impairs the glutamate–NO–cGMP pathway in the cerebellum in vivo, as shown by brain microdialysis in freely moving rats by CitationMonfort et al (2001) (). NMDA-induced increase in extracellular cGMP in the cerebellum was significantly lower in rats with portacaval anastomosis than in control rats, indicating that chronic liver failure impairs the function of the glutamate–NO–cGMP pathway in the rat cerebellum in vivo.
These results indicate that the function of the glutamate–NO–cGMP is altered in brain in vivo in animal models of chronic liver failure and of chronic hyperammonemia. Moreover, the step of the pathway mainly affected is the activation of soluble guanylate cyclase by NO.
Modulation of soluble guanylate cyclase by NO is altered in cerebral cortex and cerebellum of patients who died from HE
To assess whether activation of soluble guanylate cyclase by NO is also altered in cirrhotic patients with HE, we measured activation of soluble guanylate cyclase by the NO-generating agent SNAP in homogenates of the frontal cortex or cerebellum from controls and from cirrhotic patients who died from hepatic coma.
The activation of guanylate cyclase by the NO-generating agent SNAP was significantly lower (63%) in the cerebellum from cirrhotic patients than in controls. In contrast to the cerebellum results, activation of guanylate cyclase by NO was higher (about 200% of controls) in the frontal cortex from patients than in controls (CitationCorbalán et al 2002).
The above results show that activation of soluble guanylate cyclase by NO is altered both in the cerebral cortex and cerebellum of patients who died from hepatic coma. However, the effects are opposite in these areas, showing increased activation in the cortex and decreased activation in the cerebellum (CitationCorbalán et al 2002).
The mechanism by which the same pathological situation (liver cirrhosis) can lead to opposite effects in the cerebellum and cerebral cortex is not yet clear. The results obtained suggest that some intrinsic factor in the neurons, different in each cerebral area, may lead to opposite responses to ammonia of the modulation of guanylate cyclase by NO.
Differential modulation of other processes in the cerebellum and cortex has been previously reported. For example, activation of NMDA receptor induces choline release in cortical neurons but not in cerebellar neurons in culture and this release has been related to excitotoxic cell death (CitationGasull et al 2000). Also, the regulatory subunits of adenosine 3′,5′-cyclic monophosphate (cAMP)-dependent protein kinases are differentially expressed in cortical and cerebellar neurons leading to a differential ability of these neurons to transmit cAMP signals to the nucleus (CitationPaolillo et al 1999). Also the role of neuronal NO in the coupling between cerebral blood flow and local neuronal activity is different in these cerebral areas. NO is important in neurovascular coupling in the cerebellum but its role in the neocortex is less important, and it seems that in this area other neurotransmitters are more relevant in neurovascular coupling (CitationHayashi et al 2002). It is not surprising therefore that liver cirrhosis may affect differentially the modulation of soluble guanylate cyclase in the cerebellum and cortex.
Animal models of chronic hyperammonemia and of liver failure reproduce faithfully alterations in modulation of guanylate cyclase by NO found in brain of cirrhotic patients
To study the molecular mechanisms responsible for the alterations of modulation of guanylate cyclase by NO in the cerebral cortex and cerebellum of patients with liver cirrhosis and to test possible therapeutic treatments to reverse this alteration, it is important to have animal models that faithfully reproduce the effects found in human patients. We assessed whether animal models of hyperammonemia with or without liver failure reproduce the alterations in the modulation of guanylate cyclase by NO.
Rats with chronic liver failure due to portacaval anastomosis faithfully reproduce the increased activation of soluble guanylate cyclase by NO in the cerebral cortex and the reduced activation in the cerebellum (CitationMonfort et al 2001; CitationCorbalán et al 2002).
Rats with bile duct ligation plus hyperammonemia also reproduce the alterations in the modulation of soluble guanylate cyclase both in cerebral cortex and in the cerebellum (CitationRodrigo et al 2005). The reduced activation in the cerebellum is also reproduced in rats with chronic hyperammonemia without liver failure (CitationRodrigo et al 2005).
