Abstract
Experimental evidence supports a protective role of estrogen in the brain. According to the fact that Alzheimer’s disease (AD) is more common in postmenopausal women, estrogen treatment has been proposed. However, there is no general consensus on the beneficial effect of estrogen or selective estrogen receptor modulators in preventing or treating AD. It has to be said that several factors may markedly affect the efficacy of the treatment. A few years ago, the seladin-1 gene (for selective Alzheimer’s disease indicator-1) has been isolated and found to be down-regulated in brain regions affected by AD. Seladin-1 has been found to be identical to the gene encoding the enzyme 3-beta-hydroxysterol delta-24-reductase, involved in the cholesterol biosynthetic pathway, which confers protection against β-amyloid-mediated toxicity and from oxidative stress, and is an effective inhibitor of caspase-3 activity, a key mediator of apoptosis. Interestingly, we found earlier that the expression of this gene is up-regulated by estrogen. Furthermore, our very recent data support the hypothesis that seladin-1 is a mediator of the neuroprotective effects of estrogen. This review will summarize the current knowledge regarding the neuroprotective effects of seladin-1 and the relationship between this protein and estrogen.
Introduction
Epidemiological data, together with experimental and clinical evidence, appear to support a neuroprotective role of estrogen, Accordingly, hormonal therapy in Alzheimer’s disease (AD), the most prevalent neurodegenerative disorder in the elderly, has been suggested. However, there is no general consensus on this issue, which will be briefly summarized in this review. A debated question concerns the factors that act as downstream mediators of estrogen receptor activation in the brain. The identification of the seladin-1 gene and the finding that it protects the brain from toxic insults led us to hypothesize that this gene might represent the link between estrogen and neuroprotection. Seladin-1 was found to be identical to the gene encoding the enzyme 3-beta-hydroxysterol delta-24-reductase involved in the cholesterol biosynthetic pathway. Cell cholesterol content appears to play an important role in protecting neuronal cells from toxic insults. This review will address the current knowledge on the neuroprotective effects of seladin-1 and the relationship between this protein and estrogen.
Estrogen receptor-mediated neuroprotection
This topic has been extensively reviewed by many authors and the literature will be briefly summarized in this review. It is well known, based on in vitro evidence, that estrogen exerts neurotrophic and neuroprotective effects by stimulating the expression of neurotrophins and cell-survival factors, enhancing synaptic plasticity, and acting as an antioxidant factor (CitationBehl 2003; CitationMaggi et al 2004; CitationTurgeon et al 2006). In addition to the hypothalamus, which is the traditional site of estrogen action in the brain, both the estrogen receptor α (ERα) and β (ERβ) have been found in different brain areas such as the neocortex and the hippocampus, two areas highly involved in AD (CitationBehl 2003). AD is the most prevalent form of late-life mental failure in humans (CitationSelkoe 2001) and it has been calculated that every 72 seconds someone in the United States develops this disease. However, unfortunately there is still no reliable way of preventing or curing this disease. Experimental evidence supports a favorable estrogen effect in neurons, in agreement with the knowledge that AD is more common in women and that decreased estrogen levels after menopause are a risk factor for the disease (CitationPaganini-Hill and Henderson 1994). Thus, estrogen therapy has been considered a rationale option for the treatment of this disease. To date, despite the lack of general consensus, several studies indicated that estrogen treatment may decrease the risk or delay the onset of AD in postmenopausal women (CitationFillit 2002). Conversely, the data from the Women’s Health Initiative Memory Study (WHIMS) trial showed that hormone replacement therapy (HRT) has no benefit (CitationRapp et al 2003; CitationShumaker et al 2003). However, it has to be remembered that different factors may determine the efficacy of estrogen or HRT, such as age, the menopausal status, the route of administration and the dose, the starting cognitive function, and the presence of pre-existing risk factors (ie, smoking, apolipoprotein E genotype) (CitationMacLusky 2004; CitationTurgeon et al 2006). In particular, there seems to be a critical time for estrogen treatment. In fact, early and prolonged therapy has been found to produce the maximum benefit in terms of reduced risk for AD (CitationResnick and Henderson 2002; CitationZandi et al 2002). In addition, estrogen therapy is not the same as HRT and the type of progestogen used may determine the outcome of the therapeutic intervention (CitationSchumacher et al 2007) ().
