Abstract
Mood disorders are among the most prevalent and recurrent forms of psychiatric illnesses. In the last decade, there has been increased understanding of the biological basis of mood disorders. In fact, novel mechanistic concepts of the neurobiology of unipolar and bipolar disorders are evolving based on recent pre-clinical and clinical studies, most of which now focus on the role of signal transduction mechanisms in these psychiatric illnesses. Particular investigative emphasis has been given to the role of phosphorylating enzymes, which are crucial in regulating gene expression and neuronal and synaptic plasticity. Among the most important phosphorylating enzyme is protein kinase A (PKA), a component of adenylyl cyclase–cyclic adenosine monophosphate (AC–cAMP) signaling system. In this review, we critically and comprehensively discuss the role of various components of AC–cAMP signaling in mood disorders, with a special focus on PKA, because of the interesting observation that have been made about its involvement in unipolar and bipolar disorders. We also discuss the functional significance of the findings regarding PKA by discussing the role of important PKA substrates, namely, Rap-1, cyclicAMP-response element binding protein, and brain-derived neurotrophic factor. These studies suggest the interesting possibility that PKA and related signaling molecules may serve as important neurobiological factors in mood disorders and may be relevant in target-specific therapeutic interventions for these disorders.
Introduction
Mood disorders are among the most prevalent and recurrent forms of psychiatric illnesses. Unipolar depression affects about 17% of the population at some point in life (CitationKessler et al 1994; CitationHirschfeld 2002). Of these, about 4% of men and 8% of women are at a lifetime risk for a severe form of depression, and an additional 20% are at risk for milder forms. Major depression is often a chronic and lifelong illness, and about 80% of the depressed population experience multiple episodes. Each depressive episode greatly increases the risk of future episodes and also lessens responsiveness to treatments. About 10% of patients who experience a single or multiple episodes of depression can have a manic episode. About 1%–2% of the total population is at risk for bipolar disorder (recurrent depressive and manic episodes), and another 2%–3% may have milder forms (CitationSpearing 2001; CitationHirschfeld et al 2002). Both depression and bipolar disorders are associated with a high risk of suicide. Among teenagers, bipolar patients are at twice the risk to commit suicide as unipolar depressed patients; however, the risk of suicide is higher in elderly patients over the age of 60 who are suffering from unipolar depression. Therefore, researchers are striving for greater understanding of the neurobiology of these disorders that will lead to better treatment and prevention.
Both unipolar and bipolar disorders are complex, and no unified theory can currently satisfactorily explain this complexity. Besides the clinical manifestation, there are subtypes of unipolar and bipolar disorders, and patients with these subtypes respond differently to various treatment approaches. Although depression is a part of bipolar disorder, it still is unclear whether the neurobiology of unipolar depression is similar to that of the depressive state in bipolar disorder.
Earlier studies focused on the role of monoamines in the pathophysiology of mood disorders, hypothesizing decreased availability of monoamines in the synaptic cleft and probably a compensatory increase in monoamine receptors (CitationJans et al 2007). This was substantiated by observations of an increased availability of monoamines after treatment with antidepressants. These studies provided useful information of the involvement of monoamines and monoamine receptors in the pathophysiology of mood disorders, but they also raised several important questions. For example, monoamine depletion does not produce depressive symptoms in normal individuals. Also, not every patient responds to the antidepressants that modulate monoamines. In addition, there is a lag time between starting an antidepressant or mood stabilizing drug treatment and responsiveness. It takes about 2–4 weeks for these drugs to become fully effective and thus the immediate effects on monoamines or receptors cannot explain their mechanisms of action. In addition, a number of neurotransmitter receptors have been implicated in the pathophysiology of mood disorders, and they interact with each other in a complex fashion, mostly at the level of intracellular signal transduction mechanisms. Most neurotransmitter receptors regulate intracellular reactions by an indirect mechanism involving an intermediate transducing molecule known as guanosine 5’-triphosphate (GTP) binding protein, or G protein. Activated G proteins alter the functions of many signaling pathway downstream effectors. Most of these effectors are enzymes that produce intracellular second messengers. The second messengers trigger the signaling cascade further downstream and cause a cellular response. It is the downstream intracellular signaling mediated by these neurotransmitter receptors that co-ordinates the behavior of individual cells within the brain in various physiological processes.
Emerging evidence indicates that mood disorders are associated with altered neuronal plasticity (CitationDuman 2000; CitationDuman et al 2000). A number of studies using magnetic resonance spectroscopy, anatomical, and morphometric techniques have repeatedly shown that there are structural abnormalities in the brains of depressed or bipolar patients (CitationSoares and Mann 1997; CitationRajkowska 2000, Citation2002). Adaptive responses in intracellular molecules, together with the modulation of functional responses mediated by the phosphorylation of critical proteins, and ultimately gene expression, controlled by intracellular signaling cascades, all participate in a major way in synaptic and structural plasticity. Given the critical importance of intracellular signaling in amplifying, integrating, and regulating physiological processes and in mediating gene expression, over the last decade numerous studies have examined the role of signaling molecules in the pathophysiology of mood disorders.
One of the major intracellular signaling pathways that has been implicated in synaptic and structural plasticity is adenylyl cyclase–cyclic adenosine monophosphate (AC–cAMP) signaling. Phosphorylation mediated by the cAMP signaling pathway can be elicited by various physiological ligands in cells and is critically involved in cell growth, differentiation, apoptosis, and gene expression (CitationSkahegg and Tasken 2000). In this review, we will discuss the role of the adenylyl cyclase–cAMP signaling pathway in mood disorders in general, including the results of investigations of cAMP and adenylyl cyclase, but major emphasis will be given for the role of the enzyme protein kinase A (PKA), which mediates phosphorylation and, therefore, physiological functions in the brain.
Adenylyl cyclase–cAMP signaling pathway
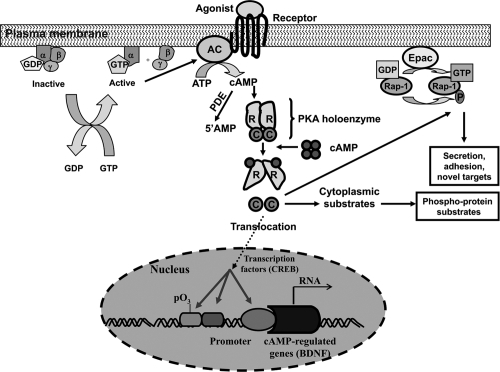
A schematic representation of the adenylyl cyclase–cAMP signaling pathway is provided in . A number of neurotransmitter receptors, including serotonergic and adrenergic, utilize this pathway to mediate their physiological functions. In this signaling pathway, an agonist binding to receptors causes the activation of heterotrimeric guanine nucleotide binding protein (G proteins), which consist of α, β, and γ subunits. The receptor-mediated activation of G proteins causes the release of guanine nucleotide diphosphate (GDP) from α subunit, allowing GTP to bind to and dissociate the α subunit from the βγ subunits. Both α and βγ subunits can then modulate activity of effectors, including adenylyl cyclase. Activation of adenylyl cyclase causes the conversion of ATP to cAMP, which serves as a second messenger. cAMP then activates the phosphorylation enzyme PKA. Once activated, PKA phosphorylates various intracellular proteins and thereby modifies hormonal and neurotransmitter responses, including receptor downregulation or desensitization, alteration of neurotransmitter release, and activation or repression of gene expression (CitationBorrelli et al 1992; CitationSpaulding 1993; CitationNestler and Greengard 1994). The negative regulation of cAMP signaling occurs through the activation of cAMP-specific phosphodiesterases, which convert cAMP to 5’AMP.
Figure 1 Overview of the adenylyl cyclase–cyclicAMP signaling system. In this signaling pathway, the binding of agonists to receptors leads to the activation of G proteins, which in turn activate adenylyl cyclase. In the inactive state the α subunit of G protein is bound to GDP and to βγ subunits. The binding of agonists to receptors causes an interaction of receptors with G proteins, which, in turn, releases GDP in exchange with GTP. This leads to generation of α-GTP and a βγ subunits dimer. Both α and βγ subunits can interact with effectors. Activation of adenylyl cyclase causes generation of cAMP from ATP. cAMP binds to the regulatory (R) subunits of tetrameric PKA holoenzymes. The binding of cAMP to an R subunit lowers its affinity for the C subunit. This causes the release of free catalytic (C) subunits. The C subunit can catalyze reversible protein phosphorylation and irreversible phosphorylation of newly synthesized peptides. Phosphorylation of substrates can occur in the cytoplasm or, after translocation of the C subunits, in the nucleus. One of the substrates of PKA is transcription factor cyclicAMP response element binding protein CREB, which regulates transcription of many neuronally expressed genes, including brain-derived neurotrophic factor (BDNF). The exchange protein activated by cAMP (Epac) is stimulated by cAMP to exchange GDP with GTP on Rap-1. Rap can also be regulated by phosphorylation by a C subunit of PKA. Deactivation of PKA is achieved through degradation of cAMP to 5’AMP by cAMP-specific phosphodiesterases.