These results show that animal models reproduce faithfully the alterations in the modulation of guanylate cyclase by NO found in brain of patients who died from HE due to liver cirrhosis. Other forms of HE such as Reye’s syndrome or Wilson’s disease are much less frequent and have been less studied, and it is not yet possible by now to compare the neurobiological alterations in these situations with those of cirrhotic patients or to assess the validity of the animal models to study these disorders.
These animal models are therefore adequate to:
study the molecular mechanisms by which liver failure alters the function of the glutamate–NO–cGMP pathway;
assess the functional consequences of this alteration and its contribution to the neurological alterations in HE, for example in learning ability;
test possible therapeutic treatments to normalize the function of the glutamate–NO–cGMP pathway and to normalize the altered neurological functions, for example learning ability.
Pharmacological manipulation of extracellular cGMP concentration in brain restores learning ability in rats with chronic liver failure or chronic hyperammonemia
The data summarized above show that:
cognitive function and learning ability are impaired in patients with liver disease and in animal models of chronic liver failure or of chronic hyperammonemia;
the glutamate–NO–cGMP pathway modulates some forms of learning;
the function of the glutamate–NO–cGMP pathway is impaired in brain in vivo in animal models of chronic hyperammonemia and of chronic liver failure and also in autopsied brain from patients who died from HE
We hypothesized that:
the alterations in the function of the glutamate–NO–cGMP pathway and the decrease in extracellular cGMP in the brain in hyperammonemia and liver disease may be responsible for the impairment in learning ability and intellectual function, and that
pharmacological modulation of extracellular cGMP concentration may restore learning ability in hyperammonemia and HE.
To assess this possibility we tried to reverse the impairment in learning ability of hyperammonemic rats by increasing cGMP. We increased extracellular cGMP by using three different treatments:
continuous intracerebral administration of zaprinast, an inhibitor of the phosphodiesterase that degrades cGMP;
chronic oral administration of sildenafil, an inhibitor of the phosphodiesterase that crosses the blood–brain barrier, and
continuous intracerebral administration of cGMP.
Continuous intracerebral zaprinast increases extracellular cGMP and restores learning ability in hyperammonemic rats
We tested whether increasing cerebral cGMP by inhibiting its degradation is able to restore learning ability in hyperammonemic rats. We administered intracerebrally zaprinast, an inhibitor of the phosphodiesterase that degrades cGMP, to control or hyperammonemic rats, continuously for 28 days, by using osmotic minipumps. Extracellular cGMP was significantly reduced in hyperammonemic rats and treatment with zaprinast increased extracellular cGMP to the same level present in control rats.
We carried out tests of conditional discrimination learning with control and hyperammonemic rats treated or not with zaprinast. Learning ability was significantly reduced in hyperammonemic rats. Continuous intracerebral administration of zaprinast to hyperammonemic rats completely restored the learning ability of these rats (CitationErceg et al 2005b). This indicates that increasing cGMP by inhibiting its degradation restores learning ability in hyperammonemic rats.
The changes in extracellular cGMP are parallel to changes in learning ability, supporting a role for cGMP levels in learning ability.
Oral sildenafil normalizes function of glutamate–NO–cGMP pathway and extracellular cGMP and restores learning ability in rats with chronic liver failure or with hyperammonemia without liver failure
As shown above, zaprinast is effective in restoring learning ability in hyperammonemic rats, but it does not cross the blood–brain barrier and is therefore not suitable for clinical treatment of patients.
We therefore studied whether oral administration of sildenafil, an inhibitor of the phosphodiesterase that degrades cGMP and crosses the blood–brain barrier, restores learning ability in rats with chronic liver failure or with chronic hyperammonemia without liver failure.
Chronic oral treatment with sildenafil normalizes the function of the glutamate–NO–cGMP pathway and extracellular cGMP in rats with portacaval anastomosis and also restored the ability of rats with portacaval anastomosis to learn the Y maze conditional discrimination task (CitationErceg et al 2005a).