Figure 1 Factors affecting the response of the brain to hormone replacement therapy.
With regard to the ER involved in neuroprotection, the observations from ERα (ERKO) and ERβ (βERKO) mice suggest a critical role for ERα. In fact, whereas 17β-estradiol exerted a protective effect in the brain of ovariectomized βERKO mice, it did not in ERKO mice (CitationDubal et al 2001). This finding appears in agreement with the reported decreased expression of ERα in hippocampal neurons of AD patients (CitationHu et al 2003). However, a possible role of ERβ in neuroprotection has been postulated, based on the evidence that βERKO mice undergo increased neuronal loss throughout life compared to wild-type controls (CitationWang et al 2001). It has to be said that, in addition to classical nuclear ERs, more recent findings suggest that the brain contains a plethora of ERs, such as ERγ and a variety of nuclear as well as cytoplasmic and plasma membrane receptors (CitationHawkins et al 2000; CitationToran-Allerand 2004; Vasudevan and Pfaff 2007; CitationArbogast 2008).
The neuroprotective role of the selective estrogen receptor modulators (SERMs) has been less extensively investigated. Nonetheless, a neuroprotective effect of tamoxifen and raloxifene has been observed (CitationDhandapani and Brann 2002) and a beneficial role of tamoxifen and raloxifene against β-amyloid toxicity has been demonstrated in rat neurons (CitationO’Neill et al 2004a, Citation2004b). There is increasing evidence that SERMs may also be neurotrophic, by increasing for instance synaptic density and stimulating neurite outgrowth (CitationDhandapani and Brann 2002). Data regarding the clinical use of SERMs in AD are very limited, so far. However, the Multiple Outcomes of Raloxifene Evaluation trial evaluated the cognitive function in more than 5000 women with osteoporosis assigned to receive raloxifene (60 mg or 120 mg) or placebo daily for three years. Compared to those taking placebo, women receiving 120 mg/day of raloxifene had a 33% lower risk of mild cognitive impairment and somewhat lower risks of Alzheimer’s disease and any cognitive impairment (CitationYaffe et al 2005).
In summary, the basic science strongly supports a neuroprotective role of estrogen/SERMs. Although there is no clear cut evidence yet that these molecules can decrease the risk or ameliorate the clinical course of AD, it is conceivable that there might be a proper space for a hormonal-based intervention in this disease. Undoubtedly, a more profound knowledge of the molecular mechanisms by which ERs activation determines neuroprotective effects may further support this conclusion.
The identification and characterization of seladin-1
The seladin-1 gene was first identified by Greeve and colleagues (2000), who used a differential mRNA display approach to identify genes that were differentially expressed in selective vulnerable brain regions in AD, such as the hippocampus, the amygdala, the inferior temporal cortex, and the enthorhinal cortex (CitationSelkoe 2001). Among the >30 genes differentially expressed in AD vulnerable brain regions versus unaffected areas, Greeve and colleagues (2000) identified a novel cDNA showing a markedly reduced expression in the inferior temporal cortex of AD patients compared to the frontal cortex, obtained shortly postmortem. Conversely, this cDNA was evenly expressed in the brain of unaffected individuals. This gene was named seladin-1 from selective Alzheimer’s disease indicator-1 and its full-length cDNA was cloned from a human brain cDNA library (GenBank accession number AF261758). The seladin-1 gene spans 46.4 kb, maps to chromosome 1p31.1–p33, and comprises nine exons and eight introns; it encodes an open reading frame of 516 amino acid residues. To localize the cellular distribution of seladin-1, human H4 neuroglioma cells were transfected with a seladin-1-green fluorescent protein (GFP) fusion construct. Seladin-1 appeared to be mainly located in the endoplasmic reticulum and, although to a lesser extent, in the Golgi apparatus. A subsequent study demonstrated that the down-regulation of seladin-1 expression in vulnerable AD brain areas is paralleled by an increase in the amount of hyperhosphorylated tau, a protein component of neurofibrillary tangles (CitationIivonen et al 2002). Apart the brain, seladin-1 mRNA has been also detected in many different human organs and the highest levels of expression have been found in the adrenal gland, the liver, the lung and the prostate.
