Abstract
Alzheimer’s disease (AD) is a leading cause of chronic dementia in the US. Its incidence is increasing with an attendant increase in associated health care costs. Since its first description in a patient by Dr. Alois Alzheimer over a century ago, a large body of biomedical literature has established a detailed clinical and molecular profile of this disorder. Amyloid β peptide (Aβ; a 39–42 amino acid molecule) is the major component of senile plaques, the lesions that are one of the pathologic hallmarks of AD (CitationWong et al 1985). Although many aspects of the biology of amyloid β have been investigated, several fundamental questions about how this peptide causes AD neuropathology remain unanswered. The key question is: How is Aβ toxic to cerebral neurons? Because plaques are extra-neuronal deposits, it is difficult to imagine a structural basis for their toxicity. As an interesting contrast the other pathognomonic feature of AD, neurofibrillary tangles, are intra-axonal structural anomalies that are composed of the hyperphosphorylated microtubule associated (MAP) protein, tau. This review will assess the current thinking that relates to a recent hypothesis of Aβ toxicity. In 1992, Hardy and Higgins reported findings that suggested a new and intriguing possibility. These authors found that Aβ peptides disrupt Ca2+ homeostasis in neurons and increase intracellular Ca2+ [Ca2+]i. This was corroborated by Mattson and his colleagues who demonstrated that Aβ exposure to human cortical neurons raised [Ca2+]i (CitationMattson, Cheng et al 1992); (CitationHardy and Higgins 1992). Finally, Nelson Arispe’s group at the NIH specifically investigated the possibility that Aβ peptides might function like Ca2+ ion channels (CitationArispe et al 1993). This and several subsequent studies have laid the foundation for a novel idea: “Aβ peptides are, in part, toxic to neurons because they form aberrant ion channels in neuronal membranes and thereby disrupt neuronal homeostasis”. In this review we shall critically examine this theory in light of classic and contemporary literature.
Introduction
Dementia of the Alzheimer’s type
Dr. Alois Alzheimer, a Bavarian psychiatrist, interviewed a patient in 1901. This was a 51 year-old woman who had declining cognitive abilities and memory. Her neurological evaluation by Alzheimer, Emil Kraeplin and others first helped define this clinicopathological syndrome of late-life mental decline. However, it was much later (in the decade of the 1960s) that the work of Tomlinson et al established Alzheimer’s disease (AD) as the most common form of senile dementia (CitationTomlinson et al 1968, Citation1970). Today, with exponential advances in fields such as neurology, pathology, biophysics, computer modeling, molecular genetics, and immunochemistry, many aspects of AD, its etiology and underlying mechanisms have been discovered. However, a robust scientific debate still continues about the fundamental mechanistic details of the disorder. It’s not surprising therefore, that a cure has not yet been discovered, even in the face of an increasing health care burden and near epidemic proportions of the epidemiology of the disease in the United States and across the western hemisphere.
Amyloid β metabolism
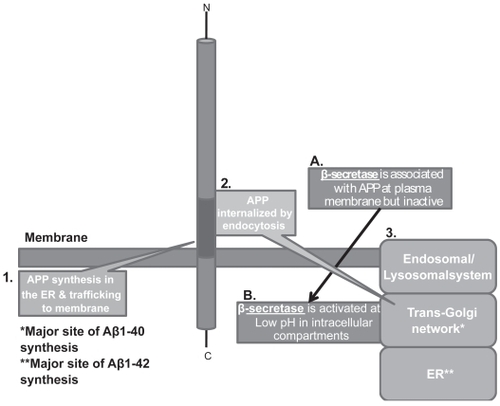
How Aβ accumulates in the brain is a complex narrative. Briefly, a locus on the human chromosome 21 (21q21.3) encodes a transmembrane glycoprotein called APP (Amyloid Precursor Protein). This 695–770 amino acid peptide does not have an established physiological function. As a consequence of metabolic processing, this molecule can generate both amyloidogenic (ie, having a propensity to produce Aβ) and non-amyloidogenic products (CitationFraser et al 1997). If APP is proteolytically cleaved by the enzyme α-secretase, the neuroprotective fragment sAPPα (for secreted APPα) is produced and Aβ formation is prevented (CitationPearson and Peers 2006). In contrast, if sequential cleavage by β- and then γ-secretases predominates, Aβ is formed. β- and γ-secretase cleavages are constitutive events in that Aβ is detectable in the pico-to-nanomolar range in normal brains (CitationHaass et al 1992; CitationSeubert et al 1993; CitationWilson et al 1999). However, relatively recent research has shown that β-secretase has optimal activity at a pH of ~4.0 (CitationLin et al 2000). The enzyme is usually located on the cell surface where the ambient pH is much higher (~7) forcing it to remain dormant (CitationLin et al 2000; CitationHe et al 2007). The proteolysis of APP by β-secretase necessitates internalization of both molecules through an endocytic mechanism into lysosomes. Here, at more optimal pH, β-secretase then cleaves APP and these intracellular sites become the major locations for the production of Aβ (CitationKoo and Squazzo 1994; CitationCook et al 1997; CitationHartmann et al 1997). Specifically, APP proteolysis following internalization occurs in:
The Endoplasmic Reticulum: This is the major site of Aβ1–42 synthesis (CitationWilson et al 1999).
The Trans-Golgi Network: Where most of the Aβ1–40 is generated (CitationWilson et al 1999).
The Endosomal/Lysosomal system: This contributes only minor amounts of Aβ (CitationWilson et al 1999).
There may be two distinct fates of secreted Aβ peptides. Indirect evidence suggests that in the normal brain these peptides are cleared from the extracellular space. In humans who inherit the Apolipoprotein E4 (ApoE4) allele (and this is known to increase their risk of developing AD), there is no apparent increase in Aβ production (eg, plasma Aβ levels in these subjects remain normal). However, in human neuropathological studies as well as mouse models, inheritance of ApoE4 causes a rise in the steady state levels of Aβ in the brain. This either happens by an enhancement of the fibrillogenic potential or be decreasing the clearance of Aβ (CitationSelkoe 2001).
Metabolic labeling studies have also demonstrated that newly formed Aβ is rapidly turned over in the brain and that Aβ-degrading proteases help regulate its levels in the brain (CitationSavage et al 1998). These enzymes are largely serine or metalloproteases and include insulin degrading enzyme (IDE), neprilysin (NEP), plasmin, plasminogen activator (uPA/tPA), endothelin enzyme-1 and matrix metalloproteinase-9 (MMP-9) (CitationSelkoe 2001).
Reports have suggested that their may be two distinct pools of Aβ. The intracellular pool remains unsecreted and is possibly degraded while the secreted pool, via the Golgi and post-Golgi vesicles and the Endosomal/Lysosomal system is secreted and this fraction is destined to contribute to the formation of senile plaques in the brains of AD patients. The peculiarities of the aging brain that predispose neurons in general to this fate of Aβ and to specific populations within the cortex, are not well understood and might hold the key to the overarching causality of AD (CitationWild-Bode et al 1997; CitationWilson et al 1999). , and schematically summarize APP metabolism, Aβ synthesis and the ultimate fates of these peptides.
Figure 1 APP proteolytic processing and major fates of the amyloid β (Aβ) fragment. Sequential cleavage by β-secretase (BACE-1) followed by γ-secretase [containing Presenilin 1 and 2 (PS1/2)] generates Aβ. This fragment has several fates. (a) It can aggregate and accumulate as extraneuronal plaques which characterize Alzheimer’s dementia. (b) It can be cleared from the extracellular space (a mechanism that may be altered in AD brains). (c) It can be degraded by a variety of proteases such as Insulin Degrading Enzyme (IDE), Neprilyisn (NEP), Plasmin, Plasminogen Activator (uPA/tPA), Endothelin Enzyme-1 or Matrix Metalloprotease-9 (MMP-9). Hypothetically, at least, it might return to the plasma membrane and insert in oligomer form as an ion channel. γ-secretase cleavage is also thought to liberate an intracellular domain (AICD) which may influence gene transcription.
![Figure 1 APP proteolytic processing and major fates of the amyloid β (Aβ) fragment. Sequential cleavage by β-secretase (BACE-1) followed by γ-secretase [containing Presenilin 1 and 2 (PS1/2)] generates Aβ. This fragment has several fates. (a) It can aggregate and accumulate as extraneuronal plaques which characterize Alzheimer’s dementia. (b) It can be cleared from the extracellular space (a mechanism that may be altered in AD brains). (c) It can be degraded by a variety of proteases such as Insulin Degrading Enzyme (IDE), Neprilyisn (NEP), Plasmin, Plasminogen Activator (uPA/tPA), Endothelin Enzyme-1 or Matrix Metalloprotease-9 (MMP-9). Hypothetically, at least, it might return to the plasma membrane and insert in oligomer form as an ion channel. γ-secretase cleavage is also thought to liberate an intracellular domain (AICD) which may influence gene transcription.](/cms/asset/c0d5347e-fc48-4e56-ab0b-60aa2289b5d5/dndt_a_12160207_f0001_b.jpg)
Figure 2 Sequential cleavage by β-secretase and γ-secretase releases Aβ fragment of varying length. β-secretase cleavage has to occur following internalization of APP-enzyme complex at pH values around 4.0 in cytosolic locations.