Adenylyl cyclase and cAMP formation in mood disorders
Since regulation of the intracellular level of a second messenger is critical for the transduction of a cellular response, many studies have examined cAMP generation in mood disorders. The intracellular level of cAMP is determined by the rate of its synthesis from ATP by adenylyl cyclase. Most earlier studies utilized peripheral tissues to examine levels of cAMP or receptor-mediated cAMP formation. For example, it was shown that the baseline level of cAMP in plasma, cerebrospinal fluid (CitationBelmaker et al 1980; CitationPost et al 1982; CitationMaj et al 1984), or in leukocytes or lymphoblast cell lines (CitationKlysner et al 1987; CitationKay et al 1993), is not altered in various mood states. On the other hand, changes in receptor-mediated cAMP formation were found in these patients. The inhibition of cAMP formation by prostaglandins (PG) has commonly been used as an index of imidazoline (I)2-adrenergic receptor responsiveness. Using this approach, CitationWang et al (1974) reported that PGE1-stimulated cAMP accumulation and norepinephrine (NE) inhibition of PGE1-stimulated cAMP accumulation were not changed in platelets of depressed patients. Similar results were reported by CitationMurphy et al (1974). On the other hand, CitationSiever et al (1984) observed decreased PGE1-stimulated cAMP accumulation and the inhibition of this response by NE in platelets of depressed patients; and CitationKanof et al (1986) observed that whereas PGE1-stimulated cAMP response was significantly decreased, NE inhibition of PGE1-stimulated cAMP formation was not changed in platelets of depressed patients (CitationKanof et al 1988). Thus, studies of the responsiveness of α2-adrenergic receptors (ARs) in platelets of depressive patients do not consistently show decreased PGE1-stimulated cAMP response in depressed patients.
Besides α-AR-mediated responsiveness, the functional significance of β-ARs in depression was out by determining agonist-stimulated cAMP formation in leukocytes or lymphocytes of depressed patients and normal controls by our group (CitationPandey et al 1979) and other investigators (CitationHealy et al 1983; CitationMann et al 1985; CitationKlysner et al 1987; CitationEbstein et al 1988). CitationPandey et al (1979) reported for the first time that NE-stimulated [3H]cAMP accumulation was significantly lower in leukocytes of depressed or bipolar patients; significantly lower isoproterenol-stimulated cAMP accumulation was also observed in these patients. In the same year, CitationExtein et al (1979) reported similar findings in the lymphocytes of a mixed group of depressed or manic patients. CitationMann et al (1985) also observed lower isoproterenol-stimulated cAMP levels in lymphocytes of patients with endogenous depression. CitationEbstein et al (1988) determined isoproterenol-, forskolin-, as well as PGE1-stimulated cAMP accumulation in lymphocytes of depressed patients and observed that the isoproterenol-stimulated cAMP level was significantly decreased in these patients. These investigators also observed significantly lower isoproterenol-stimulated cAMP accumulation in depressed nonresponders compared with responders. CitationKanof et al (1989) also observed lower isoproterenol-stimulated cAMP accumulation in depressed patients. On the other hand, a number of studies suggest the opposite changes in cAMP formation in bipolar patients. For example, CitationYoung et al (1993) reported that forskolin-stimulated adenylyl cyclase activity is significantly increased in temporal and occipital cortices of bipolar patients compared with normal control subjects. Also CitationKay et al (1993) reported that PGE1-stimulated adenylyl cyclase activity was significantly increased in lymphoblast cell lines of bipolar patients. CitationKlysner et al (1987) observed increased β2-AR responsiveness in manic-depressive patients compared with euthymic patients treated with antidepressants.
Adenylyl cyclase exists in multiple forms, each a unique gene product (CitationTang and Gilman 1991), and can be grouped into three main classes: 1) Types II, IV, and VII are activated synergistically by Gsα and Gβγ subunits; 2) Types V and VI are inhibited by Giα and Ca2+; and 3) Types VIII, I, and III are activated synergistically by Gsα together with Ca2+-calmodulin. CitationReiach et al (1999) reported that immunolabeling of specific types (Type IV) and activity of adenylyl cyclase are significantly decreased in postmortem brain of depressed suicide victims compared with normal control subjects. Although the significance of the decrease in this specific type of adenylyl cyclase is not clear, nonetheless this supports the findings of decreased cAMP signaling in depressed patients. The status of expression of adenylyl cyclase subunits in bipolar disorder has not been studied.
Overall these studies show a differential response of agonist-induced cAMP formation in response to β2-ARs activation. On the one hand, depressed patients show decreases in responsiveness of β2-ARs, adenylyl cyclase activity, and expression of a specific subtype of adenylyl cyclase; bipolar patients, on the other hand, show increased forskolin-, PGE1- or β-adrenergic-stimulated adenylyl cyclase response. Interestingly, a number of studies show that the changes in adenylyl cyclase responsivity are not accompanied by consistent changes in β-AR density or affinity (CitationMann et al 1985; CitationBerrettini et al 1987; CitationJeanningros et al 1991), which suggests that post-receptor signal transduction mechanisms may be playing an important role in mood disorders, albeit in a different manner in unipolar versus bipolar disorders.
Protein kinase A (PKA) in mood disorders
Since most of the effects of cAMP are mediated by its receptor PKA, and since a number of studies suggest that PKA is regulated by sustained activation of cAMP (CitationSpaulding 1993; CitationFrancis and Corbin 1999), studying the status of PKA can thus provide a direct evidence of altered cAMP signaling. In addition, PKA participates directly in many physiological functions in the central nervous system, and by phosphorylating the components of other signaling cascades, it provides the means for cross-talk between the AC–cAMP and other signaling systems (CitationBeebe 1994; CitationBornfeldt and Krebs 1999; CitationJordan et al 2000). Therefore, a number of recent studies have focused on the role of PKA in mood disorders.
Characteristics of PKA
As shown in , PKA is a holoenzyme composed of two genetically distinct regulatory (R) and two catalytic (C) subunits that form a tetrameric holoenzyme (R2C2). In the absence of cAMP, PKA exists as a stable inactive tetramer; the catalytic activity of cAMP is suppressed when the C subunits form a complex with the R subunits. After an increase in intracellular cAMP, the regulatory PKA subunits bind to cAMP in a co-operative manner, which results in the disassociation of the holoenzyme into an R2 (cAMP)4 dimer and two monomers of catalytically active C kinase. The R subunits remain in the cytoplasm, and the free catalytic subunits either translocate into the nucleus (CitationWolf et al 1999) or remain in the cytosol. In both locations, PKA C subunits phosphorylate serine and threonine residues of specific substrates (CitationScott et al 1990). In the nucleus, regulation of transcription by PKA is mediated by cAMP-responsive nuclear factors, which bind to and regulate the expression of genes containing a cAMP-response element binding element (CRE) consensus in their promoter region. Phosphorylation of the CRE binding protein (CREB) modulates its activity. One of the important genes whose transcription is regulated by PKA/CREB is brain derived neurotrophic factor (BDNF), described later in this review. Besides its role in gene transcription, PKA is capable of phosphorylating a large number of substrates involved in neurotransmitter release; receptor desensitization; cell growth, differentiation, survival; and synaptic plasticity (CitationBorrelli et al 1992; CitationNestler and Greengard 1994; CitationRiccio et al 1999; CitationLara et al 2003).
On the basis of the elution profile on DEAE exchange chromatography, two major forms of PKA have been identified, ie, Type I and Type II. These two types differ in their structure in the regulatory subunits incorporated, termed RI or RII, whereas their catalytic subunits are either identical or very similar. Cloning studies have revealed multiple isoforms for each regulatory and catalytic subunit. Two RI subunits, termed RIα and RIβ, and two RII subunits, termed RIIα and RIIβ, have been identified. Furthermore, three distinct catalytic subunits have been identified, termed Cα, Cβ, and Cγ. These isozymes may form either homo- or heterodimers of the R subunits yielding holoenzyme complexes of PKA composed of a number of different combinations, including RIα2C2, RIβ2C2, RIIα2C2, RIIβ2C2, and RIαRIβC2. The presence of multiple C subunits adds substantial diversity to the action of PKA. Each regulatory and catalytic subunit is a separate gene product and has a distinct expression pattern in different tissues (CitationScott et al 1987; CitationClegg and McKnight 1988; CitationSkalhegg and Tasken 2000). For example, tissue distribution studies suggest that RIIβ is predominantly expressed in brain, adrenal, and adipose tissues and is the principal mediator of cAMP activity in the mammalian CNS (CitationSarkar et al 1984), whereas Cβ is expressed primarily in the brain (CitationUhler et al 1986). On the other hand, whereas Cα is ubiquitously expressed. Cγ is present only in testis. We will discuss all those C and R subunits which are present in brain.
PKA in bipolar disorder
The Warsh group provided the first evidence that PKA may be altered in bipolar disorder. They found significantly decreased [3H]cAMP binding to regulatory subunits in the cytosolic but not in the membrane fractions of frontal, temporal, and occipital cortices, cerebellum, and thalamus of bipolar patients (CitationRahman et al 1997). They further observed greater basal and cAMP-stimulated PKA activity, and lower EC50 values for cAMP stimulation of PKA activity, in the temporal cortex of bipolar patients (CitationFields et al 1999).
Clinical studies performed in blood cells also provide evidence of abnormalities in PKA in bipolar patients. For example, CitationKarege et al (2002a; Citation2004) reported that PKA activity is significantly greater in the cytosolic fraction of cultured lymphoblasts from euthymic bipolar patients compared with normal controls. In addition, they found that [3H]cAMP binding is decreased in the cytosolic fraction of lymphoblasts of bipolar patients. The results of these studies are similar to the findings in postmortem brain of bipolar patients.