With regard to its biological effects, seladin-1 was found to confer resistance against β-amyloid and oxidative stress-induced apoptosis and to effectively inhibit the activation of caspase-3, a key mediator of the apoptotic process. However, another study indicated that this multifaced protein may have a more complex role in cell apoptosis. In fact, following oncogenic and oxidative stress, seladin-1 was found to bind P53 amino terminus domain and to displace E3 ubiquitin ligase Mdm2 from P53, thus resulting in P53 accumulation (CitationWu et al 2004).
Seladin-1 was found to have also an enzymatic activity involved in cholesterol biosynthesis, which was found to be markedly reduced in desmosterolosis, a rare autosomal recessive disorder characterized by multiple congenital anomalies (CitationFitzpatrick et al 1998). In fact, patients with desmosterolosis have elevated plasma levels of the cholesterol precursor desmosterol and this abnormality suggested a deficiency of the enzyme 3-beta-hydroxysterol delta-24-reductase (DHCR24), which catalyzes the reduction of the Δ24 double bond in desmosterol to produce cholesterol. CitationWaterham and colleagues (2001) were able to identify the human DHCR24 cDNA, which appeared identical to seladin-1. DHCR24 activity was confirmed in vitro by enzymatic assay following heterologous expression of the DHCR24 cDNA in Saccharomyces cerevisiae. Conversely, in constructs containing mutant DHCR24 alleles from patients with desmosterolosis the conversion from desmosterol into cholesterol was nil or markedly reduced.
Desmosterolosis belongs to a group of several inherited disorders, linked to enzyme defects in the cholesterol biosynthetic pathway at the post-squalene level, which have been described in recent years (CitationHerman 2003). These genetic diseases are characterized by major developmental malformations and in most cases determine severe neuropsycological alterations, suggesting an important role of cholesterol in brain homeostasis. The role of cholesterol in facilitating the onset and progression of AD is a debated and unsolved question. In fact, on one hand cholesterol may be viewed as a toxic factor. It has been reported that elevated cholesterol levels increase β-amyloid formation in in vitro systems and in most animal models of AD (CitationYanagisawa 2002; CitationPuglielli et al 2003). The identification of the ε4 allelic variant of the apolipoprotein E as a major genetic risk factor for AD is also consistent with a role of cholesterol in the pathogenesis of this disease. Accordingly, epidemiological studies suggested that statin therapy may provide protection against AD, although the clinical benefit of statins might be also due to their cholesterol-independent effects on cerebral circulation and inflammation (CitationReiss et al 2004). This hypothesis appears to be substantiated by the fact that the majority of the commercially available statins does not cross the blood–brain barrier. On the other hand, it has to be considered that the central nervous system (CNS) is a very unique organ with regard to cholesterol metabolism: in fact, although the CNS accounts for only 2% of the entire body mass, it contains about 25% of the total amount of unesterified cholesterol in the entire body. In addition, most of the CNS cholesterol is produced via local de novo synthesis. Keeping this in mind, it is not surprising that several studies pointed out the fact that the intracellular content of cholesterol, particularly the amount contained in the cell membrane, should be addressed much more than the plasma levels (CitationYanagisawa 2002). In this new scenario, an appropriate amount of membrane cholesterol would create a barrier against toxic insults, whereas a cholesterol-depleted membrane would ease the interaction with toxic factors such as β-amyloid, which may generate for instance an anomalous number of calcium channels leading to the accumulation of toxic levels of calcium (CitationArispe and Doh 2002). In addition, in membranes from AD patients or in rodent hippocampal neurons with a moderate reduction of membrane cholesterol the interaction between the amyloidogenic enzyme β-secretase and amyloid precursor protein (APP) appeared to be facilitated, thus leading to elevated production of β-amyloid (CitationAbad-Rodriguez et al 2004). These results suggest that loss of neuronal membrane cholesterol contributes both to increased membrane interaction with β-amyloid and to excessive amyloidogenesis in AD. Thus, the reduced expression of seladin-1 in AD vulnerable regions is in keeping with the “membrane integrity” theory.
