 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
A novel and integral approach to the understanding of human neurodegenerative diseases (HNDDs) and cancer based upon the disruption of the intracellular dynamics of the hydrogen ion (H+) and its physiopathology, is advanced. From an etiopathological perspective, the activity and/or deficiency of different growth factors (GFs) in these pathologies are studied, and their relationships to intracellular acid-base homeostasis reviewed. Growth and trophic factor withdrawal in HNDDs indicate the need to further investigate the potential utilization of certain GFs in the treatment of Alzheimer disease and other neurodegenerative diseases. Platelet abnormalities and the therapeutic potential of platelet-derived growth factors in these pathologies, either through platelet transfusions or other clinical methods, are considered. Finally, the etiopathogenic mechanisms of apoptosis and antiapoptosis in HNDDs and cancer are viewed as opposite biochemical and biological disorders of cellular acid-base balance and their secondary effects on intracellular signaling pathways and aberrant cell metabolism are considered in the light of the both the seminal and most recent data available. The “trophic factor withdrawal syndrome” is described for the first time in English-speaking medical literature, as well as a Darwinian-like interpretation of cellular behavior related to specific and nonspecific aspects of cell biology.
“We can only cure what we can understand first”
Introduction
Modern medicine faces an almost total lack of success both in the prevention and treatment of human neurodegenerative diseases (HNDDs), like Alzheimer’s disease (AD), multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), Hungtington’s disease (HD), retinitis pigmentosa (RP), and others. Symptomatic treatments are available for other processes, such as Parkinson’s disease (PD). The main problem seems to lie in the fact that it has not been so far possible to prevent, stop or reverse the premature and spontaneous process of progressive cell death or programmed cell death (PCD) via apoptosis or related mechanisms in HNDDs (paraptosis, autophagy, etc) (CitationHarguindey et al 2007). New conceptual approaches appear most necessary in order to discover novel therapies for the prevention and treatment of these processes (CitationSperandio et al 2000; CitationLagadic-Gossman et al 2004; CitationBröcker et al 2005).
Attempts to advance an integral and unifying hypothesis on the etiopathogenesis of HNDDs have been proposed for decades. This has mainly focused on the lack in growth factor (GF) stimulation, mainly on deficiencies in nerve growth factor (NGF) (CitationAppel 1983; CitationHefti 1983; CitationSchultze-Herbrüggen et al 2008). PCD in HNDDs, either apoptosis-mediated or not, can be induced either by: A) Depletion, spontaneous or induced, of different GFs, either from neural origin or otherwise (trophic factor withdrawal, or TFW) (CitationRebollo et al 1995; CitationKhaled et al 2001; CitationTatton et al 2002; CitationResnick et al 2004), and/or B) The activation or overexpression of mechanisms that stimulate PCD (ie, caspases, etc.), which can also be secondary to TFW-mediated mechanisms (CitationSperandio et al 2000; CitationTatton et al 2002; Liu et al 2004; CitationLavrik et al 2005;Riedl et al 2005). While acting upon mechanism B is to a great deal still controversial, mechanism A can be to a certain extent counteracted through the therapeutic utilization of a wide array of trophic and/or human GFs in order to control and/or even suppress pathological cell death programs (PCDP). The utilization of GFs, besides preventing cell death, is used to stimulate cell growth and proliferation in a wide array of clinical settings in modern clinical practice () (Varon 1981; CitationBarbin 1987; CitationKhaled et al 2001; CitationTatton et al 2002; CitationAnitua et al 2004, Citation2007b; CitationAnitua 2005). Recently, several plasma signaling proteins have been found to be significantly deficient even in preclinical and asymptomatic stages of AD, revealing a decrease in the circulation of different hematopoiesis-dependent GFs, like platelet-derived growth factor (PDGF), transforming growth factor-1 (TGF-1), epidermal growth factor (EGF), vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), hepatocyte growth factor (HGF), etc., in this disease (CitationRay et al 2007). Since several of them, like PDGF, TGF-1, EGF, VEGF, bFGF, HGF, bone morphogenetic protein-2 (BMP-2), -4, -6, connective tissue growth factor (CTGF), etc. () (CitationAnitua et al 2004; CitationKut et al 2007), are contained in high concentrations in young functional platelets and/or leukocytes, it becomes surprising that while highly sophisticated methods are being investigated to induce and increase the activity of a cohort of growth factors, either from neural origin or not, in different models of HNDDs (CitationStorkebaum et al 2004; CitationVende Velde et al 2005; CitationWagner et al 2006), at least to our knowledge no previous mention has been made on the possibility of using young and healthy platelets and, perhaps to a lesser degree, leukocyte transfusions, their most widespread natural source, in the treatment of certain HNDDs. The apparent lack of specificity in the stimulation of tissue regeneration by a cohort of several trophic factors and other cytokines contained in young and healthy platelets () (CitationAnitua et al 2004, Citation2005, Citation2006, Citation2007b, Citation2008a) led us to introduce the concept of a general “trophic factor withdrawal syndrome” in recent publications of our group (CitationAnitua et al 2007b; CitationHarguindey et al 2007, Citation2008). Finally, since epigenetic and genetic factors can primarily influence the susceptibility to neurodegeneration, the implementation of personalized treatments based on pharmacogenetic principles are also acquiring an increasing importance in order to optimize the so far limited therapeutic resources nowadays available in these complex neurodegenerative disorders (CitationCacabelos 2005; CitationCacabelos et al 2006).