In postmortem brain of bipolar patients, CitationChang et al (2003a) found that the levels of cytosolic Cα and RIIβ in temporal cortex were significantly higher in bipolar patients. This indicates that the increased PKA activity in postmortem brain and in platelets of bipolar patients could be due to increased levels of Cα. The authors argued that an increase in RIIβ level may be associated with an increase in PKA II and a decrease in PKA I holoenzyme levels. PKA RII has a much lower affinity for cAMP than the type I PKA holoenzyme. Such a putative shift in PKA I and PKA II holoenzyme levels could have accounted for the decreased [3H]cAMP binding found in bipolar patients (CitationChang et al 2003a). binding found in bipolar patients CitationPerez et al (1999) also reported increased PKA catalytic subunits in bipolar patients, although they did not examine whether this increase was in α or β subunits of PKA. Using the same brain tissues, CitationChang et al (2003b) did not find any significant differences in mRNA levels of these C and R subunits of PKA in bipolar patients, suggesting that a post-translational mechanism is responsible for the changes in protein levels of RIIβ and Cα subunits.
PKA in unipolar disorder
A number of studies in peripheral tissues of depressed patients suggest abnormalities in the cAMP signaling pathway at the level of PKA. For example, CitationShelton et al (1996) and CitationManier et al (1996) reported significantly decreased β-adrenergic receptor-stimulated PKA activity in the presence of cAMP in fibroblasts of depressed patients. CitationShelton et al (1999) later confirmed that β-adrenergic receptor-stimulated PKA activity was decreased only in melancholic depressed patients but not in other depressive subtypes. More recently, CitationAkin et al (2005) reported reduced PKA activity along with reduced expression of PKA RIIα, Cα, and Cβ subunits in fibroblasts of melancholic depressed subjects as compared with nonmelancholic and normal control subjects. Overall these studies in peripheral tissues demonstrate decreased activation of PKA in depressed patients, which could be associated specifically with melancholic subtype.
In a comprehensive study, we examined whether PKA is altered in postmortem brain of suicide subjects by comparing suicide subjects with and without major depression (CitationDwivedi et al 2002a). We observed that [3H]cAMP binding and basal and cAMP-stimulated PKA activity were significantly decreased in prefrontal cortex of suicide victims. The extent of the decrease in cAMP-stimulated PKA activity was greater than that of the endogenous PKA activity. Interestingly, we found significant changes only in those suicide victims who had major depression but not in those who had other mental disorders. We recently confirmed this finding in a large population of depressed suicide victims obtained from another cohort (CitationDwivedi et al 2004a). We further examined whether the decreases in PKA regulatory and catalytic activities are related to altered expression of specific C and/or R subunits. Interestingly, we observed that protein levels of only PKA RIIβ and Cβ were significantly lower in prefrontal cortex of depressed suicide subjects, without any changes in protein levels of the other regulatory or catalytic subunits. These decreases were associated with decreases in their respective mRNA levels. Our results thus suggest that the decreases in [3H]cAMP binding and PKA activity could be due to decreases in the expression of RIIβ and Cβ, respectively. CitationOdagaki et al (2001) found similar results in prefrontal cortex of depressed patients. In a clinical population, CitationPerez et al (2001) studied the expression of PKA R and C subunits and found that the level of regulatory subunits of type II PKA was significantly lower in platelets of untreated depressed patients compared with euthymic patients or normal controls.
Overall, studies of PKA in unipolar and bipolar patients suggest that whereas cAMP binding is decreased in both bipolar and depressed patients, PKA activity is increased in bipolar patients and decreased in depressed patients. In addition, the expression of RIIβ is increased in bipolar patients and decreased in depressed patients. On the other hand, whereas the Cβ subunit is decreased in depressed patients, the level of Cα is increased in bipolar patients. Thus, it appears that PKA is differentially regulated in depressed versus bipolar patients. The postmortem brain studies did not differentiate between melancholic and nonmelancholic subtypes, therefore, it is difficult to state that the changes in PKA in depressed subjects are specific to one subtypes as suggested in studies of peripheral tissues of depressed subjects. Further studies are required to confirm these findings.
Hypothalamic-pituitary-adrenal axis (HPA) and PKA
Effect of Stress on PKA
Stress is known to cause changes in the hypothalamic-pituitary-adrenal (HPA) axis in both humans as well as in nonhuman models. Stress or a hyperactive HPA axis is a well-known phenomenon in depression. It is, therefore, of interest to examine whether changes in PKA are related to stress. In a detailed study, we examined the effect of glucocorticoids after bilateral adrenalectomy and supplementation with exogenous glucocorticoid, as well as the effect of endogenous glucocorticoids, on various measures of PKA in rat brain (CitationDwivedi et al 2000). For this, rats were injected with various doses of corticosterone at different time intervals, and in a separate experiment, adrenalectomies were performed on rats who were then injected with various doses of corticosterone. We observed that 1 day of corticosterone treatment had no significant effect, but 4 days of corticosterone treatment decreased [3H]cAMP binding to the regulatory subunit of PKA and PKA catalytic activity in the rat cortex and hippocampus. These changes were much more profound after 14 days of corticosterone. These effects were also dose dependent. The higher dose of corticosterone was much more effective in causing changes in PKA than the lower dose. Adrenalectomy produced the direct opposite results to those of corticosterone treatment, increasing [3H]cAMP binding and PKA activity in both cortex and hippocampus in a time-dependent manner. These changes were reversed by corticosterone treatment in a dose dependent manner. The higher dose of corticosterone completely reversed the changes in PKA after adrenalectomy (CitationDwivedi et al 2000). A very interesting observation was noted when we examined the expression levels of PKA regulatory and catalytic subunits. We found that the mRNA and protein expressions of RIα, RIIβ, and Cβ isoforms were significantly decreased in cortex and hippocampus after corticosterone treatment. Removal of adrenal glands increased the expression of these subunits, and corticosterone treatment of adrenalectomized rats reversed the adrenalectomy-induced changes in PKA RIα, RIIβ, and Cβ subunits (CitationDwivedi et al 2000). These changes were very similar to those we observed in depressed suicide subjects. Our studies thus suggest that the expression of specific isoforms of PKA regulatory and catalytic subunits is under the regulation of glucocorticoids and that stress may be playing an important role in such changes.
Effect of learned helpless behavior on PKA
Since the ability to cope with stress is critical at the human level, parallel studies of the effects of uncontrollable stress have been performed in animals, with the results of proactive interference with the acquisition of escape/avoidance responding (CitationSeligman and Maier 1967). This phenomenon is termed learned helplessness (LH) and has been used extensively as an animal model of stress-induced behavioral depression (CitationPetty and Sherman, 1979; CitationSherman et al 1982). To further examine whether there were alterations in PKA in LH behavior, we studied similar paradigms in the brain of LH rats as we studied in the brain of suicide subjects. We found very similar changes in PKA, such that [3H]cAMP binding and PKA activity were significantly decreased along with selectively decreased expression of only RIIβ and Cβ subunits (CitationDwivedi et al 2004b), as we had observed in depressed suicide subjects. These changes were well correlated with the stress-induced behavioral paradigms. For example, LH behavior dissipated 4 days after the induction of LH. At the same time, in these animals, the changes in PKA also reverted to the normal level, which suggests that the changes in PKA were specific to stress-induced behavioral depression. Interestingly, very recently, CitationRao et al (2007a) reported that CBZ treatment significantly decreased cAMP-dependent PKA activity, but not basal PKA activity, compared with control values On the other hand, animal model of depression show that PKA activity is increased in frontal cortex of rats (CitationRao et al 2007, Citationb). These studies further strengthen the hypothesis that bipolar disorder could be associated with increased PKA, which may be reversed by mood stabilizing drugs.
Mechanistic and functional aspects of PKA changes
The mechanisms responsible for the selectively altered expression of PKA subunits in bipolar or depressed patients are not clear at the present time. From Chang’s study (CitationChang et al 2003b) it is clear that the alterations in RIIβ and Cα protein levels in postmortem brain of bipolar patients are due to changes at the post-translational level. In depressed patients, however, we found that mRNA levels of RIIβ and Cβ were decreased (CitationDwivedi et al 2004a). It has been demonstrated in rat Sertoli cells, that activation of the cAMP pathway stimulates mRNA levels of RIα, RIIα, Cα, and RIIβ, the increase in RIIβ being the greatest (CitationOyen et al 1988; CitationLandmark et al 1993). However, prolonged stimulation with cAMP results in a decrease in RIIβ mRNA (CitationOyen et al 1988). In depressed patients, it has been shown that the expression of stimulatory G protein (GSα) is increased and of inhibitory G protein (Giα) is decreased (CitationPachecco et al 1996; CitationDwivedi 2002b), which suggests greater stimulation of the cAMP pathway. It is quite possible that this sustained stimulation may cause an adaptive change in expression of the RIIβ subunit. Another possible explanation of the reduced expression of the RII β subunit could be related to protein kinase C (PKC), a phosphorylating enzyme in the phosphoinositide signaling pathway. It has been shown that activation of PKC inhibits cAMP induction of RIIβ mRNA and that this inhibition persists even after PKC downregulation (CitationTasken et al 1992). The explanation of the selective decrease in the Cβ subunit is more complex. It has been shown that regulatory subunits associate with catalytic subunits and protect the catalytic subunits from degradation by proteolytic enzymes (CitationSteinberg and Agard 1981). Furthermore, in brain tissues of RIIβ-mutant mice, there is a profound decrease in Cα and Cβ subunit levels due to increased degradation CitationBrandon et al (1998). However, in postmortem brain of depressed subjects, expression of only the Cβ subunit was decreased, and this decrease was present at both the transcriptional and the translational levels, eliminating the possibility of any degradation. It appears that additional factors or mechanisms may be involved in the regulation of the Cβ gene in postmortem brain of depressed suicide subjects. Irrespective of the mechanisms involved, the above-mentioned studies clearly indicate that specific catalytic and regulatory subunits of PKA are differentially regulated in unipolar and bipolar disorders.