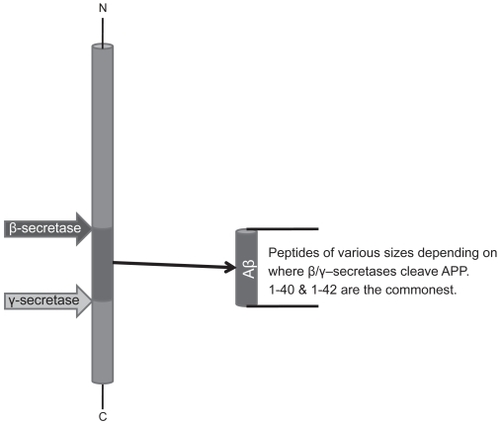
Figure 3 APP internalization and generation of Aβ. APP is trafficked through constitutive secretory pathways, undergoes post-translational modification and ultimately locates to the plasma membrane. Poorly understood mechanisms/signals then effect internalization/endocytosis of APP to intracellular sites where optimum pH exists for activation of is β-secretase (BACE-1) that process APP.
Aside from senile plaques, the other characteristic lesions of AD are neurofibrillary tangles which are intra-cellular structures composed of hyperphosphorylated tau protein. The ongoing debate in AD research is the connection between these neuron-associated anomalies and the ultimate emergence of the Alzheimer’s clinical syndrome (CitationBlessed et al 1968; CitationNeve et al 1990; CitationKatzman and Saitoh 1991). Amyloid plaques are not simple structures composed of a single molecular population (ie, Amyloid β). In fact, a variety of substances have been found in these lesions. These include:
Proteoglycans, including heparan, chondroitin, keratin and dermatan sulphate proteoglycans.
Inflammatory molecules, including acute phase proteins, cytokines, chemokines, complement proteins, complement inhibitor.
Serum related molecules, including amyloid P component.
Metal ions, including Fe, Cu, Zn.
Amyloidogenic related molecules, including non-Aβ component of AD amyloid (NAC) that represents about 10% of the non-SDS-soluble material in amyloid plaques. Apolipoprotein E, low density lipoprotein receptor-related protein], cystatin-C (gamma-trace peptide).
Protease and clearance related elements, including α1-antichymotrypsin, α1-trypsin, lysosomal proteinases, ubiquitin, α2-macroglobulin.
Antioxidant defense proteins, including ferritin, ceru-loplasmin, SOD-1 and SOD-2, HO-1 and possibly catalase.
Cholinesterases, including acetylcholinesterase and butyrylcholinesterase.
Other proteins, including the multifunctional clusterin (Apo-J, SP-40).
[For an excellent and very comprehensive overview of plaque composition and chemistry, see CitationAtwood and Martins (2002)].
This classical description of AD pathogenesis (the “amyloid hypothesis of AD”) has certain important caveats. Data has shown that the mature aggregated form of Aβ (amyloid fibrils) which are observed in cortical areas of AD patients, may not be the direct causative agents of neuronal degeneration and death (CitationEliezer 2006). In fact, typical amyloid deposits are absent in certain forms of AD and even when they are evident, may not be toxic or even help in cell survival (CitationTompkins and Hill 1997; CitationArrasate et al 2004). This deviation from conventional wisdom has, in part, led to the emergence of an alternative theory which is sometimes referred to as the “oligomer hypothesis of AD”. This theory posits that while mature amyloid fibrils may not themselves be toxic, some species formed during the process of fibrillar assembly might be the primary toxic entities. These molecules seem to be generated in the synthesis of amyloid fibrils and are Aβ oligomeric structures (CitationCaughey and Lansbury 2003; CitationWalsh and Selkoe 2004).
Physiology of Aβ peptides
Many studies suggest that Aβ might have a physiological function. In fact, Aβ peptide can be detected in human cerebrospinal fluid (CSF) in a variety of isotypes ranging in length from 38 to 42 amino acids. Typically, in healthy individuals, the predominant form of Aβ is Aβ1–40 (about 90% of the total amount) while Aβ1–42 represents approximately a 10% fraction. This ratio is dramatically altered in AD with about even representation of both isotypes (CitationMehta et al 2001; CitationPlant et al 2003). Thus Aβ toxicity might represent a disturbance of normal function.
Evidence for a physiologic role
Aβ peptide appears to be involved in synaptic signaling (CitationPearson and Peers 2006). In 2003, Kamenetz et al reported that Aβ synthesis was increased when hippocampal neurons were activated by evoked potentials. This seemed to occur through increased APP trafficking toward β-secretase sites on the cell membrane which, in turn, increased the probability of β-secretase cleavage (CitationKamenetz et al 2003). In addition to increased Aβ production, other APP fragments were also liberated in greater numbers. It has been hypothesized that this scenario is a negative feedback mechanism where Aβ can (which is also known to inhibit synaptic activity) protect against unregulated signaling and consequent excitotoxicity at the synapse. In fact, Kamenetz observed that when γ-secretase activity was blocked, the frequency of excitatory postsynaptic currents (EPSC) was increased. In addition, an earlier study has also shown that kainate-induced seizures are potentiated in APP-knockout mice (CitationSteinbach et al 1998). Neuronal activity has been shown to modulate both the basal secretion of Aβ as well as the upregulated synthesis noted in genetic APP mutations (such as the Swedish mutation) that are causally linked to certain familial forms of AD. Further and somewhat indirect evidence that supports a physiologic role for Aβ, is the mechanism through which benzodiaz-epines (by enhancing inhibitory neurotransmission) and NMDA receptor antagonists (eg, Memantine) act to slow the cognitive decline of AD (CitationFastbom et al 1998; CitationWinblad and Poritis 1999). More indirect evidence that supports this idea is the enhancement of Ca2+ and K+ ion channel activity that has been observed in rodent cortical neuronal cultures upon application of soluble forms of Aβ (CitationPrice et al 1998; CitationRamsden et al 2002). Recently it has also been reported that NMDA-receptor stimulation also upregulates APP synthesis with a concomitant increase in Aβ levels (CitationLesne et al 2005). As was described in a previous section, sequential cleavage of APP by β- and then γ-secretases creates Aβ fragments. The discovery that the Aβ1–42 fragment was the major component of plaques in AD led to an interest in developing pharmaceutical inhibitors of these enzyme complexes. Some studies have alluded to the toxicity of such inhibitors. For example, CitationPlant et al (2003) have reported that inhibition of β- or γ-secretase in neurons can compromise cell viability. These observations also support a physiological function for Aβ peptides. Indeed, in APP-knockout mice neurological and behavioral deficits are noted even though neurons can be harvested from pre-natal animals and successfully cultured suggesting that even though APP (and possibly Aβ) might have a functional role, they are not mandated for survival (CitationZheng et al 1995; CitationHarper et al 1998; CitationWhite et al 1998).
Because APP has so many proteolytic products, a compelling argument can be made that APP-processing is also a physiological process and that Aβ is part of this complex system. When the delicate balance of this process is disturbed, toxic forms of Aβ proliferate leading to neuronal degeneration (see below).
The toxicity of Aβ peptides
It is now widely accepted that the accumulation of Aβ in cerebral centers involved in cognition and memory is a common feature of AD pathology. These areas are mainly the temporoparietal and frontal cortices, the cingulate gyrus and hippocampus (CitationWenk 2003, Citation2006). Glenner first identified Aβ in meningovascular amyloid deposits in AD patients (CitationGlenner and Wong 1984). Subsequently several other laboratories reported finding this peptide in senile plaques (CitationGorevic et al 1986; CitationSelkoe et al 1986; CitationMann et al 1996) The composition of plaques is attributed largely to the Aβ1–42 form of amyloid β (CitationDickson 1997; CitationSelkoe 2001). However, which form of Aβ is most critical to AD pathogenesis remains the subject of some debate. One of the controversies is that the major component of both diffuse and senile plaques is the Aβ1–42 form and yet this 42-amino acid fragment of APP is a quantitatively minor product of processing. In addition to Aβ1–42, the amino terminus truncated form Aβx–42 is also found in plaques (CitationIwatsubo et al 1994; CitationGravina et al 1995). The heterogeneity of Aβ species is principally linked to where the fragment is terminated. Termination at amino acid 40 produces the slightly shorter Aβ1–40, while at 42, the more amyloidogenic Aβ1–42 is formed. In addition, variant splicing of the N-terminus produces the Aβ x–40 and Aβx–42 fragments (CitationWild-Bode et al 1997).