Table 1 Use of platelet-based technology in modern clinical medicine
Table 2 Growth factors contained in normal platelets
The trophic factor withdrawal syndrome (TFWS) in the etiopathogenesis of human neurodegenerative diseases (HNDDs)
Large scale apoptosis induced by trophic hormone withdrawal was first reported in hormone-dependant tumors (CitationKerr et al 1972). Microenvironmental depletion or functional inability of a wide array of growth factors whose stimulus is necessary for cell survival, such as insulin-like growth factor-1 (IGF-1), PDGF, VEGF, EGF, FGF, and neurotrophins like NGF or others (CitationBarbin 1987;Desmuck et al 1997; CitationGuo et al 1997;Counts et al 2000; CitationEstévez et al 2000; CitationPoser et al 2003; CitationChao et al 2006), apart from cytokines like interleukin-2 (IL-2), IL-3, IL-7 (CitationKhaled et al 2001), a downregulated Bcl-2 (CitationShacka et al 2005), or an upregulated p53, etc., is sufficient to induce massive neural cell apoptosis (CitationRebollo et al 1995; CitationTatton et al 2003) (). In the central nervous system (CNS), a functional deficiency of certain growth factors in the microenvironment can be secondary to: A) a systemic failure of growth factor production, this is, lack of availability; B) inadequate activity of certain trophic molecules in the cell membranes of target tissues of neural origin; C) transmembrane receptor down-regulation or binding deficiency, and/or D) abnormalities in the intracellular signaling cascade. An altered function of any of these circumstances may have a key etiopathogenic role in the pathological cell death of different HNDDs (CitationPoliti et al 2001; CitationTatton et al 2002; CitationAlvarez et al 2007; CitationCorzo et al 2007; CitationRay et al 2007). Apoptotic or parapototic cell suicide can be initiated by removal of different growth factors, while trophic factor withdrawal is a key factor in inducing an apparently sine qua non intracellular acidification that seems necessary in mediating the different cell death programs in both neural cells and cancer cells alike (CitationBurns et al 1983; CitationVincent et al 1999). Thus, the systematic clinical utilization of different GFs, alone or in combination, opens new possibilities in the prevention and treatment of HNDDs (CitationBröker et al 2005). This review is an exercise in both translational and transversal research between neurology, hematology, and oncology research in order to propose new pathways towards a better understanding of the pathogenesis of HNDDs to discover new therapeutic approaches through the clinical utilization of GFs and other similar measures in both the prevention and treatment of these processes.
Table 3 Opposed trends of apoptotic-related parameters and therapeutic directions in human neurodegenerative diseases (HNDDs) and cancer
Mediating mechanisms in the pathogenesis of HNDDs. The universal role of Na+/H+ exchange and intracellular pH in the action of growth and trophic factors. Cellular Darwinism and the law of specificity and nonspecificity in biology
The role of Na+/H+ exchange and intracellular pH as universal mediators and in the activation and inhibition of growth and trophic factors in HNNDs and cancer
Some of the main areas of cancer research, from etiopathogenesis to treatment at both the basic and clinical levels (malignant transformation, growth and proliferation, cell migration, angiogenesis, the metastatic process, multiple drug resistance to chemotherapy (MDR), oncogene expression, growth factor activity, tumor glycolysis, cell cycling, DNA synthesis, apoptosis, etc.), have been recently integrated under an Unitarian perspective based upon the dynamics of the hydrogen ion and Na+/H+ antiporter (CitationHarguindey et al 2005). Similar aspects of cancer have lead some researchers to consider that “cancer Achilles’s heel” can hide within the frame of H+ dynamics and its secondary effects on cellular metabolism (J. Poúyssegür, personal communication). Furthermore, the most active proton pump inhibitors (PPI) of the amiloride series and beyond, increasingly appear as cancer “magic bullet” because their selective potential to induce a selective metabolic collapse and cell death, apoptosis-mediated or otherwise, of cancer cells and tissues irrespective of origin and cell lineage (CitationRich et al 2000; CitationTorigoe et al 2002; CitationIzumi et al 2003; CitationHarguindey at al 2005; CitationPoüyssegúr et al 2006).