As mentioned earlier, at the functional level, PKA is involved in myriads of physiological functions in the brain, including neurotransmitter synthesis and release, gene expression, synaptic plasticity, memory, and cell growth and differentiation. Furthermore, PKA participates in neurite outgrowth (CitationSong and Poo 1999), neuronal differentiation (CitationLiesi et al 1983), and cell survival (CitationRydel and Greene 1988; CitationLi et al 2000). The major mechanism of PKA-mediated function is through the phosphorylation of specific substrates, which include CREB, nuclear receptors, and high mobility group-containing proteins, thus influencing their dimerization or DNA binding properties (CitationFimia and Sassone-Corsi 2001). Any abnormality in the expression or catalytic activation state of PKA would alter the functional capacity of the substrates that are activated in response to PKA activation. Some of the substrates whose functions are altered in mood disorders are discussed in the next sections.
Another important aspect of PKA is its involvement in the cross-talk between different signaling mechanisms. For example, PKA interacts at various levels with other signaling pathways, including inactivation of phospholipase C (PLC)β (CitationLiu and Simon 1996); phosphorylation of inositol 1,4,5-trisphosphate (IP3) receptors, thereby modulating Ca2+ influx (CitationBugrim 1999); phosphorylation of G proteins, with a consequent decrease in PI hydrolysis and Ca2+ release (CitationWen et al 1992); and phosphorylation of calcium-calmodulin kinase (CitationMatsushita and Nairn 1999). Through these actions PKA also profoundly affects physiological functions mediated by other signaling pathways.
The functions of each specific catalytic and regulatory subunit of PKA are not clear; however, CitationLudvig et al (1990) showed that RIIβ immunolabeling is associated with post-synaptic structures, suggesting that this subunit is involved in several post-synaptic neuronal functions. Also a few studies indicate that each catalytic and regulatory subunit has distinct functions in the CNS. For example, CitationConstantinescu et al (2002) reported that in NG108-15 cells, which contain type I PKA (CαRIβ) primarily in cytosol and type II PKA (CαRIIβ) in the particulate and nuclear fractions, forskolin- or ethanol-stimulated activation of CREB and gene activation are differentially regulated by the two different types of PKAs. Exposure of these cells to ethanol or forskolin caused translocation of only Cα, but not Cβ, to the nucleus. They have proposed that Type II PKA is responsible for the induction of CREB. Interestingly, it has been shown that RIIβ can bind to and activate transcription via CRE (CitationSrivastava et al 1998). Binding of RIIβ to CRE is enhanced by cAMP, and mutant mice lacking the autophosphorylation site exhibit reduced capacity to bind CRE (CitationSrivastava et al 1998). On the other hand, Type I PKA is activated in the cytoplasm, and activates other transcription cofactors, whcih interact with phosphorylated CREB to induce gene transcription. Further evidence is derived from a study showing that Type I PKA controls cell growth whereas Type II PKA participates in growth arrest and induction of apoptosis (CitationCho-Chung 1990). Also, many studies have demonstrated that RIIβ and Cβ subunits may be specifically involved in neuronal and behavioral functions. For example, RIIβ-mutant mice exhibit defective motor behavior (CitationBrandon 1998) and Cβ-mutant mice show impaired hippocampal plasticity (CitationQi et al 1996). Targeted disruption of the RIβ subunit gene results in mice that exhibit defects in long-term depression and depolarization, which suggests a deficit in a learning-related form of synaptic plasticity (CitationBrandon et al 1998). It has also been shown that RIβ deficiency produces selective defects in mossy fiber long-term potentiation (CitationBrandon et al 1998).
Whether the observed selective abnormalities in expression of catalytic and regulatory subunits pertain to a specific disease state, such as unipolar or bipolar disorder, is not clear at the present time. However, given the significance of PKA in many biological actions in the brain, together with emerging studies demonstrating specific roles for its regulatory and catalytic subunits in physiological and behavioral manifestations, the observations of differential regulatory and catalytic activities, along with differential selective expression of selective regulatory and catalytic subunits, in mood disorder patients suggest that these abnormalities in PKA may be of critical importance in the pathophysiology of mood disorders.
Target substrates of PKA: Role in mood disorders
Rap-1 in mood disorders
Rap-1 is a member of the Ras family of small guanine nucleotide triphosphates with the highest homology to Ras and is highly expressed in the CNS (CitationKitayama et al 1989; CitationDrugan et al 2000; CitationBos et al 2001). Rap-1 has been shown to be activated through the stimulation of various transmembrane receptors, including receptor tyrosine kinases, G protein-coupled receptors, cytokine receptors, and cell-adhesion molecules (CitationHattori and Minato 2003). A number of studies suggest that Rap-1 is activated directly by PKA or indirectly by cAMP through Epac (cAMP-regulated guanine nucleotide exchange factor) (Citationde Rooij et al 1998; CitationBos et al 2001; CitationBerruti 2003) and thereby modulates various pathways, including the cell survival extracellular signal-regulated kinase (ERK) and the phosphoinositide (PI) 3-kinase pathways. This leads to the modulation of physiological functions, such as cell proliferation, differentiation, adhesion, and neurite outgrowth.
To examine the functional response of altered cAMP, initially, CitationPerez et al (1995) and CitationZanardi et al (1997) studied cAMP-stimulated phosphorylation of a PKA substrate in depressed or bipolar patients and found that the cAMP-stimulated phosphorylation of a 22-kDa protein (which was confirmed as Rap-1) was increased in platelets of euthymic bipolar patients. Recently, CitationPerez et al (2000) found that cAMP-mediated endogenous phosphorylation of Rap-1 was significantly increased, along with an increase in level of Rap-1, in platelets of euthymic bipolar patients. In another study, CitationPerez et al (1999) reported that phosphorylation of Rap-1 was significantly increased in euthymic, depressed, or manic patients. On the other hand, they found that the level of Rap-1 was significantly lower in platelets of depressed patients (CitationPerez et al 2001). Recently, we investigated Rap-1 in postmortem brain of depressed suicide subjects and found that Rap-1 activation was significantly reduced in prefrontal cortex and hippocampus of these subjects. This was associated with significant reductions in Rap-1 mRNA and protein levels in these brain areas (CitationDwivedi et al 2006). These studies indicate differences in activation and expression in Rap-1 in unipolar versus bipolar disorders. The findings of altered Rap-1 in depressed or bipolar patients not only provide the functional link to altered PKA but also indicate that abnormalities in Rap-1 may be crucial in the pathophysiology of mood disorders.
Transcription factor CREB in mood disorders
The functional significance of various signal transduction mechanisms depends upon the activation/repression of transcription factors that regulate the expression of genes involved in physiological functions. One such transcription factor is CREB, whose activity is regulated by PKA. CREB is a member of the basic leucine zipper subfamily of transcription factors (CitationBorrelli et al 1992). Phosphorylation of CREB at serine 133 leads to its dimerization and activation by binding to the promoter region of target genes known as cAMP-response elements (CREs) at the consensus motif 5′TGACGTCA3′, which is found in many neuronally expressed genes (CitationMontiminy et al 1990). It has been shown that CREB can bind to CRE even in the unphosphorylated form; however, it does not stimulate transcription unless it is phosphorylated at the serine 133 residue. CREB phosphorylation matches very well with the stimulation of transcription of CRE-containing genes. After serine 133 phosphorylation by PKA, CREB binds to the transcriptional co-activator CREB binding protein (CBP), which leads to stimulation of the transcription of many cAMP-responsive genes that are involved in several aspects of neuronal functioning, including the excitation of nerve cells (CitationMoore et al 1996; CitationMarshall and Dragunow 2000), central nervous sytem (CNS) development, long-term synaptic plasticity (CitationSilva et al 1998), and cell survival.
A number of direct and indirect studies demonstrate that CREB could be involved in mood disorders (reviewed by CitationSulser et al 2002). Studies in rodents demonstrate that overexpression of CREB in hippocampus causes antidepressant-like effects in forced swim stressed or learned helpless rats (CitationChen et al 2001). On the other hand, in the temporal cortex of depressed patients, CitationDowlatshahi et al (1998) reported decreased CREB immunoreactivity. CitationYamada et al (2003) examined the level of CREB and of its active form, phospho-CREB, in orbitofrontal cortex of antidepressant-free patients with major depression. They found that both CREB and phospho-CREB were significantly decreased in depressed subjects. In another study, CitationManier et al (2000) found that the isoproterenol-stimulated increase in phospho-CREB was significantly decreased in fibroblasts of depressed patients. CitationAkin et al (2005) recently reported decreased phospho-CREB in fibroblasts of depressed subjects and showed that this decrease is associated with the melancholic subtype. In a postmortem brain study, we demonstrated that expression and functional characteristics of CREB were decreased in prefrontal cortex and hippocampus of a group of suicide subjects that included depressed suicide subjects (CitationDwivedi et al 2003a) A role for CREB in depression is further supported by a recent study demonstrating a linkage of CREB with major depression. In this study CitationZubenko et al (2002) performed a linkage analysis of six polymorphic markers located in a 15 cM region of chromosome 2q33-35 and unipolar depression. They found significant linkage of unipolar depression to a 451 Kb region of 2q33-34, which contains the CREB1 gene. This suggests that the CREB1 gene may be an attractive candidate for a susceptibility gene for unipolar depression. These described studies thus indicate decreased expression/functional response of CREB in depression.