Mice with a targeted disruption of the DHCR24 gene have been generated (CitationWechsler et al 2003). As expected, plasma and tissues of DHCR24-/- mice contained virtually no cholesterol, whereas desmosterol accumulation was observed. These animals were around 25% smaller in size than DHCR24+/+ and DHCR24+/- littermates at birth. In contrast to initial observations, it was subsequently demonstrated that these animals show a lethal dermopathy at birth and die within a few hours (CitationMirza et al 2006). This finding is in agreement with the severe phenotype observed in humans affected by desmosterolosis.
Seladin-1 as a new effector of ER-mediated neuroprotection
As mentioned previously, there is experimental evidence that estrogen and SERMs may confer neuroprotection. However, in most of the studies, that have been performed so far, animal models have been used. In other cases human cells, yet transformed or of neoplastic origin, have been used. Recently, we addressed the neuroprotective effects of estrogen/SERMs using a unique human cell model, ie, GnRH-secreting neuroblast long-term cell cultures from human fetal (8–12 weeks of gestational age) olfactory epithelium. These fetal neuroepithelial cells (FNC) were established, cloned and propagated previously by CitationVannelli and colleagues (1995) at the Department of Anatomy, Histology, and Forensic Medicine of the University of Florence, Italy. They show unique features, because they express both neuronal and olfactory markers that are typical of maturing olfactory receptor neurons (CitationVannelli et al 1995). FNC cells are electrically excitable and following exposure to a number of different aromatic chemicals show a specific increase in intracellular cAMP, indicating some degree of functional maturity. Thus, FNC cells appear to originate from the stem cell compartment that generates mature olfactory receptor neurons. In addition, they express both ERα and β (CitationBarni et al 1999). Therefore, they represented a suitable human in vitro model, that could be of help in i) providing further information on the role of estrogen in neurons, and in ii) answering the question whether seladin-1 might be an effector of ERs activation.
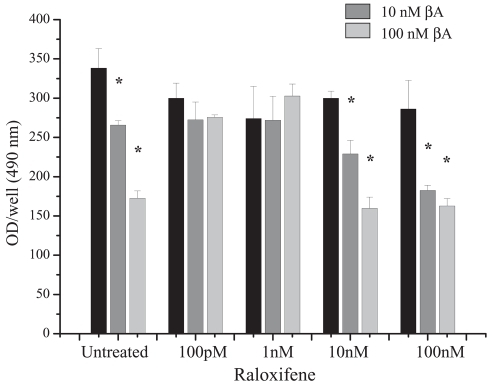
Our findings confirmed the protective role of estrogen/SERMs in the brain. In fact, we observed that, whereas in the absence of estrogen pre-incubation β-amyloid (10 and 100 nM) and H2O2 (200 μM) significantly reduced cell viability, the pre-treatment with 17β-estradiol (100 pM–100 nM) effectively counteracted β-amyloid- or oxidative stress-induced toxicity (CitationBenvenuti et al 2005). In agreement with estrogen, also the SERM tamoxifen (100 pM–100 nM) effectively protected FNC cells from the toxic effects of β-amyloid, whereas partially different results were observed using raloxifene. In fact, cell viability after exposure to β-amyloid was preserved at low concentrations of raloxifene (100 pM and 1 nM). Conversely, 10 and 100 nM did not exert protective effects (). In addition, we demonstrated that the protective action of estrogen in FNC cells was associated to a counteracting effect against β-amyloid-induced apoptosis, as demonstrated by the strong inhibition of the activation of caspase-3.