These facts suggest that the central anomaly in AD might be an alteration of the ratio between the normally predominant form (Aβ1–40) and Aβ1–42 Aβ has intriguingly been observed not only in the classic lesions of AD but also in diffuse plaques that are recognized by amyloid antibodies but do not take on classic amyloid stains (CitationLevites et al 2006). Other Aβ species (most prominently Aβ1–40 and Aβx–40) are also present, especially in cerebrovascular amyloid deposits that are notably seen in the aging brain (CitationGolde et al 2000). Soluble oligomers referred to as Aβ-derived diffusible ligands (ADDLs) are also capable of disturbing neuronal function in rodents and they are observed to accumulate in the brains of AD patients (CitationKlein et al 2004; CitationWalsh and Selkoe 2004). In contrast Aβ can be detected in human brains not affected by AD. For example, Aβ can be measured in the CSF of non-demented human subjects (Aβ1–42) and in the media of normal neuronal cultures (isoform not identified) (CitationHaass et al 1992; CitationTamaoka et al 1997). Thus, it is not merely the presence of any Aβ isoform that pushes the brain toward cognitive decline. More specifically the relative levels of Aβ1–42 predispose to neuronal dysregulation and dementia (CitationYounkin 1998). In addition, Aβ1–42 has been shown to accumulate as amyloid fibrils as well as soluble intermediates more readily than Aβ1–40 (CitationJarrett et al 1993; CitationLevites et al 2006). Exogenous Aβ causes neuronal degeneration in primary cultured neurons (CitationYankner et al 1990). Exogenous Aβ causes the same phenomenon when injected into the brains of adult rats (CitationKowall et al 1991; CitationDe Ferrari et al 2003; CitationMorgan et al 2004; CitationReyes et al 2004). Synthesized Aβ peptides cause both trophic and toxic changes in cultured neurons (CitationWhitson et al 1989; CitationKoh et al 1990; CitationYankner et al 1990; CitationMattson et al 1992; CitationLorenzo and Yankner 1994). Research has also suggested that Aβ might not be directly toxic itself. It may transform the neuron into becoming more susceptible to other background noxious agents. For example, these might be excitatory amino acids or free radicals (CitationKoh et al 1990; CitationMiranda et al 2000). Aβ’s toxicity also appears to be related to its aggregation state. Thus, aggregated forms of Aβ are toxic while solubilized (generally newly synthesized) forms are not (CitationPike et al 1993). Talafous has suggested that the monomeric form of Aβ (in α-helix conformation) is neurotrophic and that when this changes to the oligomeric, β-sheet conformation, the peptide becomes neurotoxic. This hypothesis needs further evaluation (CitationTalafous et al 1994). In an interesting study, CitationRoher and colleagues (1996) were able to quantify the proportion of Aβ monomers, dimmers and trimers as 55:30:15, respectively. Further, monomers and dimmers exhibited toxicity in culture only in the presence of microglia. Without these cells, even a 10-fold increase in dose has failed to exhibit toxic effects. Therefore it is now widely held that the toxicity of Aβ peptides is, in part, related to their aggregation state. This elegantly explains the various deviations in published literature discussed here (CitationMorgan et al 2004).
Intraneuronal Aβ
Another intriguing aspect of Aβ species is that they are also synthesized in subcellular compartments such as the endosomes and lysosomes, particularly but not exclusively in non-neuronal cell systems (CitationHaass et al 1992; CitationWilson et al 2002). Both major forms of Aβ peptide appear to be synthesized in specific subcellular locations. Aβ1–40 is formed in the transgolgi network (TGN) while Aβ1–42 is produced in the endoplasmic reticulum/immediate compartment (ER/IC). The Aβ1–42 is retained inside the cell in both neurons and other cell types and is therefore not destined to be secreted into the extracellular matrix with the possible fate of becoming part of a plaque deposit (CitationCook et al 1997; CitationHartmann et al 1997; CitationWild-Bode et al 1997; CitationXu et al 1997; CitationSkovronsky et al 1998; CitationGreenfield et al 1999). Are these intracellular (or with more relevance, intraneuronal) Aβ fractions toxic as well? There is mounting evidence that suggests this might be the case. The following is a selection of current literature supporting this idea:
Takashi and colleagues (2002) have observed that intra-neuronal Aβ accumulates in multi-vesicular bodies and late endosomes in both the brains of AD patients as well as APP transgenic mice. In a more recent study, CitationOakley and colleagues (2006) have found that Aβ accumulates near the axon hillock in APP/PS1 double transgenic mice and suggest that this might potentially interfere with axonal transport and possibly action potential generation as well.
More directly, investigations of intracellular Aβ have shown that the peptides can reduce presynaptic and post-synaptic markers, cause abnormal neuronal morphology and elevation of p25 [activator of cyclin-dependent kinase 5 (cdk5); an early marker of neuronal degeneration] (CitationUrbanc et al 2002; CitationCruz and Tsai 2004).
It has also been suggested that an over accumulation of Aβ1–42 inside the neuron causes lysis and amyloid plaques represent the sites where this happens. In other words, these lesions are the remnants of lysed neuronal populations (CitationWirths et al 2004). This hypothesis is not without some empirical support. For example:
In neurons in culture, over time there is an increase in intracellular Aβ1–42 as well as an increase in the Aβ1–42/Aβ1–40 ratio. This increase is higher than secreted Aβ (CitationTurner et al 1996; CitationSkovronsky et al 1998).
As noted above, neurofibrillary tangles are the other hallmark lesion of AD and are composed of hyper-phosphorylated tau protein. The protein is observed to be about 20–25 nm wide with periodic twisting and is therefore also referred to as paired helical filaments (PHF). PHF accumulation is one of the earliest events in AD pathology. Intracellular Aβ appears to accumulate even before PHF becomes detectable and, more interestingly, declines as cognitive dysfunction progresses and plaque deposition accelerates (CitationGouras et al 2000; CitationFernandez-Vizarra et al 2004).
In several AD animal models, intraneuronal Aβ correlates with early synaptic dysfunction prior to plaque deposition and the appearance of tangles (CitationTakahashi et al 2002; CitationWirths et al 2002; CitationOddo et al 2003; CitationSchmitz et al 2004).
In animal models in which βAPP, Presenilin-1 (PS1) or Presenilin-2 (PS2) genes are mutated, the intracellular Aβ1–42/Aβ1–40 ratio is increased (CitationWang et al 2006).
Overall, these tantalizing pieces of evidence strongly suggest that Aβ peptides are not an isolated extracellular phenomenon. Instead, there seems to be a dynamic relationship between two pools of Aβ, one inside and the other external to neurons in critical brain regions. Further details of the temporal relationship between these two pools needs further study and should reveal fascinating insights into AD pathology.
Aβ triggers apoptosis or necrosis?
Imaging and molecular studies on human brains affected by AD show ample evidence that there is both focal and diffuse degeneration and loss of neuronal tissue. What is less clear is whether this represents necrosis of cells in the brain or a manifestation of apoptosis. For example CitationBehl and colleagues (1994) observed that PC12 cells when exposed to Aβ (25–35) underwent necrosis (electron microscopy data). In addition and quite interestingly, evidence has been found in humans carrying the presenilin-1 mutation E280A, that neurons undergo necrosis (CitationVelez-Pardo et al 2001). In contrast, several other recent studies seem to implicate apoptosis as the underlying mechanism of neural dysfunction and loss. For example, increased expression of p53 (a widely used marker of apoptotic DNA damage) was noted in neurons exposed to Aβ (CitationCopani et al 2002). More specifically, studies have shown that the expression of the pro-apoptotic effector bax is upregulated and the anti-apoptotic molecule, bcl2 is decreased in AD (CitationParadis et al 1996; CitationTamagno et al 2003). Tamagno’s group have also published results that demonstrate recruitment of p53 and cytosolic release of cytochrome-c as well as activation of caspase-3 (all excellent markers of apoptosis), in Aβ exposed neurons (CitationTamagno et al 2003).
Another confounding aspect of deciding between which of these two phenomena dominate in a given situation is that the distinction between the two has become increasingly less certain. This is evident, for example, from a sharing of signaling pathways between the two processes (CitationMcHugh and Turina 2006). In addition, one of the best studied AD causing gene mutations is in the Presenilin-1 locus on chromosome 14. This has been shown to increase neuronal vulnerability to kainic-acid induced necrosis by one of this review’s co-authors (CitationGuo et al 1999). Furthermore, the intensity of the same type of insult can become decisive in whether apoptosis or necrosis will supervene (CitationDenecker et al 2001). Other situational peculiarities also seem to make a difference. For example a depletion of cellular ATP has been shown to shift the outcome from apoptosis to necrosis (CitationEguchi et al 1997; CitationLeist et al 1997).
Neuronal Ca2+ homeostasis and the role of amyloid β
From the evolutionary standpoint, Ca2+ homeostasis is affected through an ancient cellular system of ion channels, intracellular stores and signaling cascades. This is not surprising because the ion is intimately involved in a wide range of physiological reactions and processes (CitationVerkhratsky 1998; CitationToescu and Verkhratsky 2000). This system utilizes the very large transmembrane Ca2+ gradient which far exceeds that of all the other physiologically relevant ions. Therefore even small changes in Ca2+ conductance in the cell membrane result in large effects on intracellular Ca2+ concentrations [Ca2+]i. Thus the system is one with a very high signal-to-noise ratio.