Hyperammonemia is one of the main factors contributing to the neurological alterations in HE. To assess the role of hyper-ammonemia in the alterations in learning ability, extracellular cGMP, and function of the glutamate–NO–cGMP pathway observed in rats with portacaval anastomosis, we carried out experiments similar to those reported above using rats with chronic moderate hyperammonemia without liver failure.
Chronic hyperammonemia significantly reduced the ability of rats to learn the conditional discrimination task. Treatment with sildenafil normalized the function of the glutamate–NO–cGMP pathway and extracellular cGMP in hyperammonemic rats without liver failure and restored their ability to learn the conditional discrimination task (CitationErceg et al 2005a).
Continuous intracerebral administration of cGMP restores learning ability in hyperammonemic rats
To further confirm that changes in extracellular cGMP are responsible for the changes in learning ability, we tested whether increasing only extracellular cGMP without affecting intracellular cGMP is also able to restore learning ability in hyperammonemic rats. To do this we administered cGMP intracerebrally to control or hyperammonemic rats, continuously for 28 days, by using osmotic minipumps. cGMP is not able to cross the cellular membrane and therefore increases cGMP only in the extracellular fluid. We carried out tests of conditional discrimination learning with control and hyperammonemic rats treated or not with cGMP. Continuous intracerebral administration of cGMP to hyperammonemic rats completely restored the learning ability of hyperammonemic rats.
The results summarized above clearly point out a role for extracellular cGMP concentration in the ability of rats to learn the Y maze task. Changes in extracellular cGMP are parallel to changes in learning ability, supporting a correlation between cGMP levels and learning ability.
The possible mechanisms by which extracellular cGMP would modulate learning are not yet clear. Only a few reports suggest some physiological role for extracellular cGMP as a neuroprotector against excitotoxicity (CitationMontoliu et al 1999) and as a modulator of sodium uptake in kidney (CitationSasaki et al 2004). However, the mechanisms involved remain unclear.
Conclusion
In conclusion, the above data indicate that the impairment of learning ability in hyperammonemic rats with or without chronic liver failure is due to impairment of the glutamate–NO–cGMP pathway. As the function of this pathway is also altered in the brain of patients with liver cirrhosis, this alteration should also contribute to the cognitive impairment in these patients.
Moreover, increasing extracellular cGMP by pharmacological means may be a new therapeutic approach to improve learning and memory performance in individuals in whom cognitive abilities are impaired for different reasons, for example, in patients with evident HE and also in patients with minimal (subclinical) HE who present reduced performance in psychometric tests.
The modulation of extracellular cGMP can be achieved by different means: modulation of its synthesis by soluble or particulate guanylate cyclase or of its degradation by phosphodiesterases. There are already pharmacological tools to apply these treatments to patients. Several companies are selling inhibitors of phosphodiesterases that may increase extracellular cGMP and are also investigating new molecules that may modulate cGMP levels. The inhibitors of cGMP-degrading phosphodiesterase (mainly of phosphodiesterase 5) used in the animal studies described above are currently being used for the clinical treatment of erectile dysfunction and of pulmonary hypertension. In fact, sildenafil is the active component of Viagra®.
However, in the case of cirrhotic patients, caution must be taken considering the possible deleterious increase in the existing vasodilatation in liver disease by sildenafil (CitationTzathas et al 2002). It has also been reported that in patients with ascites and cirrhosis, inhibition of phosphodiesterase 5 leads to increased plasma levels of the rennin–angiotensin–aldosterone system (CitationThiesson et al 2005). Therefore additional studies seem to be required to find some safe procedure to increase extracellular cGMP in the brain without inducing deleterious effects in peripheral tissues. Pharmacological manipulation of cGMP in the brain by safe procedures may be a useful treatment to restore cognitive and intellectual functions in patients with overt or minimal HE.
Acknowledgments
This work was supported by grants from the Ministerio de Ciencia y Tecnología (SAF2002-00851) and from Ministerio de Sanidad (Red G03-155) of Spain and by grants from Consellería de Empresa, Universidad y Ciencia, Generalitat Valenciana (Grupos03/001) and GV04B-055 of Generalitat Valenciana.