Figure 2 Effect of different concentrations of raloxifene on β-amyloid (βA) (10 and 100 nM) toxicity, as assessed by MTS assay (Promega Corp., Madison, WI).
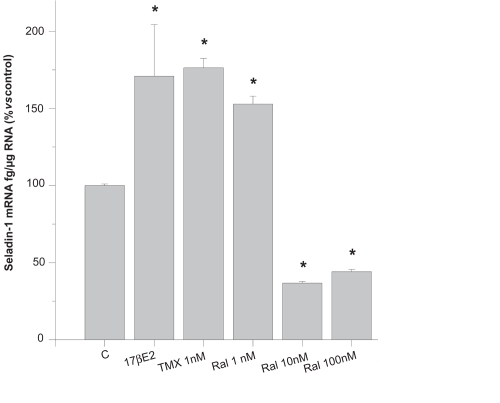
Finally, in order to answer the question whether estrogen and/or SERMs have an effect on seladin-1 expression, we evaluated the expression of seladin-1 mRNA in FNC cells, treated or not with 17β-estradiol, tamoxifen or raloxifene, by quantitative real-time RT-PCR based on TaqMan technologies. We found that FNC cells constitutively express seladin-1 (112 ± 2.26 fg/μg total RNA, mean ± SE). Noticeably, 17β-estradiol (10 pM–100 nM) significantly increased the amount of seladin-1 mRNA. 1 nM tamoxifen or raloxifene determined a similar increase of seladin-1 mRNA, compared to an equal concentration of 17β-estradiol. However, higher concentrations of raloxifene (10–100 nM) determined a marked reduction of seladin-1 expression, in keeping with the observed lack of a neuroprotective effect at these concentrations (). The effect of a selective ERα (propylpyrazole-triol, PPT) or a selective ERβ (diarylpropionitrile, DPN) agonist was also tested. We found that PPT determined a significant increase of the amount of seladin-1 mRNA at a concentration (10 nM) which has been reported to induce evident transcriptional activity (CitationHarrington et al 2003), whereas DPN produced a weaker effect. These additional findings suggested a predominant role of ERα in mediating the stimulatory effect of estrogen on seladin-1 expression. In conclusion, this study led us to hypothesize that seladin-1 might be a mediator of the neuroprotective effects of estrogen/SERMs. In particular, the parallelism between the concentrations of raloxifene that conferred neuroprotection on one hand, and stimulated seladin-1 expression on the other hand, was highly predictive that this was true.
Figure 3 Amount of seladin-1 mRNA, assessed by real-time RT-PCR, in untreated control FNC cells (C), in cells treated with 1 nM 17β-estradiol (17βE2), 1 nM tamoxifen (TMX) or raloxifene (Ral) (1–100 nM).
This hypothesis was supported by very recent additional findings. In fact, we demonstrated that, upon silencing seladin-1 expression by small interfering RNA methodology, the protective effect against β-amyloid and oxidative stress toxicity exerted by 17β-estradiol was lost. Furthermore, a computer-assisted analysis revealed the presence of half-palindromic estrogen responsive elements (EREs) upstream the coding region of the seladin-1 gene. A region spanning around 1500 bp was cloned in a luciferase reporter vector, which was transiently co-transfected with the estrogen receptor α in CHO cells. The exposure to 17β-estradiol, as well as to raloxifene and tamoxifen increased luciferase activity, thus suggesting a functional role for the half EREs of the seladin-1 gene (CitationLuciani et al 2008). Admittedly, these data provide a direct demonstration that seladin-1 is a fundamental mediator of the neuroprotective effects of estrogen.