The influx of Ca2+ ions through voltage-gated plasma membrane channels (VDCC) is a critical signal in neurotransmitter release from the presynaptic terminal as well as subsequent responses by the postsynaptic cell (CitationYuste et al 2000; CitationBurnashev and Rozov 2005; CitationHartmann and Konnerth 2005). The resting neuron maintains a large Ca2+ gradient between the extracellular space and the cytosol. Specific signals such as voltage change or ligand-receptor interactions, effect opening of a variety of Ca2+ channels. These conduits are found both on the plasma membrane (the L, N and some R-type channels) as well as on the surface of intracellular Ca2+ stores (eg, the RyR channels on the SER) (CitationAkita and Kuba 2000; CitationVanterpool et al 2005). These channels cause a dramatic increase in intracellular Ca2+ concentration ([Ca2+]i) and are (depending on the magnitude of the Ca2+ transient) evoke complex buffering systems {eg, Ca2+ buffering proteins, smooth ER Ca2+-ATPases (SERCA), mitochondrial Ca2+ uptake, plasmalemmal Ca2+-ATPases (PMCA) and the Na+/Ca2+ exchanger}. These corrections tend to restore [Ca2+]i to resting levels (CitationWuytack et al 2002; CitationBuchholz et al 2007). The transient rise in [Ca2+]i under normal conditions is very short lived (typically on the order of seconds to minutes). This rise in Ca2+ in turn sets up downstream signaling for a variety of cellular functions such as neurite growth, synaptogenesis, synaptic transmission and plasticity and cell survival (CitationMattson 2007). However in pathological states (such as AD) and even in normal aging, Ca2+ flux and cellular load is poorly controlled. The mechanisms of these disturbances are varied and include oxidative stress (particularly membrane associated oxidative stress; MAOS) and disturbed energy metabolism (CitationMattson 2007). MAOS impairs the function of ion-motive ATPases and glutamate and glucose transporters and this in turn promote membrane depolarization, further Ca2+ influx and glutamate excitotoxicity through over activation of glutamate receptors (CitationMattson 1998; CitationArundine and Tymianski 2004; CitationMattson 2007). provides a summary of native neuronal ion channels.
Table 1 Summary of native neuronal ion channels
Why is Ca2+ so critical to the neuron? More specifically, why is the aging brain of an AD patient more sensitive to Ca2+ perturbations in general and the impact of Aβ in particular? These fascinating questions have direct bearing if one is to be convinced that abnormal ion channels (especially those showing selectivity for cations like Ca2+) might be part of the toxic mechanism behind AD pathology. In brief the following summary of investigation helps to understand why this might be the case:
Evidence suggests that larger Ca2+-dependent after hyperpolarization and increased activity of L-type Ca2+ channels occurs in the aging brain.
Increased Ca2+ release from intracellular stores appears to contribute to cell death and vulnerability in several models of neuronal toxicity.
Some AD-associated gene mutations (eg, Presenilin mutations) appear to alter the expression of intracellular Ryanodine receptors (RyR) which mediate Ca2+ release from intracellular stores.
In hippocampal slices from the aging rat brain, Ca2+-induced Ca2+ release (CICR) seems to be upregulated.
[For a review see CitationThibault et al 2007].
In view of the significant correlation between intra-neuronal accumulation of Aβ and the critical role of Ca2+ ion channels in subcellular structures (such as the ER), it is seductive to think that Aβ’s ion channel forming ability might also extend to intracellular sites. Specifically, what if these peptides were also capable of inserting as aberrant channels in intracellular Ca2+ stores? Could this possibility amplify the perturbations Aβ peptides can induce on the surface of neurons? Not only is this a fascinating idea, it is also very challenging to prove. Some of the difficulty stems from the technical issues of electrophysiological recordings from intracellular organelles. In fact, it was not until 1997 that Jonas and Kaczmarek reported successful results of patch-clamp recordings from intracellular membranes from a variety of cell types including Chinese hamster ovary cells, the giant presynaptic terminal of the squid and rat microglial cells. Prior to this report, electrophysiological recordings from intracellular structures were limited to artificial lipid bilayers or preparations of isolated organelles. However, in addition to the technical difficulty of achieving consistent giga-ohm seal patch-clamps on intracellular membranes, the additional difficulty in getting meaningful electrophysiological data is that the ionic composition of intracellular compartments is not objectively measurable in such experimental models. Thus, short of creating an excised patch and then studying ionic flow across (for instance in an inside-in configuration) the subcellular membrane, convincing characterization of ion channels (gating, conductance, selectivity, etc) cannot be ascertained (CitationJonas et al 1997). Intracellular patch-clamping is a technique in its infancy. In the future, advances in both recording hardware and computer software are likely to advance our understanding of intracellular ion channels in general, and Aβ induced channels in particular.
A selection of literature that supports Ca2+ ion perturbation as a central aspect of AD pathology is presented:
The levels of the calcium binding protein calsenilin are elevated in the cortex of AD patients. This is also noted in the neocortex and hippocampus regions of βAPP mutant transgenic mice as well as neurons in culture (CitationJo et al 2004).
In neurons that are burdened by PHF, increases in Ca2+-dependent proteases and Ca2+-activated kinases have been demonstrated (CitationXiao et al 1996; CitationGrynspan et al 1997).
In both familial and sporadic forms of AD as well as transgenic animal models of the disease, disruptions of Ca2+ homeostatic mechanisms have been reported (CitationEckert et al 2001).
These are very selective examples of a fast growing body of literature that unambiguously implicates Ca2+ and its cellular regulation in AD pathogenesis (CitationMattson et al 1992). In addition Mattson’s group have clearly shown that the aggregation state of Aβ and loss of neuronal Ca2+ homeostasis are directly related (CitationMattson et al 1993). This last point is an important one for the purpose of this review since it lays the foundation for examining the ion channel hypothesis of AD pathophysiology in greater detail.
Aβ toxicity as a result of the formation of abnormal ion channels
Shortly after Mattson et al and Hardy et al had reported perturbed neuronal Ca2+ homeostasis under the influence of Aβ, Nelson Arispe first reported that Aβ formed ion channels in model membranes (CitationHardy and Higgins 1992; CitationMattson et al 1992; CitationArispe et al 1993). The hypothesis that Arispe et al tested was based, principally on Mattson and Hardy’s reports of disturbances in Ca2+ homeostasis. Briefly, certain conditions or triggers (eg, genetic influences, aging, etc) predispose APP proteolysis to produce more Aβ1–42 fragments. This peptide can oligomerize and insert into the cell membrane as a cation-selective ion channel. These channels allow unregulated Ca2+ movement into the cell leading to a breakdown of ionic homeostasis (see below).
Calcium ion alterations induced by Aβ
Since Ca2+ is critical to cell function and therefore tightly regulated, disorders or disruptions in the Ca2+ signaling machinery of the neuron has many deleterious consequences. One of the earliest reports in the literature that demonstrated Ca2+ toxicity with reference to AD, was by Hardy and Higgins. These researchers reported that Aβ disrupted Ca2+ homeostasis and increased [Ca2+]i (CitationHardy and Higgins 1992). Further confirmation of this idea was provided by CitationYoung et al (1986) who reported that [Ca2+]i rose when Aβ was applied to neurons in culture. In an attempt to explain these findings, Nelson Arispe’s group posited a new hypothesis basing it on the assumption that Aβ peptides could be forming Ca2+ conducting channels in cell membranes. This would readily explain Ca2+ anomalies reported by Hardy et al. They tested the idea rigorously and published their findings in 1993. Arispe and colleagues created a de novo planar bilayer of palmitoy-loleolylphosphatidylethanolamine and phosphatidylserine in a plexiglass chamber where channel currents could be recorded across the membrane. In this setup, when millimolar concentrations of Aβ1–40 were applied to the membrane, discrete channel currents were consistently observed within minutes. Standard electrophysiological techniques (such as an analysis of reversal potentials) suggested that the Aβ peptide formed cation-selective channels and that at transmembrane potential negative to −4 mV, Ca2+ moved across the membrane and the kinetics of this translocation was consistent with transport through a Ca2+ channel. These data were very compelling and heralded subsequent work by many other investigators who validated the phenomenon of ion flow (especially Ca2+) across artificial membranes under the influence of Aβ (CitationArispe et al 1993) (see below).
The Aβ ion channel hypothesis tested
Soon after publishing data on ion flow in artificial membranes, Arispe collaborated with the Pollard and Rojas group at the NIH. Together, these investigators modeled the molecular structure of Aβ ion channels. This mathematical/computer simulation theorized that Aβ could form ion channels if it assembled into subunits. Alone, a single Aβ peptide sequence could not be visualized to form an ion pore given the relatively large conductances that had been observed in empirical data (5 nS) and the relatively small size of Aβ (about 40 or 42 amino acids) (CitationDurell et al 1994) The model suggested that oligomeric Aβ subunits could form an ionophore-like assembly that closely resembled native cation channels.