References
- AguilarMAMinarroJFelipoV2000Chronic moderate hyperammonemia impairs active and passive avoidance behavior and conditional discrimination learning in ratsExp Neurol1617041310686089
- AmodioPQueroJCDel PiccoloF1996Diagnostic tools for the detection of subclinical hepatic encephalopathy:comparison of standard and computerized psychometric tests with spectral-EEGMetab Brain Dis11315278979251
- AmodioPDel PiccoloFMarchettiP1999Clinical features and survival of cirrhotic patients with subclinical cognitive alterations detected by the number connection test and computerized psychometric testsHepatology291662710347105
- AmodioPMarchettiPDel PiccoloF1999Spectral versus visual EEG analysis in mild hepatic encephalopathyClin Neurophysiol11013344410454268
- AmodioPDel PiccoloFPettenoE2001Prevalence and prognostic value of quantified electroencephalogram (EEG) alterations in cirrhotic patientsJ Hepatol35374511495040
- AmodioPMontagneseSGattaA2004Characteristics of minimal hepatic encephalopathyMetab Brain Dis192536715554421
- ApelqvistGHindfeltBAnderssonG1998Diurnal and gender effects by chronic portacaval shunting in rats on spontaneous locomotor and rearing activities in an open-fieldBehav Brain Res9325329659983
- ApelqvistGHindfeltBAnderssonG1999Altered adaptive behaviour expressed in an open-field paradigm in experimental hepatic encephalopathyBehav Brain Res1061657310595432
- AzorinIMinanaMDFelipoV1989A simple animal model of hyperammonemiaHepatology10311142759549
- BengtssonFNobinAFalckB1986Portacaval shunt in the rat: selective alterations in behavior and brain serotoninPharmacol Biochem Behav241611162426719
- BengstssonFBuggeMHallH1989Brain 5-HT1 and 5-HT2 binding sites following portacaval shunt in the ratRes Exp Med18924956
- BernabeuRSchmitzPFaillaceM1996Hippocampal cGMP and cAMP are differentially involved in memory processing of inhibitor avoidance learningNeuroreport758588730835
- BernabeuRSchroderNQuevedoJ1997Further evidence for the involvement of a hippocampal cGMP/cGMP-dependent protein kinase cascade in memory consolidationNeuroreport8222149243615
- ChanCYHuangSWWangTF2004Lack of detrimental effects of nitric oxide inhibition in bile duct-ligated rats with hepatic encephalopathyEur J Clin Invest34122814764075
- ChenJZhangPZuoP1997Memory-related changes of nitric oxide synthase activity and nitrite level in rat brainNeuroreport8177149189930
- CorbalánRChatauretNBehrendsS2002Region selective alterations of soluble guanylate cyclase content and modulation in brain of cirrhotic patientsHepatology3611556212395325
- CordobaJDupuisJGottsteinJ1997Stenosis of a portacaval anastomosis affects circadian locomotor activity in the rat:a multivariable analysisAm J Physiol273G1218259435546
- CordobaJCabreraJLataifL1998High prevalence of sleep disturbance in cirrhosisHepatology27339459462628
- DanyszWZajaczkowskiWParsonsCG1995Modulation of learning processes by ionotropic glutamate receptor ligandsBehav Pharmacol64557411224354
- DelayER1996Effects of MK-801 on acquisition, retention and cross-modal transfer of active avoidance behaviour in ratsPsychobiology24195201
- ElithornALunzerMWeinmanJ1975Cognitive deficit associated with chronic hepatic encephalopathy and their response to levodopaJ Neurol Neurosurg Psych387948
- ErcegSMonfortPHernandez-ViadelM2005aOral administration of sildenafil restores learning ability in rats with hyperammonemia and with portacaval shuntsHepatology4129930615660436
- ErcegSMonfortPHernandez-ViadelM2005bRestoration of learning ability in hyperammonemic rats by increasing extracellular cGMP in brainBrain Res10361152115725408