Many different classes of antidepressants upregulate CREB expression in rat hippocampus (CitationNibuya et al 1996), which suggests that CREB participates in the mechanism of action of antidepressants. Recently, CitationKoch et al (2002) investigated phosphorylation of CREB in T-lymphocytes of depressed patients before and after antidepressant treatment. They found that responders showed a significant increase in phosphorylation of CREB compared with nonresponders, suggesting that CREB may be a molecular state marker for antidepressant response. Recently, CitationThome et al (2000) studied the functional relevance of increased CREB by antidepressants, and observed that in transgenic mice containing the CRE-Lac Z reporter, activation of the CRE site led to increased CRE-mediated gene transcription.
In contrast to what occurs with antidepressants, CitationStewart et al (2001) found a lower CREB level in temporal cortex of bipolar patients treated with mood stabilizing drugs. In rat cortex and hippocampus, similar findings were noted by CitationChen et al (1999), who reported that lithium, but not valproate, decreased the level of phospho-CREB. An in vitro study also suggested that lithium decreases cAMP-dependent CREB phosphorylation and DNA binding activity in neuroblastoma cells (CitationWang et al 1999). These studies thus suggest that mood stabilizing drugs and antidepressants behave differently in regulating CREB. Whether this mean that CREB expression is increased in bipolar patients is not clear at the present time, however; these studies do point in this direction.
BDNF in mood disorders
Among the epigenetic factors that may influence the development and survival of neurons in the CNS are neurotrophins. The most important and widely distributed member of the neurotrophin family in the brain is BDNF. It has been shown that PKA/CREB activation increases BDNF transcription through a Ca2/CRE within exon III of BDNF (CitationFinkbeiner 2000). BDNF-mediated activation of its cognate receptor, tyrosine receptor kinase (Trk) B, influences neurite outgrowth, phenotypic maturation, morphological plasticity, and synthesis of proteins for differentiated functioning of neurons and synaptic functioning (CitationHuang and Reichardt 2001). Several studies suggest that BDNF is also involved in nerve regeneration, structural integrity, and maintenance of neuronal plasticity in the adult brain, including activity-dependent regulation of synaptic activity, and in neurotransmitter synthesis (CitationThoenen 1995). A pathological alteration of the neurotrophic factor system thus may not only lead to altered neural maintenance and regeneration, and therefore structural abnormalities in the brain, but also to reduced neural plasticity. The result could be an impairment of an individual’s ability to adapt to crisis situations.
Emerging studies suggest that mood disorders and stress may both be associated with structural abnormalities in the brain. For example, patients with affective disorders show reductions in hippocampal volume, in density and size of cortical neurons in the dorsolateral PFC and orbitofrontal cortex, and in density of nonpyramidal neurons. There is also a layer-specific reduction in interneurons in the anterior cingulate cortex and in nonpyramidal neurons in the hippocampal formation, and a marked decrease in glial cell number and density in PFC of mood disorder patients (CitationRajkowska 2000; CitationBenes et al 2001; CitationMiguel-Hidalgo et al 2002). Similarly, several studies show that stress or glucocorticoid administration causes neuronal atrophy (CitationSapolsky 1996; CitationBrown et al 1999; CitationMcEwen 1999), a decrease in the number or length of apical dendrites (CitationWoolley et al 1990; CitationWatanabe et al 1992), and even loss of hippocampal neurons (CitationSapolsky 2000). Among the epigenetic factors that may influence the development and survival of neurons in the CNS are neurotrophins. The most important and widely distributed member of the neurotrophin family in the brain is BDNF. BDNF-mediated activation of tyrosine receptor kinase (Trk) B influences neurite outgrowth, phenotypic maturation, morphological plasticity, and synthesis of proteins for differentiated functioning of neurons and synaptic functioning (CitationHuang and Reichardt 2001). Recent studies suggest that BDNF is also involved in nerve regeneration, structural integrity, and maintenance of neuronal plasticity in adult brain, including activity-dependent regulation of synaptic activity, and in neurotransmitter synthesis (CitationHuang and Reichardt 2001; CitationThoenen 1995). A pathological alteration of the neurotrophic factor system thus may not only lead to altered neural maintenance and regeneration, and therefore structural abnormalities in the brain, but also to reduced neural plasticity. The results could be impairment of an individual’s ability to adapt to crisis situations.
BDNF in unipolar disorder
There is a growing body of evidence that demonstrates the involvement of BDNF in unipolar disorder. For example, CitationSiuciak et al (1996) studied the behavioral effects of midbrain infusion of BDNF in rats. They found that rats who received BDNF showed greatly reduced learned helpless behavior compared with rats who were given saline. CitationShirayama et al (2002) also reported that bilateral infusion of BDNF into the dentate gyrus of rats produced an antidepressant-like effect in both learned-helpless and forced-swim-test models of depression. Further evidence of the role of BDNF in depression comes from studies of BDNF expression in rat brain after different types of stresses. Immobilization stress and glucocorticoids significantly decrease BDNF expression in limbic areas (CitationBarbany and Persson 1992; CitationNibuya et al 1995; CitationSmith et al 1995a, Citation1995b; CitationDuman et al 1997; CitationUeyama et al 1997). Chronic treatment with antidepressants prevents the stress-induced lowering of BDNF (CitationNibuya et al 1995; CitationDuman 2002). More direct evidence is derived from studies showing that serum BDNF levels were significantly decreased in drug-free depressed patients and were negatively correlated with Montgomery-Asberg-Depression Rating Scale scores (CitationKarege et al 2002b). Recently, CitationShimizu et al (2003) reported that the serum level of BDNF was significantly decreased in antidepressant-free depressed patients compared with those medicated with antidepressants or normal controls. They also reported that the serum BDNF level was negatively correlated with HDRS scores. More recently, we observed that expression of BDNF and of the active form of TrkB, ie, full-length TrkB, is significantly decreased in prefrontal cortex and hippocampus of depressed and nondepressed suicide subjects (CitationDwivedi et al 2003b). At the genetic level, CitationSen et al (2003) has reported that the val allele of the BDNF val66met polymorphism is associated with neuroticism, a heritable risk factor for depression, further emphasizing the role of BDNF in depression.
Other evidence of the role of BDNF in depression comes from studies suggesting that BDNF is a target of antidepressants. For example, chronic administration of several classes of antidepressants or electroconvulsive shock upregulates the expression of BDNF in rat hippocampus (CitationSmith et al 1995b; CitationNibuya et al 1995). Electroconvulsive shock also increases the expression of TrkB in rat hippocampus and blocks the downregulation of BDNF in response to restraint-induced stress (CitationNibuya et al 1995; CitationLindeforts et al 1995). A consecutive 10-day daily exposure to ECS also increases the expression of BDNF, not only in hippocampus, but also in parietal cortex, entorhinal cortex, frontal cortex, neostriatum, and septum (CitationAltar et al 2003). Pretreatment with antidepressants blocks the stress-induced downregulation of BDNF in hippocampus (CitationLindeforts et al 1995). A postmortem brain study of depressed subjects who were being treated with antidepressants at the time of death also suggests an increase in the level of BDNF in hippocampal regions (CitationChen et al 2001).
BDNF in bipolar disorder
BDNF may also play an important role in the pathophysiology of bipolar disorder. For example, chronic lithium or valproate treatment of rats increases the expression of BDNF in hippocampus and temporal and frontal cortices (CitationFukumoto et al 2001). A number of genetic studies in humans indicate a linkage of BDNF to bipolar disorder. Chromosome 11p13-14 is a putative locus for the genes responsible for the development of bipolar disorder. The BDNF gene is located in this region of the chromosome. Recently, family-based association studies for the dinucleotide repeat polymorphism at position -1040 bp showed that allele A3 was preferentially transmitted to the affected individuals. The results of the val66met single-nucleotide polymorphisms (SNP) showed a significant association for allele G. Transmission/disequilibrium test haplotype analysis showed a significant result for the G-allele combination, which suggests that a DNA varient in the vicinity of the BDNF locus confers susceptibility to bipolar disorder (CitationNeves-Pereira et al 2002). CitationSklar et al (2002) studied the association between 76 candidate genes and bipolar disorder by genotyping 90 SNPs in these genes and identified BDNF as a potential risk allele for bipolar disorder; although CitationNakata et al (2003) found no association for two polymorphisms (-1360C > T and 196G > A) in a Japanese population with bipolar disorder. Very recently, CitationTsai (2004) suggested the possibility that BDNF overactivity may be associated with the manic state on the basis of studies showing: 1) there is a positive association between genetic polymorphism of BDNF and bipolar disorder, 2) agents that induce mania increase BDNF (CitationPeet and Peters 1995; CitationVollenweider et al 1998; CitationMeredith et al 2002), 3) atypical antipsychotics, often used for the treatment of mania, decrease the level of BDNF in frontal and occipital cortices, and hippocampus (CitationAngelucci et al, 2000), and 4) there is increased mossy fiber staining in the supragranular layer in bipolar patients, which is associated with BDNF action. Further studies are required to explore such an interesting idea.