In 1997 Sanderson and Ingram reported that at micromolar concentrations, Aβ25–35 could induce voltage-gated Ca2+ currents in the hNT human teratoma cell line. This was an odd finding because of the different Aβ isoform that was used (25–35 rather than 1–42 or 1–40) and that the authors were interpreting their results to show voltage-gating. This is difficult to reconcile since the current understanding of ion channel voltage sensors is that they are structures formed by pore loops and the S4 transmembrane segment of channel subunits (CitationBezanilla 2000). An analogous structure has not been shown in Aβ channels or channel models. Despite these caveats the Sanderson paper is still an important one because it suggests that channel pore formation might not be limited to only one isotype of Aβ.
In 1997, Arispe and his colleagues further added to data in support the of the “Aβ ion channel” paradigm. In an immortalized human hypothalamic cell line (GT1–7), they used the patch-clamp technique to record currents under voltage-clamp conditions. Aβ1–40 and the reverse sequence 40–1 were both tested in these experiments. Only Aβ1–40 was demonstrated to form cation-selective channels in this cell type. In the previous year (1996), Arispe had already reported that Aβ channel activity could be modulated by Zn2+. Specifically, Zn2+ altered channel kinetics (at 250 μM concentrations) and blocked channel activity at higher concentrations (CitationArispe et al 1996). Subsequently, these investigators were also able to show that Zn2+ could block (Aβ) channel currents when applied to the intracellular face of the cell membrane. This fact was a strong argument in favor of discrete channel formation since such blockade was only thought possible by the metal ion physically obstructing a channel pore (CitationKawahara et al 1997). In 1998, Seung and Lal reported on a collaboration between the University of California at Santa Barbara and Yeungnam University in Korea. These authors looked at Aβ1–42 activity in reconstituted phospholipid vesicles and found that when the vesicles were reconstituted with Aβ1–42, there was a 4-fold increase in radio-labeled Ca2+ and this was prevented in the presence of Tris and, most interestingly, Zn2+ (CitationRhee et al 1998). Zn2+ however, was not the only reported channel blocker in these and similar experiments reported by others. Bruce Kagan’s lab at the University of California Los Angeles published findings in planar bilayers that both Aβ1–40 and Aβ1–42 induced cationic currents and these were not only blocked by Zn2+ but also by the diazo dye, congo red (CitationHirakura et al 1999). These results were further confirmed by Lal’s group in 1999 (CitationLin et al 1999). The next year, Bhatia and Lal further tested Aβ1–40, 1–42 and 25–35 in cultured endothelial cells. Even in this cell system, the Aβ ion channel idea was reinforced. In this study, Aβ1–42 was shown to cause morphologically proven cellular degeneration more readily than the other isoforms and that this effect was concentration dependent. This was also dependent on the presence of Ca2+. As predicted, micromolar amounts of Zn2+ afforded protection from peptide induced cell damage in the study (CitationBhatia et al 2000).
In recent years, more and more work appears to substantiate the data of the 1980s and 90s. For example, data showing an increase in [Ca2+]i in a phaeochromocytoma cell line (PC12) by nanomolar concentrations of Aβ25–35 have been published. This effect was reported to be dependent on extracellular Ca2+ levels. Using a variety of classic Ca2+ channel blockers as well as other compounds, these authors effectively argued that the rise in [Ca2+]i was through Aβ induced channel pores and not secondary to Ca2+ entry through L-type or ligand-gated channels (CitationHuang et al 2000). In reconstituted planar lipid bilayers, Lin and Lal reported that Aβ1–42 formed oligomeric channel like structures (visualized by atomic force microscopy). They further reported that at physiologic nanomolar concentrations Aβ1–42 caused neurite loss in mouse neuroblastoma cells and at micromolar levels this effect was amplified and led to cell death. Their argument was strengthened by experiments where Ca2+ dependence of these Aβ1–42 effects was compellingly demonstrated and protection by the presence of Zn2+ was also established (CitationLin et al 2001).
In 2003 Bahadi et al added Cu2+ to the growing list of substances that could significantly modulate the currents induced by Aβ. Again working with reconstituted artificial membranes as well as liposomes, this group reported that 150 μM of Cu2+ could block outward currents quite effectively (CitationBahadi et al 2003). In 2004, Arispe reported that Aβ1–40 induced currents in lipid bilayers could be blocked by specific peptides that had been designed to block the pore region of the Aβ channel. In this novel paper, Arispe et al designed simple peptides that conformed to the Aβ oligomeric structure of ion channel models (specifically, the pore region). These peptides were typically only 7–11 residues in length and required relatively simple protein chemistry equipment to synthesize. Several of these in-house manufactured proteins were tested in Arispe’s laboratory and some were found to block channel currents. One of several peptides that were tested (designated NA4 by the authors) proved to be the most effective blocker of channel currents. This series of experiments lent strength to the hypothesis that Aβ could insert into planar bilayers and form channel-like pores whose structure could be predicted and activity blocked by specific peptides (CitationArispe 2004). Arispe has continued to publish data in support of the ion channel hypothesis even more recently. For example, in 2006 his lab has investigated Aβ’s toxic effects in cultured cell lines (PC12) with particular reference to apoptosis. His findings can be summarized as follows: (i) increase in [Ca2+]i by nanomolar concentrations of Aβ1–42; (ii) blockade of this effect by NA4 [Arispe’s previously designed Aβ channel blocking peptide]. The creation and testing of several “blocking peptides” in this study was based on the hypothesis that if Aβ was forming channel-like structures on artificial membranes, then complimentary peptide sequences of adjacent Aβ subunits in this channel-like structure would interact with each other and that these would also interact with synthesized peptide sequences designed to complement those on the subunits. Several different peptides were tested in these experiments; (CitationArispe 2004)]; (iii) increased permeability of PC12 cell membrane when exposed to Aβ1–42; (iv) retention of Aβ1–42 on cell surface even after the cell culture medium (that initially introduced the peptide) was removed. This suggested stable insertion of Aβ in the membrane, possibly as an ionophore; (v) reversal of toxic effects to the cells upon removal of Aβ1–42 (CitationSimakova and Arispe 2006).
The basic conflict in literature which supports the “ion channel” hypothesis is clear. The predominant evidence is indirect, is limited to in vitro systems and appears to implicate more that one isotype of amyloid β (the bulk of the evidence comes from studies that have used commercially sourced, exogenous Aβ1–40). The gold standard of electrophysiological studies is the whole-cell, patch-clamp experiment. In the ideal sense, if neuronal cells could be isolated from relevant regions of the brain in AD animal models (especially a hypothetical one that would have a true AD phenotype) and all the native ion channels on such a hypothetical cell could be blocked without altering the physiology of the cell followed by recording ionic currents form these neurons under clamp conditions, one could expect empirical evidence that would be direct, widely accepted and most useful to the understanding of AD. This, however, remains an elusive goal. summarizes important literature presenting data in support of Amyloid β ion channel formation and activity. As is clear from an overview of this body of literature, the experimental design, nature of model systems and results and conclusions of these studies is not consistent. Specific questions that are very critical have not been answered. For example, it is widely accepted that Aβ1–42 is probably the only toxic entity or in the very least, is more toxic to neurons in AD. Its more abundant companion, 1–40, appears to contribute less or even not at all to AD neuronal damage and loss. Which of these has more ion channel forming capabilities?
Table 2 Summary of selected key publications which support, directly or indirectly, the ion channel hypothesis of AD pathogenesis. Note relative dominance of in vitro systems and that the majority of studies utilized exogenous Amyloid β1–40
If Aβ1–40 forms channels more readily then this would ostensibly fly in the face of received wisdom that Aβ1–42 is more toxic. Why might certain cortical areas be prone to aberrant channel formation? APP is not unique to neurons or even to CNS. Why do its proteolytic products not manifest toxicity elsewhere? These questions have been posed by many but remain unanswered.
The counter argument
A strong argument against the idea of Aβ peptides forming ion channels has emerged in very recent literature. James Hall’s laboratory at the University of California at Irvine and their collaborators at the NIH have reported that amyloid oligomers breakdown or significantly reduce the normal dielectric barrier in lipid bilayers. This is in sharp contrast to the ion channel idea because it implies that the plasma membrane might become “leaky” to cations and would then induce qualitatively the same effects as have been reported in recent years (CitationSokolov et al 2006). Sokolov’s interpretation of the data is that Aβ oligomers increase the conductance of lipid bilayers and this has been tested by both anionic as well as cationic probes. These investigators saw no single channel activity in their model. It is difficult to be compelled by a single set of experiments even though Sokolov’s experimental design is perfectly valid. However, more persuasive are the points the authors make in discussing the implications of their work. Quite correctly they identify the two major hurdles that obstruct wider acceptances of the Aβ ion channel hypothesis:
The single channel currents that have been reported in Aβ literature must successfully predict the bulk conductance properties of neuronal membranes.
Current blockade by various agents must also be validated in the context of whole membranes.