- FelipoVButterworthRF2002Neurobiology of ammoniaProg Neurobiol672597912207972
- FerenciPPappasSCMunsonPJ1984Changes in glutamate receptors on synaptic membranes associated with hepatic encephalopathy or hyperammonemia in the rabbitHepatology42596141134
- FerenciPLockwoodAMullenK2002Hepatic encephalopathy–definition, nomenclature, diagnosis, and quantification: final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998Hepatology357162111870389
- Garcia-MorenoLMConejoNMGonzalez-PardoH2005Evaluation of two experimental models of hepatic encephalopathy in ratsBraz J Med Biol Res381273215665999
- GarfinkelDZisapelN1996Liver cirrhosis and circadian rhythmAnn Intern Med1251548678376
- GasullTDeGregorio-RocasolanoNZapataA2000Choline release and inhibition of phosphatidylcholine synthesis precede excitotoxic neuronal death but not neurotoxicity induced by serum deprivationJ Biol Chem27518350710748226
- GilberstadtSJGilberstadtHZieveL1980Psychomotor performance defects in cirrhotic patients without overt encephalopathyArch Intern Med140519217362383
- GroenewegMQueroJCDe BruijnI1998Subclinical hepatic encephalopathy impairs daily functioningHepatology284599657095
- HartmannIJGroenewegMQueroJC2000The prognostic significance of subclinical hepatic encephalopathyAm J Gastroenterol9520293410950053
- HayashiTKatsumiYMukaiT2002Neuronal nitric oxide has a role as a perfusion regulator and a synaptic modulator in cerebellum but not in neocortex during somatosensory stimulation—an animal PET studyNeurosci Res441556512354630
- HermenegildoCMontoliuCLlansolaM1998Chronic hyperammonemia impairs glutamatenitric oxide-cyclic GMP pathway in cerebellar neurons in culture and in the rat in vivoEur J Neurosci10320199786213
- HuertaPTSunLDWilsonMA2000Formation of temporal memory requires NMDA receptors within CA1 pyramidal neuronsNeuron254738010719900
- IguchiHKatoKIIbayashiH1982Melatonin serum levels and metabolic clearance rate in patients with liver cirrhosisJ Clin Endocrinol Metab54102577061695
- IngramDKSpanglerELKametaniH1998Intraventricular injection of N-nitro-L-arginine in rats impairs learning in a 14-unit T- mazeEur J Pharmacol34111169489850
- IngramDKSpanglerELMeyerRC1998bLearning in a 14-unit T-maze is impaired in rats following the systemic treatmentwith N-nitro-L-argininaEur J Pharmacol341199489849
- LockwoodAHYapEWWongWH1991Cerebral ammonia metabolism in patients with severe liver disease and minimal hepatic encephalopathyJ Cereb Blood Flow Metab11337411997506
- LopezLGonzalez-PardoHCimadevillaJM2002Cytochrome oxidase activity of the suprachiasmatic nucleus and pineal gland in rats with portacaval shuntExp Neurol1732758211822891
- LozevaVValjakkaALecklinA2000Effects of the histamine H(1) receptor blocker, pyrilamine, on spontaneous locomotor activity of rats with long-term portacaval anastomosisHepatology313364410655255
- LozevaVTuominenRKMannistoPT2002Effect of repeated L-histidine administration on plasma prolactin and growth hormone levels in ratsInflamm Res51S44512013404
- McCreaMCordobaJVesseyG1996Neuropsychological characterization and detection of subclinical hepatic encephalopathyArch Neurol53758638759982
- McHughTJBlumKITsienJZ1996Impaired hippocampal representation of space in CA1-specific NMDAR1 knockout miceCell871339498980239
- MarchesiniGBianchiGAmodioP2001Factors associated with poor health-related quality of life of patients with cirrhosisGastroenterology120170811208726
- MartinJROettingerRBattigK1986Behavioral effects of experimental portacaval anastomosis measured in Dashiell and radial tunnel maze configurationsPhysiol Behav382143786498