Conclusion
In this review we have critically discussed the findings pertaining to various components of adenyly cyclase–cAMP signaling relevant to mood disorders. We have briefly included studies pertaining to cAMP formation and adenylyl cyclase, however; given the importance of PKA in mediating crucial physiological functions in the brain, especially emphasized is the role of this phosphorylating enzyme in mood disorders (). Although not consistently, the majority of studies suggest that the changes in agonist-stimulated cAMP formation and in catalytic activation of adenylyl cyclase are direct opposites in unipolar versus bipolar states, such that cAMP formation and adenylyl cyclase activity are decreased in unipolar and increased in bipolar disorder. The most consistent findings were observed in PKA, and significant conclusions can be drawn from these findings. For example, bipolar disorder patients show increased PKA catalytic activity, which is associated with selectively increased expression of catalytic (Cα) and regulatory (RIIβ) subunits. In contrast, unipolar depression is associated with decreased PKA activity and decrease expression of catalytic Cβ and regulatory RII β subunits. These observations have been further substantiated by studies in rodents which show that corticosterone administration or induction of learned helplessness behavior causes similar changes in PKA as were found in postmortem brain of depressed subjects. The opposite findings involving cAMP formation and adenylyl cyclase and PKA activities in unipolar and bipolar disorders are quite important, and the findings of the differential regulation of PKA C and R subunits in these two disorders are especially intriguing. Whether these particular PKA subunits are involved in specific behavioral and clinical manifestations, remains to be elucidated.
Figure 2 PKA catalytic (C) and regulatory (R) subunit isoforms expressed in brain and decrease (↓) or increase (↑) in expression of specific C and R subunits and of CREB and BDNF in unipolar (UP) and bipolar (BP) disorders. PKA R subunits are divided into two subtypes, RI and RII. RI and RII subtypes contain two isoforms each, ie, α and β. All four R subunit isoforms are expressed in brain. On the other hand, brain expresses α and β isoforms of C subunit. The indicated decreases and increases in PKA subunits are based on human postmortem and peripheral studies in unipolar and bipolar disorder patients. The question mark (?) indicates the possible upregulation of BDNF and CREB in bipolar disorder patients based on preclinical studies.
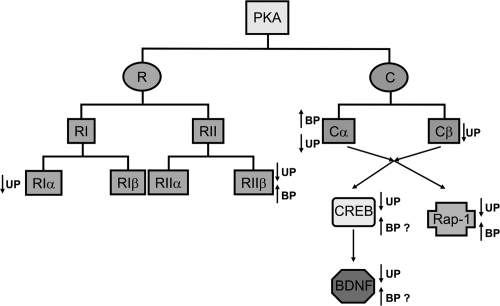
In addition, interesting findings about the substrates for PKA, namely, Rap-1, CREB, and BDNF, have emerged. For example, changes in Rap-1 also appear to be the opposite in unipolar and bipolar disorders, as has been for the changes in PKA. Similarly, an intriguing hypothesis has been put forward suggesting an induction of BDNF expression in the manic phase but reduction in its expression in the depressed phase. A systematic study in a large population of mood disorder patients is required to confirm these findings. Overall, in view of the current findings, a novel concept of the neurobiology of mood disorders is evolving in which disruptions in PKA/CREB/Rap-1/BDNF signaling could be seen as important biological risk factors that may be useful in delineating the etiology of these two disorders and eventually result in site-specific therapeutic interventions.
Acknowledgements
This work is funded by the National Institute of Mental Health (RO1 MH68777 and KO1 MH01836), American Foundation for Suicide Prevention, and NARSAD to Dr. Yogesh Dwivedi and by the National Institute of Mental Health (R01 MH48153 and RO1 MH56528) to Dr. Pandey.
References
- AkinDManierDHSanders-BushESignal transduction abnormalities in melancholic depressionInt J Neuropsychopharmacology20058516
- AltarCAWhiteheadREChenREffects of electroconvulsive seizures and antidepressant drugs on brain-derived neurotrophic factor protein in rat brainBiol Psychiatry200354703914512210
- AngelucciFMatheAAAloeLBrain-derived neurotrophic factor and tyrosine kinase receptor TrkB in rat brain are significantly altered after haloperidol and risperidone administrationJ Neurosci Res2000607839410861791
- BarbanyGPerssonHRegulation of neurotrophin mRNA expression in the rat brain by glucocorticoidsEur J Neurosci1992439640312106347
- BeebeSJThe cAMP-dependent protein kinases and cAMP signal transductionSemin Cancer Biol19945285947803765
- BelmakerRHZoharJEbsteinRPCyclic nucleotides in mental disorderAdv Cyclic Nucleotide Res198012187986250353
- BenesFMVincentSLTodtenkopfMThe density of pyramidal and nonpyramidal neurons in anterior cingulate cortex of schizophrenic and bipolar subjectsBiol Psychiatry20015039540611566156
- BerrettiniWHCappellariCBNurnbergerJIβ-adrenergic receptors on lymphoblasts. A study of manic-depressive illnessNeuropsychobiology1987171582819764
- BerrutiGcAMP activates Rap1 in differentiating mouse male germ cells: a new signaling pathway mediated by the cAMP-activated exchange factor Epac?Cell Mol Biol200349381812887090
- BornfeldtKEKrebsEGCrosstalk between protein kinase A and growth factor receptor signaling pathways in arterial smooth muscleCell Signal1999114657710405757
- BorrelliEMontmayeurJPFoulkesNSSignal transduction and gene control: The cAMP pathwayCrit Rev Oncog19923321381329990
- BosJLde RooijJReedquistKARap1 signalling: adhering to new modelsNat Rev Mol Cell Biol200123697711331911
- BrandonEPLogueSFAdamsMRDefective motor behavior and neural gene expression in RIIβ-protein kinase A mutant miceJ Neurosci1998183639499570795
- BrownESRushAJMcEwenBSHippocampal remodeling and damage by corticosteroids: implications for mood disordersNeuropsychopharmacology1999214748410481830
- BugrimAERegulation of Ca2+ release by cAMP-dependent protein kinase A mechanism for agonist-specific calcium signaling?Cell Calcium1999252192610378083
- ChangALiPPWarshJJAltered cAMP-dependent protein kinase subunit immunolabeling in postmortem brain from patients with bipolar affective disorderJ Neurochem2003a847819112562522
- ChangALiPPWarshJJcAMP-dependent protein kinase (PKA) subunit mRNA levels in postmortem brain from patients with bipolar affective disorder (BD)Mol Brain Res2003b116273712941458
- ChenACShirayamaYShinKHExpression of the cAMP response element binding protein (CREB) in hippocampus produces an antidepressant effectBiol Psychiatry2001497536211331083
- ChenBDowlatshahiDMacQueenGMIncreased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medicationBiol Psychiatry200150260511522260
- ChenBWangJFHillBCLithium and valproate differentially regulate brain regional expression of phosphorylated CREB and c-FosMol Brain Res199970455310381542
- Cho-ChungYSRole of cyclic AMP receptor proteins in growth, differentiation, and suppression of malignancy: new approaches to therapyCancer Res19905070931002224844
- CleggCHMcKnightGSGenetic characterization of a brain-specific form of the type I regulatory subunit of cAMP-dependent protein kinaseProc Natl Acad Sci U S A198885370373375237
- ConstantinescuAGordonASDiamondIcAMP-dependent protein kinase types I and II differentially regulate cAMP response element-mediated gene expressionJ Biol Chem200227718810611886856
- de RooijJZwartkruisFJVerheijenMHEpac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMPNature199839647479853756
- DowlatshahiDMacQueenGMWangJFIncreased temporal cortex CREB concentrations and antidepressant treatment in major depressionLancet1998352175459848357
- DruganJKRogers-GrahamKGilmerTThe Ras/p120 GTPase-activating protein (GAP) interaction is regulated by the p120 GAP pleckstrin homology domainJ Biol Chem200027535021710954709
- DumanRSHeningerGRNestlerEJA molecular and cellular theory of depressionArch Gen Psychiatry1997545976069236543
- DumanRSMalbergJNakagawaSNeuronal plasticity and survival in mood disordersBiol Psychiatry200048732911063970
- DumanRSStructural alterations in depression: cellular mechanisms underlying pathology and treatment of mood disordersCNS Spectr20027140215220856
- DwivediYConleyRRRobertsRC[3H]cAMP binding sites and protein kinase A activity in the prefrontal cortex of suicide victimsAm J Psychiatry2002a159667311772692
- DwivediYMondalACRizaviHSDifferential and brain region-specific regulation of Rap-1 and Epac in depressed suicide victimsArch Gen Psychiatry2006636394816754837
- DwivediYMondalACShuklaPKAltered protein kinase A in brain of learned helpless rats: effects of acute and repeated stressBiol Psychiatry2004b56304015219470
- DwivediYPandeyGNAdrenal glucocorticoids modulate [3H]cyclic AMP binding to protein kinase A (PKA), cyclic AMP-dependent PKA activity, and protein levels of selective regulatory and catalytic subunit isoforms of PKA in rat brainJ Pharmacol Exp Ther2000294103610871301
- DwivediYRaoJSRizaviHSAbnormal expression and functional characteristics of cyclic adenosine monophosphate response element binding protein in postmortem brain of suicide subjectsArch Gen Psychiatry2003a602738212622660