Neither of these two phenomena has been tested thus far. Therefore, the overall limitations of the evidence that has been published from many different groups working in parallel in several different institutions may be summarized as:
Much of the evidence that centers on the demonstration of single channel currents, have been recorded either in artificial membranes or in immortalized cell lines.
Ionic conductances of bulk membranes have neither been predicted by single channel properties nor have these been tested.
In vivo experiments on cortical neurons in primary culture from some of the standard animal models of AD, could effectively demonstrate classical properties of cation-selective ion channels. Such experiments have not been performed.
Unequivocal evidence that the reversal of aberrant ionic flow (eg, with selective channel blockers) through hypothesized Aβ ion channels can also attenuate the neuronal toxicity or prevent apoptosis or enhance cell survival in AD models has not been demonstrated.
Clear demonstration of neuronal cell surface expression of oligomerized, channel forming Aβ in vivo has not been done.
Conclusion
The ion channel hypothesis of AD pathophysiology is a rational extrapolation of many sets of data published over several decades. Collectively, the evidence suggests that amyloid β peptides cause neuronal degeneration and apoptosis in AD by inserting in a stable manner into the neuronal membrane as unregulated cationic channels. These abnormal channels ultimately compromise ionic homeostasis of affected neurons.
There are several inherent challenges that will continue to confound investigators who pursue this direction in AD research. As discussed above, Aβ ion channels are an unproven phenomenon at this time. In many ways, it does or potentially can, predict the phenomena observed in the neuropathology of the AD brain. In order to strengthen the “ion channel hypothesis” investigators will have to overcome several distinct challenges. These are:
Though there are many AD animal models, none are entirely satisfactory. For example, the large scale loss of neurons observed in human sufferers of AD has not been duplicated in transgenic models. Overall, a relatively modest degree of cell death has been observed in just one transgenic model (APP transgenic; ~14% neuronal loss) and this too was limited to the CA1 region of the hippocampus (CitationStalder et al 1999; CitationGotz et al 2004). In addition, the exact spatial and temporal sequence of plaque deposition and tangle formation has also not been duplicated with fidelity in these animals. Neuronal culture is the most accessible method of testing native neuronal populations and such experiments are not proximate simulations of what might be happening in the human AD brain. Furthermore, classic single-cell electrophysiological experiments (ie, with the whole-cell patch-clamp technique) is challenging and best suited for studying native channel populations.
Electrophysiological studies in neurons in culture are technically demanding and similar experiments on sub-cellular structures is only at a nascent stage and needs further development (CitationHamill et al 1981; CitationMazzanti et al 1990; CitationKeller and Hedrich 1992; CitationEhrlich et al 1994; CitationJonas et al 1997).
In studies that have been detailed in this review, the methodology involved dissolution of Aβ in water. Dissolved monomeric peptide is then added to solutions bathing lipid bilayers (in setups modified from patch-clamp amplifiers and equipment). Alternatively, the peptide is sonicated in the presence of lipids to form proteoliposomes which are then fused into the bilayer. In addition different peptide fragments are used and the consequent variability of data between laboratories should not be surprising.
The required proof of establishing abnormal ion channels is necessarily stringent. The literature claims that these channels are cation selective and can be blocked. Carrying the concept from artificial membranes to living cells is the required step that needs to be carefully and consistently demonstrated. The criteria for identification might flow through the following scheme:
Identification of single channel currents in relevant model systems.
Testing the channel in a heterologous system, such as Xenopus.
The use of advanced techniques such as reverse genetic approaches and nucleic acid microarray methods to modulate the behavior of these putative structures.
Despite these daunting obstacles, in our opinion, there is a clear advantage in pursuing the “Aβ ion channel hypothesis” because if validated it has the potential of providing an innovative framework upon which AD therapy, especially through pharmacological means, can be designed. Currently the only approved treatments for AD are either cholinesterase inhibitors (such as Aricept®) or the glutamate antagonist, Memantine®. At best these are quite limited in their efficacy to halt or attenuate the progression of Alzheimer’s dementia.
There has been enthusiastic interest in a new pharmaceutical strategy in AD treatment. These are drugs that directly attenuate Amyloid β levels in brains of AD patients. Two of these compounds are now in phase III trials. R-FLURBIPROFEN, one of these, is a single enantiomer of the NSAID, Flurbiprofen. Although this molecule lacks any activity against COX (cyclooxygenase), it is a potent attenuator of Amyloid β (2005 Mar). The other compound is TRAMIPROSATE (ALZHEMED™). In the brain, interaction between Amyloid β and proteoglycans promotes tissue deposition of the peptide. Tramiprosate (3-amino-1-propane-sulfonic acid) was developed as a glycosaminoglycan (GAG) mimetic that would interact with the GAG-binding region of Aβ thereby antagonizing fibrillation. This compound is also in phase III trials (CitationAisen 2005). Another compound that is generating interest is the γ-secretase inhibitor, LY450139. This drug is in Phase II trials. In 2004, working with this compound, Gitter and colleagues demonstrated that LY450139 was a potent, stereoselective inhibitor of Aβ secretion in SWEAPP293 cells (derived from a HEK cell line which overexpressed APP carrying the Swedish mutation, SWEAPP293). In this system, LY450139 was seen to inhibit Aβ via antagonism of γ-secretase activity (CitationGitter et al 2004). If successful in trials, drugs like LY450139 have significant potential to mature into novel treatments of AD and provide additional evidence that the Aβ neuronal load is the direct cause of dementia in this disorder.
AD remains a disease with an increasing health care burden on the US economy. The Alzheimer’s Association reports that 5 million Americans are currently afflicted (of a total population of 300 million) and AD and other dementias impact the economy to the tune of some $148 billion annually by 2007. Therefore any investment of financial or intellectual resources in this area of AD investigation is very likely to pay dividends.
It is also possible that the ion channel hypothesis is either flawed or limited in its scope. In other words, AD is either completely unrelated to abnormal ion channels or ionic flow (the Sokolov paper in 2006 is a case in point) or these anomalies only play a minor role in the mechanism of neuronal degeneration or dysfunction. If the hypothesis remains unproven or is further challenged by contradictory data, it is likely that such a shift in paradigm might unravel the mystery of Aβ toxicity to the extent that an effective treatment of AD becomes a reality.
References
- Alzheimer’s Association2007Alzheimer’s Facts and Figures.” [online]Accessed on March 1st, 2007 URL: http://www.alz.org/alzheimers_disease_alzheimer_statistics.asp
- AisenPS2005The development of anti-amyloid therapy for Alzheimer’s disease: from secretase modulators to polymerisation inhibitorsCNS Drugs19129899616332141
- AkitaTKubaK2000Functional triads consisting of ryanodine receptors, Ca(2+) channels, and Ca(2+)-activated K(+) channels in bullfrog sympathetic neurons. Plastic modulation of action potentialJ Gen Physiol116569772011055998
- ArispeN2004Architecture of the Alzheimer’s A beta P ion channel poreJ Membr Biol1971334815014916
- ArispeNPollardHB1993Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [A beta P-(1–40)] in bilayer membranesProc Natl Acad Sci U S A90221057377504270
- ArispeNPollardHB1996Zn2+ interaction with Alzheimer amyloid beta protein calcium channelsProc Natl Acad Sci U S A934171058643694
- ArrasateMMitraS2004Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal deathNature43170108051015483602
- ArundineMTymianskiM2004Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injuryCell Mol Life Sci6166576815052409
- AtwoodCSMartinsRN2002Senile plaque composition and posttranslational modification of amyloid-beta peptide and associated proteinsPeptides23713435012128091
- BahadiRFarrellyPV2003Cu2+-induced modification of the kinetics of A beta(1–42) channelsAm J Physiol Cell Physiol2854C8738012814914
- BehlCDavisJB1994Amyloid beta peptide induces necrosis rather than apoptosisBrain Res6451–2253648062088