- MauriceTSuTPParishDWNabeshim1994PRE-084, a sigma selective PCP derivative, attenuates MK-801-induced impairment of learning in micePharmacol Biochem Behav49859697886099
- MeyerRCKnoxJPurwinDA1998Combined stimulation of the glycine and polyamine sites of the NMDA receptor attenuates NMDA blockade-induced learning deficits of rats in a 14-unit-T-mazePsychopharmacology13529059498733
- MonfortPCorbalánRMartínezL2001Altered content and modulation of soluble guanylate cyclase in the cerebellum of rats with portacaval anastomosisNeuroscience10411192511457595
- MontoliuCLlansolaMKosenkoE1999Role of cyclic GMP in glutamate neurotoxicity in primary cultures of cerebellar neuronsNeuropharmacology3818839110608283
- MoroniFLombardiGMonetiG1983The release and neosynthesis of glutamic acid are increased in experimental models of hepatic encephalopathyJ Neurochem4085046131108
- MorrisRGHaganJJRawlinsJN1986Allocentric spatial learning by hippocampectomised rats:a further test of the “spatial mapping” and “working memory” theories of hippocampal functionQ J Exp Psychol B38365953809580
- MyslivecekJ1997Inhibitory learning and memory in new-born ratsProg Neurobiol533994309421830
- MurrayTKRidleyRM1997The effect of dizocilpine (MK-801) on conditional discrimination learning in the ratBehav Pharmacol838389832977
- PackardMGTeacherLA1997Posttraining injections of MK-801 procedure a time- dependent impairment of memory in two water maze tasksNeurobiol Learn Mem6842509195588
- PaolilloMFelicielloAPorcelliniA1999The type and the localization of cAMP-dependent protein kinase regulate transmission of cAMP signals to the nucleus in cortical and cerebellar granule cellsJ Biol Chem27465465210037748
- Parada-TurskaJTurskiWA1990Excitatory amino acid antagonists and memory:effect of drugs acting at N-methyl-D-aspartate receptors in learning and memory tasksNeuropharmacology291111162149871
- Parsons-SmithBGSummerskillWHJDawsonAM1957The electroencephalograph in liver diseaseLancet28677113482229
- PatelNSpanglerELGreigNH1998Phenserine, a novel acetylcho-linesterase inhibitor, attenuates impaired learning of rats in a 14-unit T-maze induced by blockade of the N-methyl-D-aspartate receptorNeuroreport917169592071
- PrickaertsJSteinbuschHWSmitsJF1997Possible role of nitric oxide –cyclic GMP pathway in object recognition memory:Effects of 7-nitroindazole and zaprinastEur J Pharmacol337125369430406
- PrickaertsJDe venteJOngW2002cGMP, but not cAMP, in rat hippocampus is involved in early stages of object memory consolidationEur J Pharmacol43683711834250
- RamponCTsienJZ2000Genetic analysis of learning behaviour-induced structural plasticityHippocampus10605911075831
- RedolatRCarrascoMCSimonVM1998Efectos de la administración aguda de MK-801, antagonista no competitivo de los receptores NMDA, sobre la evitación activa en ratonesPsicothema1013541
- RehnstromSSimertGHanssonJA1977Chronic hepatic encephalopathy A psychometrical studyScand J Gastroenterol1230511866992
- RiekkinenMStefanskiRKuitunenJ1996Effects of combined block of alpha 1-adrenoceptors and NMDA receptors on spatial and passive avoidance behaviour in ratsEur J Pharmacol3009168741159
- RikkersLJenkoPRudmanDFreidesD1978Subclinical hepatic encephalopathy:detection, prevalence, and relationship to nitrogen metabolismGastroenterology754629680502
- RodrigoRJoverRCandelaA2005Bile duct ligation plus hyper-ammonemia in rats reproduces the alterations in the modulation of soluble guanylate cyclase by nitric oxide in brain of cirrhotic patientsNeuroscience1304354315664700