- DwivediYRizaviHSConleyRRmRNA and protein expression of selective alpha subunits of G proteins are abnormal in prefrontal cortex of suicide victimsNeuropsychopharmacology2002b2749951712377388
- DwivediYRizaviHSConleyRRAltered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjectsArch Gen Psychiatry2003b608041512912764
- DwivediYRizaviHSShuklaPKProtein kinase A in postmortem brain of depressed suicide victims: altered expression of specific regulatory and catalytic subunitsBiol Psychiatry2004a552344314744463
- EbsteinRPLererBShapiraBCyclic AMP second-messenger signal amplification in depressionBr J Psychiatry198815266592844354
- ExteinITallmanJSmithCCChanges in lymphocyte beta-adrenergic receptors in depression and maniaPsychiatry Res197911917233157
- FieldsALiPPKishSJIncreased cyclic AMP-dependent protein kinase activity in postmortem brain from patients with bipolar affective disorderJ Neurochem19997317041010501218
- FimiaGMSassone-CorsiPCyclic AMP signalingJ Cell Sci20011141971211493633
- FinkbeinerSCalcium regulation of the brain-derived neurotrophic factor geneCell Mol Life Sci20005739440110823240
- FrancisSHCorbinJDCyclic nucleotide-dependent protein kinases: intracellular receptors for cAMP and cGMP actionCrit Rev Clin Lab Sci19993627532810486703
- FukumotoTMorinobuSOkamotoYChronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brainPsychopharmacology2001158100611685390
- HattoriMMinatoNRap1 GTPase: functions, regulation, and malignancyJ Biochem20031344798414607972
- HealyDCarneyPALeonardBEMonoamine-related markers of depression - changes following treatmentJ Psychiatr Res198317251606821276
- HirschfeldRMWeissmanMMDavisKLCharneyDCoyleJTNemeroffCRisk factors for major depression and bipolar disorderNeuropsychopharmacology – the fifth generation of progress2002PhiladelphiaLippincott Williams and Wilkins101725
- HuangEJReichardtLFNeurotrophins: Roles in neuronal development and functionAnnu Rev Neurosci20012467773611520916
- JansLAWRiedelWJMarkusCRSerotonergic vulnerability and depression: assumptions, experimental evidence and implicationsMol Psychiatry2007125224317160067
- JeanningrosRMazzolaPAzorinJMβ-adrenoceptor density of intact mononuclear leukocytes in subgroups of depressive disordersBiol Psychiatry199129789981675894
- JordanJDLandauEMIyengarRSignaling networks: the origins of cellular multitaskingCell200010319320011057893
- KanofPDCoccaroEFJohnsCACyclic-AMP production by polymorphonuclear leukocytes in psychiatric disordersBiol Psychiatry198925413202539205
- KanofPDJohnsCDavidsonMProstaglandin receptor sensitivity in psychiatric disordersArch Gen Psychiatry198643987933019269
- KanofPDJohnsCADavidsonMPlatelet alpha2-adrenergic receptor function in psychiatric disordersPsychiatry Res19882311222834762
- KaregeFPerretGBondolfiGDecreased serum brain-derived neurotrophic factor levels in major depressed patientsPsychiatry Res2002b109143811927139
- KaregeFSchwaldMCisseMThe cAMP-PKA activity and BDNF expression in lymphoblast cells of bipolar disorder patientsInt J Neuropsychopharmacol2002a5S59
- KaregeFSchwaldMPapadimitriouPThe cAMP-dependent protein kinase A and brain-derived neurotrophic factor expression in lymphoblast cells of bipolar affective disorderJ Affect Disord2004791879215023493
- KayGSargeantMMcGuffinPThe lymphoblast beta-adrenergic receptor in bipolar depressed patients: characterization and down-regulationJ Affect Disord199327163728386731
- KesslerRCMcGonagleKAZhaoSLifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the National Comorbidity SurveyArch Gen Psychiatry1994518198279933
- KitayamaHSugimotoYMatsuzakiTA ras-related gene with transformation suppressor activityCell19895677842642744
- KlysnerRGeislerARosenbergREnhanced histamine and beta-adrenoreceptor-mediated cyclic AMP formation in leukocytes from patients with endogenous depressionJ Affect Disord198713227322824589
- KochJMKellSHinze-SelchDChanges in CREB-phosphorylation during recovery from major depressionJ Psychiatr Res2002363697512393305
- LandmarkBFOyenOSkalheggBSCellular localization and age-dependent changes of the regulatory subunits of cAMP-dependent protein kinase in rat testisJ Reprod Fertil199399323348107013
- LaraJKusanoKHouseSInteractions of cyclic adenosine monophosphate, brain-derived neurotrophic factor, and glial cell line-derived neurotrophic factor treatment on the survival and growth of postnatal mesencephalic dopamine neurons in vitroExp Neurol2003180324512668147
- LiMWangXMeintzerMKCyclic AMP promotes neuronal surival by phosphorylation of glycogen synthase kinase 3βMol Cell Biol20002093566311094086
- LiesiPRechardtLWartiovaaraJNerve growth factor induces adrenergic neuronal differentiation in F9 teratocarcinoma cellsNature198330626576139754
- LindefortsNBrodinEMetsisMSpatiotemporal selective effects on brain-derived neurotrophic factor and trkB messenger RNA in rat hippocampus by electroconvulsive shockNeuroscience199565661707609868
- LiuMSimonMIRegulation by cAMP-dependent protein kinase of a G-protein-mediated phospholipase CNature19963828378657310
- LudvigNRibakCEScottJDImmunocytochemical localization of the neural-specific regulatory subunit of the type II cyclic AMP-dependent protein kinase to postsynaptic structures in the rat brainBrain Res1990520901022207649
- MajMArianoMGArenaFPlasma cortisol, catecholamine and cyclic AMP levels, response to dexamethasone suppression test and platelet MAO activity in manic-depressive patients. A longitudinal studyNeuropsychobiology198411168736089030
- ManierDHEiringASheltonRC(-adrenoceptor-linked protein kinase A (PKA) activity in human fibroblasts from normal subjects and from patients with major depressionNeuropsychopharmacology199615555618946429
- ManierDHSheltonRCEllisTCHuman fibroblasts as a relevant model to study signal transduction in affective disordersJ Affect Disord20006151811099740
- MannJJBrownRPHalperJPReduced sensitivity of lymphocyte beta-adrenergic receptors in patients with endogenous depression and psychomotor agitationN Eng J Med198531371520
- MarshallRDragunowMIs CREB a key to neuronal survival?Trends Neurosci200023485310652539
- MatsushitaMNairnACInhibition of the Ca2+/calmodulin-dependent protein kinase I cascade by cAMP-dependent protein kinaseJ Biol Chem1999274100869310187789
- McEwenBSStress and hippocampal plasticityAnnu Rev Neurosci1999221052210202533
- MeredithGCallenSScheuerDBrain-derived neurotrophic factor expression is increased in the rat amygdala, piriform cortex and hypothalamus following repeated amphetamine administrationBrain Res20029492182712213320
- Miguel-HidalgoJJRajkowskaGMorphological brain changes in depression. Can antidepressants reverse them?Curr Opin20021636172
- MontminyMRGonzalezGAYamamotoKKRegulation of cAMP-inducible genes by CREBTrends Neurosci19901318481693237
- MooreANWaxhamMNDashPKNeuronal activity increases the phosphorylation of the transcription factor cyclic AMP response element binding protein (CREB) in rat hippocampus and cortexJ Biol Chem199627114214208662977
- MurphyDLDonnellyCMoskowitzJCatecholamine receptor functions in depressed patientsAm J Psychiatry19741311389944372899
- NakataKUjikeHSakaiAAssociation study of the brain-derived neurotrophic factor (BDNF) gene with bipolar disorderNeurosci Lett2003337172012524161
- NestlerEJGreengardPSiegelGJAlbersRWAgranoffBWMolinoffPProtein phosphorylation and the regulation of neuronal functionBasic neurochemistry: molecular, cellular, and medical aspects1994BostonLittle Brown44974
- Neves-PereiraMMundoEMugliaPThe brain-derived neurotrophic factor gene confers susceptibility to bipolar disorder: evidence from a family-based association studyAm J Hum Genet200271651512161822
- NibuyaMMorinobuSDumanRSRegulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatmentsJ Neurosci1995157539477472505
- NibuyaMNestlerEJDumanRSChronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampusJ Neurosci1996162365728601816
- OdagakiYGarcia-SevillaJAHugueletPCyclic AMP-mediated signaling components are upregulated in the prefrontal cortex of depressed suicide victimsBrain Res20018982243111306008
- OyenOSandbergMEskildWDifferential regulation of messenger ribonucleic acids for specific subunits of cyclic adenosine 3’, 5’-monophosphate (cAMP)-dependent protein kinase by cAMP in rat Sertoli cellsEndocrinology19881222658662836170
- PachecoMAStockmeierCMeltzerHYAlterations in phosphoinositide signaling and G-protein levels in depressed suicide brainBrain Res199672337458813380
- PandeyGNDyskenMWGarverDLBeta-adrenergic receptor function in affective illnessAm J Psychiatry19791366758219719
- PeetMPetersSDrug-induced maniaDrug Saf199512146537766338
- PerezJTarditoDMoriSAbnormalities of cyclic adenosine monophosphate signaling in platelets from untreated patients with bipolar disorderArch Gen Psychiatry1999562485310078502
- PerezJTarditoDMoriSAltered Rap1 endogenous phosphorylation and levels in platelets from patients with bipolar disorderJ Psychiatr Res2000349910410758250