- BezanillaF2000The voltage sensor in voltage-dependent ion channelsPhysiol Rev8025559210747201
- BhatiaRLinH2000Fresh and globular amyloid beta protein (1–42) induces rapid cellular degeneration: evidence for AbetaP channel-mediated cellular toxicityFaseb J14912334310834945
- BlessedGTomlinsonBE1968The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjectsBr J Psychiatry1145127978115662937
- BuchholzJ NBehringerEJ2007Age-dependent changes in Ca2+ homeostasis in peripheral neurones: implications for changes in functionAging Cell632859617517039
- BurnashevNRozovA2005Presynaptic Ca2+ dynamics, Ca2+ buffers and synaptic efficacyCell Calcium3754899515820398
- CatterallWA1998Structure and function of neuronal Ca2+ channels and their role in neurotransmitter releaseCell Calcium245-63072310091001
- CaugheyBLansburyPT2003Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystandersAnnu Rev Neurosci262679812704221
- CookDGFormanMS1997Alzheimer’s A beta(1–42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cellsNat Med39102139288730
- CopaniASortinoMA2002Erratic expression of DNA polymerases by beta-amyloid causes neuronal deathFaseb J16142006812397084
- CruzJCTsaiLH2004Cdk5 deregulation in the pathogenesis of Alzheimer’s diseaseTrends Mol Med109452815350898
- De FerrariGVChaconMA2003Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by beta-amyloid fibrilsMol Psychiatry8219520812610652
- DeneckerGVercammenD2001Death receptor-induced apoptotic and necrotic cell death: differential role of caspases and mitochondriaCell Death Differ888294011526436
- DicksonDW1997The pathogenesis of senile plaquesJ Neuropathol Exp Neurol564321399100663
- DurellSRGuyHR1994Theoretical models of the ion channel structure of amyloid beta-proteinBiophys J6762137457535109
- EckertASchindowskiK2001Alzheimer’s disease-like alterations in peripheral cells from presenilin-1 transgenic miceNeurobiol Dis823314211300728
- EguchiYShimizuS1997Intracellular ATP levels determine cell death fate by apoptosis or necrosisCancer Res57101835409157970
- EhrlichBEKaftanE1994The pharmacology of intracellular Ca(2+)-release channelsTrends Pharmacol Sci15514597754532
- EliezerD2006Amyloid ion channels: a porous argument or a thin excuse?J Gen Physiol1286631317130516
- FastbomJForsellY1998Benzodiazepines may have protective effects against Alzheimer diseaseAlzheimer Dis Assoc Disord1211479539405
- Fernandez-VizarraPFernandezAP2004Intra- and extracellular Abeta and PHF in clinically evaluated cases of Alzheimer’s diseaseHistol Histopathol1938234415168346
- FraserS PSuhYH1997Ionic effects of the Alzheimer’s disease beta-amyloid precursor protein and its metabolic fragmentsTrends Neurosci20267729023874
- Flurizan2005Mar. Molecule of the month. MPC-7869Drugs News Perspect182141
- FuruichiTMikoshibaK1995Inositol 1, 4, 5-trisphosphate receptor-mediated Ca2+ signaling in the brainJ Neurochem643953607861177
- GeverJRCockayneDA2006Pharmacology of P2X channelsPflugers Arch45255133716649055
- GitterBDCDLiWDieckmanDKBenderMHNissenJSMabryTEYinTBoggsLNMcClureDBLittleSPJohnstoneEMAudiaJEMayPCHyslopPA2004Stereoselective Inhibition of Amyloid Beta Peptide Secretion by LY450139, A Novel Functional Gamma Secretase InhibitorNeurobiol Aging25Suppl 2S571
- GlennerGGWongCW1984Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid proteinBiochem Biophys Res Commun1203885906375662
- GoldeTEEckmanCB2000Biochemical detection of Abeta isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer’s diseaseBiochim Biophys Acta150211728710899442
- GorevicPDGoniF1986Isolation and partial characterization of neurofibrillary tangles and amyloid plaque core in Alzheimer’s disease: immunohistological studiesJ Neuropathol Exp Neurol456647643772397
- GotzJStrefferJR2004Transgenic animal models of Alzheimer’s disease and related disorders: histopathology, behavior and therapyMol Psychiatry976648315052274
- GourasGKTsaiJ2000Intraneuronal Abeta42 accumulation in human brainAm J Pathol1561152010623648
- GravinaSAHoL1995Amyloid beta protein (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43)J Biol Chem27013701367706234
- GreenfieldJPTsaiJ1999Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptidesProc Natl Acad Sci U S A96274279892704
- GrynspanFGriffinWB1997Calpains and calpastatin in SH-SY5Y neuroblastoma cells during retinoic acid-induced differentiation and neurite outgrowth: comparison with the human brain calpain systemJ Neurosci Res483181919160241
- GuoQFuW1999Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in miceNat Med5110169883847
- HaassCKooEH1992Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragmentsNature357637850031608449
- HaassCSchlossmacherMG1992Amyloid beta-peptide is produced by cultured cells during normal metabolismNature359639332251383826
- HamillOPMartyA1981Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patchesPflugers Arch3912851006270629
- HardyJAHigginsGA1992Alzheimer’s disease: the amyloid cascade hypothesisScience256505418451566067
- HarperSJBilslandJG1998Mouse cortical neurones lacking APP show normal neurite outgrowth and survival responses in vitroNeuroreport913305389804315
- HartmannJKonnerthA2005Determinants of postsynaptic Ca2+ signaling in Purkinje neuronsCell Calcium3754596615820394
- HartmannTBiegerSC1997Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptidesNat Med391016209288729
- HeXCooleyK2007Apolipoprotein receptor 2 and X11 alpha/beta mediate apolipoprotein E-induced endocytosis of amyloid-beta precursor protein and beta-secretase, leading to amyloid-beta productionJ Neurosci271540526017428983
- HirakuraYLinMC1999Alzheimer amyloid abeta1–42 channels: effects of solvent, pH, and Congo RedJ Neurosci Res5744586610440895
- HofmannFBielM1994Molecular basis for Ca2+ channel diversityAnnu Rev Neurosci173994188210181
- HollmannMHeinemannS1994Cloned glutamate receptorsAnnu Rev Neurosci17311088210177
- HuangHMOuHC2000Antioxidants prevent amyloid peptide-induced apoptosis and alteration of calcium homeostasis in cultured cortical neuronsLife Sci661918799210809185
- HuangHMOuHC2000Blockage of amyloid beta peptide-induced cytosolic free calcium by fullerenol-1, carboxylate C60 in PC12 cellsLife Sci661615253310794500
- IwatsuboTOdakaA1994Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43)Neuron13145538043280
- JarrettJTBergerEP1993The C-terminus of the beta protein is critical in amyloidogenesisAnn N Y Acad Sci69514488239273
- JarrettJTBergerEP1993The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s diseaseBiochemistry3218469378490014
- JoDGLeeJY2004Induction of pro-apoptotic calsenilin/DREAM/KChIP3 in Alzheimer’s disease and cultured neurons after amyloid-beta exposureJ Neurochem8836041114720210
- JonasEAKnoxRJ1997Giga-ohm seals on intracellular membranes: a technique for studying intracellular ion channels in intact cellsNeuron1917139247259
- KamenetzFTomitaT2003APP processing and synaptic functionNeuron3769253712670422
- KatzmanRSaitohT1991Advances in Alzheimer’s diseaseFaseb J53278862001787
- KawaharaMArispeN1997Alzheimer’s disease amyloid beta-protein forms Zn(2+)-sensitive, cation-selective channels across excised membrane patches from hypothalamic neuronsBiophys J73167759199772
- KellerBUHedrichR1992Patch clamp techniques to study ion channels from organellesMethods Enzymol207673811382206
- KleinWLStineWBJr2004Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer’s diseaseNeurobiol Aging2555698015172732
- KohJYYangLL1990Beta-amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damageBrain Res5332315202289145
- KooEHSquazzoSL1994Evidence that production and release of amyloid beta-protein involves the endocytic pathwayJ Biol Chem269261738698021238
- KowallNWBealMF1991An in vivo model for the neurodegenerative effects of beta amyloid and protection by substance PProc Natl Acad Sci U S A88167247511714596
- LeistMSingleB1997Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosisJ Exp Med1858148169126928
- LesneSAliC2005NMDA receptor activation inhibits alpha-secretase and promotes neuronal amyloid-beta productionJ Neurosci254193677716221845
- LevitesYDasP2006Anti-Abeta42- and anti-Abeta40-specific mAbs attenuate amyloid deposition in an Alzheimer disease mouse modelJ Clin Invest116119320116341263
- LinHBhatiaR2001Amyloid beta protein forms ion channels: implications for Alzheimer’s disease pathophysiologyFaseb J151324334411689468
- LinHZhuYJ1999Amyloid beta protein (1–40) forms calcium-permeable, Zn2+-sensitive channel in reconstituted lipid vesiclesBiochemistry3834111899610460176
- LinXKoelschG2000Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor proteinProc Natl Acad Sci U S A97414566010677483
- LorenzoAYanknerBA1994Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo redProc Natl Acad Sci U S A91251224377991613
- MannDMIwatsuboT1996Atypical amyloid (A beta) deposition in the cerebellum in Alzheimer’s disease: an immunohistochemical study using end-specific A beta monoclonal antibodiesActa Neuropathol (Berl)916647538781665
- MattsonMP1998Modification of ion homeostasis by lipid peroxidation: roles in neuronal degeneration and adaptive plasticityTrends Neurosci2125379498297