Conclusions
ER-mediated neuroprotection has been assessed by a number of studies and justifies the attempts that have been proposed to prevent of treat AD with estrogen or SERMs. The partially disappointing results from previous clinical trials may have been determined by several confounding factors, such as the rather advanced age of the patients, the cognitive function at baseline, the presence of pre-existing risk factors and the progestogen used. Our data reinforce the role of estrogen as a protective factor for the brain and address seladin-1 as a mediator of this effect. Thus, we feel that further trials involving estrogen or SERMs in neuroprotection may be encouraged, provided that the patients are carefully selected and the treatment regimen is appropriately chosen.
Acknowledgements
The work performed by Drs. Susanna Benvenuti, Paola Luciani, Cristiana Deledda, Ilaria Cellai, Silvana Baglioni, Fabiana Rosati, Giovanna Danza, Gabriella Barbara Vannelli, Francesca Dichiara, Matteo Morello, Stefania Gelmini, and Elisa Franceschi is acknowledged. The experimental studies presented in this review were partially supported by a grant from Ente Cassa di Risparmio di Firenze and by a grant from Ministero dell’Istruzione, dell’Università e della Ricerca. The authors wish to thank Eli Lilly for kindly providing raloxifene. The authors report no conflicts of interest in this work.
References
- Abad-RodriguezJLedesmaMDCraessaertsKNeuronal membrane cholesterol loss enhance amyloid peptide generationJ Cell Biol20041679536015583033
- ArbogastLAEstrogen genomic and membrane actions at an intersectionTrends Endocrinol Metab2008191218023201
- ArispeNDohMPlasma membrane cholesterol controls the cytotoxicity of Alzheimer’s disease AbetaP (1–40) and (1–42) peptidesFASEB J20021615263612374775
- BarniTMaggiMFantoniGSex steroids and odorants modulate gonadotropin-releasing hormone secretion in primary cultures of human olfactory cellsJ Clin Endocrinol Metab19998442667310566683
- BehlCEstrogen can protect neurons: modes of actionJ Steroid Biochem Mol Biol20038319519712650716
- BenvenutiSLucianiPVannelliGBEstrogen and SERMs exert neuroprotective effects and stimulate the expression of seladin-1, a recently discovered anti-apoptotic gene, in human neuroblast long-term cell culturesJ Clin Endocrinol Metab20059017758215585566
- DhandapaniKMBrannDWProtective effects of estrogen and selective estrogen receptor modulators in the brainBiol Reprod20026713798512390866
- DubalDBZhuHYuJEstrogen receptor alpha, not beta, is a critical link in esradiol-mediated protection against brain injueryProc Natl Acad Sci USA20019819525711172057
- FillitHMThe role of hormone replacement therapy in the prevention of Alzheimer’s diseaseArch Intern Med200216219344212230415
- FitzpatrickDRKeelingJWEvansMJClinical phenotype of desmosterolosisAm J Med Genet199875145529450875
- GreeveIHermans-BorgmeyerIBrellingerCThe human DIMINUTO/DWARF1 homolog seladin-1 confers resistance to Alzheimer’s disease-associated neurodegeneration and oxidative stressJ Neurosci20012073455211007892
- HarringtonWRShengSBarnettDHActivities of estrogen receptor alpha- and beta-selective ligands at diverse estrogen responsive gene sites mediating transactivation or transrepressionMol Cell Endocrinol2003206132212943986
- HawkinsMBThorntonJWCrewsDIdentification of a third distinct estrogen receptor and reclassification of estrogen receptors in teleostsProc Natl Acad Sci USA20009710751611005855
- HermanGEDisorders of cholesterol biosynthesis: prototypic metabolic malformation syndromesHum Mol Genet200312Spec No 1R758812668600
- HuXYQinSLuYPDecreased estrogen receptor-α expression in hippocampal neurons in relation to hyperphosphorylated tau in Alzheimer patientsActa Neuropathol20031062132012819990