- Romero-GomezMBozaFGarcia-ValdecasasMS2001Subclinical hepatic encephalopathy predicts the development of overt hepatic encephalopathyAm J Gastroenterol9627182311569701
- SakimuraKKutsuwadaTItoIManabe1995Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunitNature37315157816096
- SasakiSSiragyHMGildeaJJ2004production and role of extracellular guanosine cyclic 3',5' monophosphate in sodium uptake in human proximal tubule cellsHypertension432869114718358
- SchomerusHHamsterW1998Neuropsychological aspects of portal-systemic encephalopathyMetab Brain Dis133617710206827
- SchomerusHHamsterW2001Quality of life in cirrhotics with minimal hepatic encephalopathyMetab Brain Dis16374111726087
- SchomerusHHamsterWBlunckH1981Latent portasystemic encephalopathy. I. Nature of cerebral functional defects and their effect on fitness to driveDig Dis Sci26622307249898
- SmithRDGrzelakMECoffinVL1997Reduction of dizocilpine and scopolamine-induced deficits in avoidance responding by SCH 54388, a metabolite of felbamatePharmacol Biochem Behav58657649329055
- SmithSDringenbergHCBennettBM2000A novel nitrate ester reverses the cognitive impairment caused by scopolamine in the Morris water mazeNeuroreport113883611117508
- SrivastavaAMehtaRRothkeSP1994Fitness to drive in patients with cirrhosis and portal-systemic shunting:a pilot study evaluating driving performanceJ Hepatol21102387699223
- SteindlPEFinnBBendokB1995Disruption of the diurnal rhythm of plasma melatonin in cirrhosisAnn Intern Med12327477611593
- SteindlPECoyDLFinnB1996A low-protein diet ameliorates disrupted diurnal locomotor activity in rats after portacaval anastomosisAm J Physiol271G555608897872
- TangYPShimizuEDubeGRRampon1999Genetic enhancement of learning and memory in miceNature40163910485705
- TarterREHegedusAMVan ThielDH1984Nonalcoholic cirrhosis associated with neuropsychological dysfunction in the absence of overt evidence of hepatic encephalopathyGastroenterology86142176714571
- TarterREArriaAMCarraJ1987Memory impairments concomitant with nonalcoholic cirrhosisInt J Neurosci3285393596928
- ThiessonHCJensenBLJespersenB2005Inhibition of cGMP-specific phosphodiesterase type 5 reduces sodium excretion and arterial blood pressure in patients with NaCl retention and ascitesAm J Physiol288F104452
- TsienJZHuertaPTTonegawaS1996The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memoryCell871327388980238
- TzathasCChristidouALadasSD2002Sildenafil (viagra) is a risk factor for acute variceal bleedingAm J Gastroenterol97185612135063
- Van der RijtCCSchalmSW1992Quantitative EEG analysis and evoked potentials to measure (latent) hepatic encephalopathyJ Hepatol1414121500675
- WangVSaabS2003Ammonia levels and the severity of hepatic encephalopathyAm J Med114237812637141
- WeissenbornKHeidenreichSEnnenJ2001Attention deficits in minimal hepatic encephalopathyMetab Brain Dis16131911726083
- WeissenbornKHeidenreichSGiewekemeyerK2003Memory function in early hepatic encephalopathyJ Hepatol39320512927916
- XuX1997NMDA receptor antagonist MK-801 selectively impairs learning of the contiguity of the conditioned stimulus and unconditioned stimulus in goldfishPharmacol Biochem Behav5849169300610
- YamadaKHiramatsuMNodaY1996Role of nitric oxide and cyclic GMP in the dizocilpine-induced impairment of spontaneous alternation behavior in miceNeuroscience74365748865189
- ZeePCMehtaRTurekFW1991Portacaval anastomosis disrupts circadian locomotor activity and pineal melatonin rhythms in ratsBrain Res56017221760725
- ZouL-BYamadaKTanakaT1998Nitric oxide synthase inhibitors impair reference memory formation in a radial arm maze task in ratsNeuropharmacology37323309681930