- MattsonMP2007Calcium and neurodegenerationAging Cell633375017328689
- MattsonMPChengB1992beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicityJ Neurosci122376891346802
- MattsonMPTomaselliKJ1993Calcium-destabilizing and neuro-degenerative effects of aggregated beta-amyloid peptide are attenuated by basic FGFBrain Res621135498221072
- MazzantiMDeFeliceLJ1990Ion channels in the nuclear envelopeNature343626076472304551
- McHughPTurinaM2006Apoptosis and necrosis: a review for surgeonsSurg Infect (Larchmt)71536816509786
- MehtaPDPirttilaT2001Amyloid beta protein 1–40 and 1–42 levels in matched cerebrospinal fluid and plasma from patients with Alzheimer diseaseNeurosci Lett3041–2102611335065
- MirandaSOpazoC2000The role of oxidative stress in the toxicity induced by amyloid beta-peptide in Alzheimer’s diseaseProg Neurobiol6266334810880853
- MorganCColombresM2004Structure and function of amyloid in Alzheimer’s diseaseProg Neurobiol7463234915649580
- NeveRLDawesLR1990Genetics and biology of the Alzheimer amyloid precursorProg Brain Res86257672150887
- OakleyHColeSL2006Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formationJ Neurosci2640101294017021169
- OddoSCaccamoA2003Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunctionNeuron3934092112895417
- ParadisEDouillardH1996Amyloid beta peptide of Alzheimer’s disease downregulates Bcl-2 and upregulates bax expression in human neuronsJ Neurosci1623753398922409
- PearsonHAPeersC2006Physiological roles for amyloid beta peptidesJ Physiol575Pt 151016809372
- PikeCJBurdickD1993Neurodegeneration induced by beta-amyloid peptides in vitro: the role of peptide assembly stateJ Neurosci1341676878463843
- PlantLDBoyleJP2003The production of amyloid beta peptide is a critical requirement for the viability of central neuronsJ Neurosci23135531512843253
- PriceSAHeldB1998Amyloid beta protein increases Ca2+ currents in rat cerebellar granule neuronesNeuroreport93539459512403
- RamsdenMHendersonZ2002Modulation of Ca2+ channel currents in primary cultures of rat cortical neurones by amyloid beta protein (1–40) is dependent on solubility statusBrain Res95622546112445693
- ReyesAEChaconMA2004Acetylcholinesterase-Abeta complexes are more toxic than Abeta fibrils in rat hippocampus: effect on rat beta-amyloid aggregation, laminin expression, reactive astrocytosis, and neuronal cell lossAm J Pathol164621637415161650
- RheeSKQuistAP1998Amyloid beta protein-(1–42) forms calcium-permeable, Zn2+-sensitive channelJ Biol Chem2732213379829593665
- RoherAEChaneyMO1996Morphology and toxicity of Abeta-(1–42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer’s diseaseJ Biol Chem271342063158702810
- SavageMJTruskoSP1998Turnover of amyloid beta-protein in mouse brain and acute reduction of its level by phorbol esterJ Neurosci1851743529464999
- SchmitzCRuttenBP2004Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer’s diseaseAm J Pathol1644149550215039236
- SelkoeDJ2001Alzheimer’s disease: genes, proteins, and therapyPhysiol Rev8127416611274343
- SelkoeDJ2001Clearing the brain’s amyloid cobwebsNeuron3221778011683988
- SelkoeDJAbrahamCR1986Isolation of low-molecular-weight proteins from amyloid plaque fibers in Alzheimer’s diseaseJ Neurochem4661820343517233
- SeubertPOltersdorfT1993Secretion of beta-amyloid precursor protein cleaved at the amino terminus of the beta-amyloid peptideNature361640926037678698
- SimakovaOArispeNJ2006Early and late cytotoxic effects of external application of the Alzheimer’s Abeta result from the initial formation and function of Abeta ion channelsBiochemistry451859071516669633
- SkovronskyDMDomsRW1998Detection of a novel intraneuronal pool of insoluble amyloid beta protein that accumulates with time in cultureJ Cell Biol1414103199585420
- SokolovYKozakJA2006Soluble amyloid oligomers increase bilayer conductance by altering dielectric structureJ Gen Physiol12866374717101816
- StalderMPhinneyA1999Association of microglia with amyloid plaques in brains of APP23 transgenic miceAm J Pathol154616738410362792
- SteinbachJPMullerU1998Hypersensitivity to seizures in beta-amyloid precursor protein deficient miceCell Death Differ5108586610203685
- TakahashiRHMilnerTA2002Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathologyAm J Pathol161518697912414533
- TalafousJMarcinowskiKJ1994Solution structure of residues 1–28 of the amyloid beta-peptideBiochemistry33257788967516706
- TamagnoERobinoG2003H2O2 and 4-hydroxynonenal mediate amyloid beta-induced neuronal apoptosis by activating JNKs and p38MAPKExp Neurol18021445512684028
- TamaokaASawamuraN1997Amyloid beta protein 42(43) in cerebrospinal fluid of patients with Alzheimer’s diseaseJ Neurol Sci14814159125389
- ThibaultOGantJC2007Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: minding the storeAging Cell633071717465978
- ToescuECVerkhratskyA2000Parameters of calcium homeostasis in normal neuronal ageingJ Anat, 197 Pt45639
- TomlinsonBEBlessedG1968Observations on the brains of non-demented old peopleJ Neurol Sci72331565707082
- TomlinsonBEBlessedG1970Observations on the brains of demented old peopleJ Neurol Sci113205425505685
- TompkinsMMHillWD1997Contribution of somal Lewy bodies to neuronal deathBrain Res7751–22499439824
- TurnerRSSuzukiN1996Amyloids beta40 and beta42 are generated intracellularly in cultured human neurons and their secretion increases with maturationJ Biol Chem271158966708621541
- UrbancBCruzL2002Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer’s diseaseProc Natl Acad Sci U S A992213990512374847
- VanterpoolCKPearceWJ2005Advancing age alters rapid and spontaneous refilling of caffeine-sensitive calcium stores in sympathetic superior cervical ganglion cellsJ Appl Physiol9939637115845773
- Velez-PardoCArroyaveST2001Ultrastructure evidence of necrotic neural cell death in familial Alzheimer’s disease brains bearing prese-nilin-1 E280A mutationJ Alzheimers Dis3440941512214045
- VerkhratskyAShmigolA1996Calcium-induced calcium release in neuronesCell Calcium1911148653752
- VerkhratskyATECVerkhratskyATEC1998Integrative Aspects of Calcium SignallingLondonPlenium Press428
- WalshDMSelkoeDJ2004Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegenerationProtein Pept Lett1132132815182223
- WangRWangB2006Wild-type presenilin 1 protects against Alzheimer disease mutation-induced amyloid pathologyJ Biol Chem2812215330616574645
- WenkGL2003Neuropathologic changes in Alzheimer’s diseaseJ Clin Psychiatry, 64 Suppl9710
- WenkGL2006Neuropathologic changes in Alzheimer’s disease: potential targets for treatmentJ Clin Psychiatry67Suppl 337 quiz 2316649845
- WhiteARZhengH1998Survival of cultured neurons from amyloid precursor protein knock-out mice against Alzheimer’s amyloid-beta toxicity and oxidative stressJ Neurosci18166207179698314
- WhitsonJSSelkoeDJ1989Amyloid beta protein enhances the survival of hippocampal neurons in vitroScience24348971488902928783
- Wild-BodeCYamazakiT1997Intracellular generation and accumulation of amyloid beta-peptide terminating at amino acid 42J Biol Chem272261608589195901
- WilsonCADomsRW1999Intracellular APP processing and A beta production in Alzheimer diseaseJ Neuropathol Exp Neurol5887879410446803
- WilsonCADomsRW2002Presenilins are not required for A beta 42 production in the early secretory pathwayNat Neurosci598495512145638
- WinbladBPoritisN1999Memantine in severe dementia: results of the 9M-Best Study (Benefit and efficacy in severely demented patients during treatment with memantine)Int J Geriatr Psychiatry1421354610885864
- WirthsOMulthaupG2004A modified beta-amyloid hypothesis: intraneuronal accumulation of the beta-amyloid peptide – the first step of a fatal cascadeJ Neurochem9135132015485483
- WirthsOMulthaupG2002Intraneuronal APP/A beta trafficking and plaque formation in beta-amyloid precursor protein and presenilin-1 transgenic miceBrain Pathol1232758612146796
- WongCWQuarantaV1985Neuritic plaques and cerebrovascular amyloid in Alzheimer disease are antigenically relatedProc Natl Acad Sci U S A82248729322934737
- WuytackFRaeymaekersL2002Molecular physiology of the SERCA and SPCA pumpsCell Calcium325-627930512543090
- XiaoJPerryG1996alpha-calcium-calmodulin-dependent kinase II is associated with paired helical filaments of Alzheimer’s diseaseJ Neuropathol Exp Neurol559954638800091
- XuHSweeneyD1997Generation of Alzheimer beta-amyloid protein in the trans-Golgi network in the apparent absence of vesicle formationProc Natl Acad Sci U S A9483748529108049
- YanknerBADuffyLK1990Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptidesScience2504978279822218531
- YoungJDCohnZA1986The ninth component of complement and the pore-forming protein (perforin 1) from cytotoxic T cells: structural, immunological, and functional similaritiesScience2334760184902425429
- YounkinSG1998The role of A Beta 42 in Alzheimer’s diseaseJ Physiol (Paris)922892929789825
- YusteRMajewskaA2000From form to function: calcium compartmentalization in dendritic spinesNat Neurosci37653910862697
- ZhengHJiangM1995beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activityCell814525317758106