- IivonenSHiltunenMAlafuzoffISeladin-1 transcription is linked to neuronal degeneration in Alzheimer’s diseaseNeuroscience20021133011012127087
- MacLuskyNJEstrogen and Alzheimer’s disease: the apolipoprotein connectionEndocrinology20041453062415198969
- MaggiACianaPBelcreditoSEstrogens in the nervous system:mechanisms and nonreproductive functionsAnn Rev Physiol20046629131314977405
- MirzaRHayasakaSTakagishiYDHCR24 gene knockout mice demonstrate lethal dermopathy with differentiation and maturation defects in the epidermisJ Invest Dermatol20061266384716410790
- LucianiPDeleddaCRosatiFSeladin-1 is a fundamental mediator of the neuroprotective effects of estrogen in human neuroblast long-term cell culturesEndocrinology2008 Equb ahead of print
- O’NeillKChenSBrintonRDImpact of the selective estrogen receptor modulator, raloxifene, on neuronal survival and outgrowth following toxic insults associated with aging and Alzheimer’s diseaseExp Neurol2004a1856380
- O’NeillKChenSBrintonRDImpact of the selective estrogen receptor modulator, tamoxifen, on neuronal outgrowth and survival following toxic insults associated with aging and Alzheimer’s diseaseExp Neurol2004b18826878
- Paganini-HillAHendersonVWEstrogen deficiency and risk of Alzheimer’s disease in womenAm J Epidemol199414025661
- PuglielliLTanziREKovacsDMAlzheimer’s disease: the cholesterol connectionNat Neurosci200363455112658281
- RappSREspelandMAShumakerSAEffect of estrogen plus progestin on global cognitive function in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trialJAMA200328926637212771113
- ReissABSillerKARahmanMMCholesterol in neurologic disorders of the elderly: stroke and Alzheimer’s diseaseNeurobiol Aging2004259778915212822
- ResnickSMHendersonVWHormone therapy and risk of Alzheimer diseaseJAMA20022882170212413378
- SchumacherMGuennounRGhoumaruANovel perspectives for progesterone in hormone replacement therapy, with special reference to the nervous systemEnd Rev200728387439
- SelkoeDJAlzheimer’s disease: genes, proteins, and therapyPhysiol Rev2001817417611274343
- ShumakerSALegaultCRappSREstrogen plus progestin and the incidence of dementia and mild cognitive impairment in post-menopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trialJAMA200328926516212771112
- Toran-AllerandCDMinireview: a plethora of estrogen receptors in the brain:where will it end?Endocrinology200414510697414670986
- TurgeonJLCarrMCMakiPMComplex actions of sex steroids in adipose tissue, the cardiovascular system, and brain: insights from basic science and clinical studiesEnd Rev200627576605
- VannelliGBEnsoliFZonefratiRNeuroblast long-term cell cultures from human fetal olfactory epithelium respond to odorsJ Neurosci199515428294
- WangLAnderssonSWarnerMMorphological abnormalities in the brains of estrogen receptor beta knockout miceProc Natl Acad Sci USA2001982792611226319
- WaterhamHRKosterJRomeijnGJMutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesisAm J Hum Genet2001696859411519011
- WechslerABrafmanAShafirMGeneration of viable cholesterol-free miceScience2003302208714684813
- WuCMiloslavskayaIDemontisSRegulation of cellular response to oncogenic and oxidative stress by Seladin-1Nature2004432640515577914
- YaffeKKruegerKCummingsSREffect of raloxifene on prevention of dementia and cognitive imparirment in older women: the Multiple Outcome of Raloxifene Evaluation (MORE) randomized trialAm J Psychiatry20051626839015800139
- YanagisawaKCholesterol and pathological processes in Alzheimer’s diseaseJ Neurosci Res200270361612391598
- ZandiPPCarlsonMCPlassmanBLHormone replacement therapy and incidence of Alzheimer disease in older womenJAMA20022882123912413371