- PerezJTarditoDRacagniGProtein kinase A and Rap1 levels in platelets of untreated patients with major depressionMol Psychiatry2001644911244484
- PerezJZanardiRMoriSAbnormalities of cAMP-dependent endogenous phosphorylation in platelets from patients with bipolar disorderAm J Psychiatry1995152120467625472
- PettyFShermanADReversal of learned helplessness by imipramineCommun Psychopharmacol197933713575771
- PostRMGoldPRubinowDRPeptides in the cerebrospinal fluid of neuropsychiatric patients: an approach to central nervous system peptide functionLife Sci1982311156180277
- QiMZhuoMSkalheggBSImpaired hippocampal plasticity in mice lacking the Cβ catalytic subunit of cAMP-dependent protein kinaseProc Natl Acad Sci U S A199693157168643673
- RahmanSLiPPYoungLTReduced [3H]cyclic AMP binding in postmortem brain from subjects with bipolar affective disorderJ Neurochem1997682973048978738
- RajkowskaGCell pathology in mood disordersSemin Clin Neuropsychiatry200272819212382210
- RajkowskaGPostmortem studies in mood disorders indicate altered numbers of neurons and glial cellsBiol Psychiatry200048766711063973
- RaoJSBazinetRPRapoprtSILeeHJChronic administration of carbamazepine downregulates AP-2 DNA-binding activity and AP-2alpha protein expression in rat frontal cortexBiol Psychiatry2007611546116806101
- RaoJSErtleyRNLeeHJDeMarJCJrArnoldJTRapoportSIBazinetRPn-3 polyunsaturated fatty acid deprivation in rats decreases frontal cortex BDNF via a p38 MAPK-dependent mechanismMol Psychiatry200712364616983391
- ReiachJSLiPPWarshJJReduced adenylyl cyclase immunolabeling and activity in postmortem temporal cortex of depressed suicide victimsJ Affect Disord1999561415110701471
- RiccioAAhnSDavenportCMMediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neuronsScience199928623586110600750
- RydelREGreeneLAcAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factorProc Natl Acad Sci USA1988851257612829221
- SapolskyRMStress, glucocorticoids and damage to the nervous system: the current state of confusionStress199611119807058
- SapolskyRMThe possibility of neurotoxicity in the hippocampus in major depression: a primer on neuron deathBiol Psychiatry2000487556511063972
- SarkarDErlichmanJRubinCSIdentification of a calmodulin-binding protein that co-purifies with the regulatory subunit of brain protein kinase IIJ Biol Chem1984259984066086653
- SeligmanMEMaierSFFailure to escape traumatic shockJ Exp Psychol196774196032570
- ScottJDGlaccumMBZollerMJThe molecular cloning of a type II regulatory subunit of the cAMP-dependent protein kinase from rat skeletal muscle and mouse brainProc Natl Acad Sci USA198784519263037538
- ScottJDStofkoREMcDonaldJRType II regulatory subunit dimerization determines the subcellular localization of the cAMP-dependent protein kinaseJ Biol Chem19902652156162147685
- SenSNesseRMStoltenbergSFA BDNF coding variant is associated with the NEO personality inventory domain neuroticism, a risk factor for depressionNeuropsychopharmacology20032839740112589394
- SheltonRCManierDHPetersonCSCyclicAMP-dependent protein kinase in subtypes of major depression and normal volunteersInt J Neuropsychopharmacol199921879211281988
- SheltonRCManierDHSulserFcAMP-dependent protein kinase activity in major depressionAm J Psychiatry19961531037428678172
- ShermanADSacquitneJLPettyFSpecificity of the learned helplessness model of depressionPharmacol Biochem Behav198216449547200610
- ShimizuEHashimotoKOkamuraNAlterations of serum levels of brain-derived neurotrphic factor (BDNF) in depressed patients with or without antidepressantsBiol Psychiatry20035470512842310
- ShirayamaYChenACHNakagawaSBrain-derived neurotrophic factor produces antidepressant effects in behavioral models of depressionJ Neurosci20022232516111943826
- SieverLJKafkaMSTargumSPlatelet alpha-adrenergic binding and biochemical responsiveness in depressed patients and controlsPsychiatry Res1984112873026330782
- SilvaAJKoganJHFranklandPWCREB and memoryAnnu Rev Neurosci199821127489530494
- SiuciakJALewisDRWiegandSJAntidepressant-like effect of brain derived neurotrophic factor (BDNF)Pharmacol Biochem Beh1996561317
- SkalheggBSTaskenKSpecificity in the cAMP/PKA signaling pathway. Differential expression, regulation, and subcellular localization of subunits of PKAFront Biosci20005D6789310922298
- SklarPGabrielSBMcInnisMGFamily-based association study of 76 candidate genes in bipolar disorder: BDNF is a potential risk locus. Brain-derived neutrophic factorMol Psychiatry200275799312140781
- SmithMAMakinoSKimSYStress increases brain-derived neurotropic factor messenger ribonucleic acid in the hypothalamus and pituitaryEndocrinology1995a1363743507649080
- SmithMAMakinoSKvetnanskyREffects of stress on neurotrophic factor expression in the rat brainAnn N Y Acad Sci1995b77123498597402
- SoaresJCMannJJThe anatomy of mood disorders-review of structural imaging studiesBiol Psychiatry199741861068988799
- SongHJPooMMSignal transduction underlying growth cone guidance by diffusible factorsCurr Opin Neurobiol199993556310395576
- SpauldingSWThe ways in which hormones change cyclic adenosine 3’5’-monophosphate-dependent protein kinase subunits, and how such changes affect cell behaviorEndocr Rev199314632508262010
- SpearingMBipolar disorder2001Washington, DC NIH Publication No. 02-3679
- SrivastavaKLeeYNNoguchiKThe RIIβ regulatory subunit of protein kinase A binds to cAMP response element: an alternative cAMP signaling pathwayProc Natl Acad Sci USA1998956687929618473
- SteinbergRAAgardDATurnover of regulatory subunit of cyclic AMP-dependent protein kinase in S49 mouse lymphoma cells. Regulation by catalytic subunit and analogs of cyclic AMPJ Biol Chem19812561073146270127
- StewartRJChenBDowlatshahiDAbnormalities in the cAMP signaling pathway in postmortem brain tissue from the Stanley Neuropathology ConsortiumBrain Res Bull200155625911576759
- SulserFThe role of CREB and other transcription factors in the pharmacotherapy and etiology of depressionAnn Med2002343485612452479
- TangWJGilmanAGType-specific regulation of adenylyl cyclase by G protein β gamma subunitsScience1991254150031962211
- TaskenKAKnutsenHKEikvarLProtein kinase C activation by 12-O-tetradecanoylphorbol 13-acetate modulates messenger ribonucleic acid levels for two of the regulatory subunits of 3’,5’-cyclic adenosine monophosphate-dependent protein kinases (RII alpha and RI alpha) via multiple and distinct mechanismsEndocrinology19921301271801311233
- ThoenenHNeurotrophins and neuronal plasticityScience199527059387570017
- ThomeJSakaiNShinKcAMP response element-mediated gene transcription is upregulated by chronic antidepressant treatmentJ Neurosci2000204030610818138
- TsaiSJIs mania caused by overactivity of central brain-derived neurotrophic factor?Med Hypotheses200462192214728999
- UeyamaTKawaiYNemotoKImmobilization stress reduced the expression of neurotrophins and their receptors in the rat brainNeurosci Res199728103109220467
- UhlerMDChriviaJCMcKnightGSEvidence for a second isoform of the catalytic subunit of cAMP-dependent protein kinaseJ Biol Chem19862611536033023318
- VollenweiderFXMaguireRPLeendersKLEffects of high amphetamine dose on mood and cerebral glucose metabolism in normal volunteers using positron emission tomography (PET)Psychiatry Res199883149629849724
- WangJFAsghariVRockelCCyclic AMP responsive element binding protein phosphorylation and DNA binding is decreased by chronic lithium but not valproate treatment of SH-SY5Y neuroblastoma cellsNeuroscience199991771610366032
- WangYCPandeyGNMendelsJPlatelet adenylate cyclase responses in depression: implications for a receptor defectPsychopharmacologia1974362913004367922
- WatanabeYGouldEMcEwenBSStress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neuronsBrain Res199258834151393587
- WenYAnwerKSinghSPProtein kinase A inhibits phospholipase C activity and alters protein phosphorylation in rat myometrial plasma membranesEndocrinology19921311377821324160
- WolfSMartinezCMajzoubJAInducible binding of cyclic adenosine 3′,5′-monophosphate (cAMP)-responsive element binding protein (CREB) to a cAMP-responsive promoter in vivoMol Endocrinol1999136596910319317
- WoolleyCSGouldEMcEwenBSExposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neuronsBrain Res1990531225311705153
- YamadaSYamamotoMOzawaHReduced phosphorylation of cyclic AMP-responsive element binding protein in the postmortem orbitofrontal cortex of patients with major depressive disorderJ Neural Transm20031106718012768362
- YoungLTLiPPKishSJCerebral cortex Gs alpha protein levels and forskolin-stimulated cyclic AMP formation are increased in bipolar affective disorderJ Neurochem19936189088395565
- ZanardiRRacagniGSmeraldiEDifferential effects of lithium on platelet protein phosphorylation in bipolar patients and healthy subjectsPsychopharmacology (Berl)19971294479122362
- ZubenkoGSHughesHB3rdMaherBSGenetic linkage of region containing the CREB1 gene to depressive disorders in women from families with recurrent, early-onset, major depressionAm J Med Genet2002114980712457397