While human and trophic growth factors have different origins and specific target cells, their mediating mechanisms of action at the level of the cell membrane and intracellular signaling show common final pathways in all kinds of cells (CitationHarguindey et al 2008). First, pro- and antiapoptotic intracellular signaling pathways are already known to a considerable extent, and maps of those intracellular pathways as possible therapeutic targets in different pathologies are already available (CitationHanahan 2000; CitationReed 2004; CitationLavrik et al 2005). Secondly, we have learned from experimental and translational oncology research that a key feature mediating the molecular mechanisms of action in the stimulation by the majority of, if not all, growth factors, is an increase in the rate of exchange of Na+ and H+. An over-expressed extrusion of H+ ions, which is mainly mediated by the membrane-bound Na+-H+ exchanger, induces an intracellular (IC) alkalinization and the disruption of both the IC–extracellular (EC) homeostasis of the cell (CitationBurns et al 1983; CitationMoolenar et al 1983; CitationHarguindey et al 1995, Citation2005; CitationReshkin et al 2000, Citation2003; CitationKhaled et al 2001;Harguindey 2003; CitationOrive et al 2003; CitationCardone et al 2005; CitationDi Sario et al 2007). This phenomenon is recognized as a key and specific feature not only in malignant cell transformation but also in the growth and invasion of cancer cells of all lineages and origins, as well as in the activation the metastatic process, through the creation of an abnormal IC–EC proton gradient (the so called “H+-gradient reversal”) (CitationBelaud-Rotureau et al 2000; CitationReshkin et al 2000; CitationKhaled et al 2001; CitationCardone et al 2005; CitationHarguindey et al 2005; CitationPoüysségur et al 2006).
The NHE, mainly the NHE-1 isoform, plays an essential and pivotal role in the IC signal transduction pathways of a multiplicity of different hormones, membrane signals, as well as trophic and growth factors (for a review, see CitationHarguindey et al 2005). The stimulation of this final common pathway by all the different kinds of mitogenic factors results in the activation of the Na+/H+ antiporter and its secondary induction of an elevation of intracellular pH (CitationBurns et al 1983; CitationDi Sario et al 2001; CitationCardone et al 2005; CitationHarguindey et al 2005). This phenomenon has been known for some time to represent an early and essential step in mediating cell proliferation and DNA synthesis (CitationBurns et al 1983; CitationMoolenar et al 1983; CitationL’ Allemain et al 1984; CitationGrinstein et al 1989; CitationHarguindey et al 1995; CitationReshkin et al 2000). Indeed, trophic and growth factors with clear-cut antiapoptotic activity always stimulate the NHE-1 and increase pHi, this being a fundamental homeostatic mechanism that protects cells against a pathological fall in pHi (CitationBurns et al 1983; CitationDi Sario et al 2007; CitationHarguindey et al 2007). In this way, cell death programs, either through apoptosis and/or parallel mechanisms, can be counteracted () (CitationRich et al 2000; CitationKhaled et al 2001; CitationReshkin et al 2003; CitationHarguindey et al 2005). Among these antiapoptotic–antiacidification factors are the Bcl-2 family, Bax deletion, certain oncogenes, a dysfunctional p53 suppressor gene, etc. (CitationDiGiammarino et al 2000; CitationMarches et al 2001;Thangaraju et al 2001). The main bulk of available literature points to the conclusion that cellular neuroprotection is also mediated by a pHi-sustaining effect on IC metabolism and homeostasis of an acid-base nature (therapeutic antiapoptosis) (CitationVincent et al 1999), in a similar manner that the survival mechanisms described for cancer antiapoptosis, mainly in multiple drug resistance to chemotherapy (MDR) (pathological antiapoptosis) (CitationHamilton et al 1993; CitationRoepe et al 1993; CitationGottlieb et al 1996; CitationVincent et al 1999;Simon 2000; CitationHarguindey et al 2005). Most significantly, in trophic under-stimulation and/or in growth factor deprivation the cell signals to detect IC acidification appear to be the same ones as for spontaneous apoptosis (CitationHamilton et al 1993; CitationRoepe et al 1993; CitationGottlieb et al 1996; CitationFurlong et al 1997; CitationFamulski et al 1999; CitationPark et al 1999; CitationThangaraju et al 1999; CitationBelaud-Rotureau et al 2000; CitationMatsuyama et al 2000; CitationRich et al 2000; CitationDi Sario et al 2001; CitationMarches et al 2001; CitationSimon 2001; CitationLagadic-Gossmann et al 2004; CitationDi Sario 2007) (). In this line, antiapoptotic Bcl-2 or a deleted pro-apoptotic Bax protein have been shown to directly prevent cellular acidification and thus, cellular injury (CitationPark et al 1999; CitationThangaraju et al 1999; CitationBelaud-Rotureau et al 2000; CitationBrodski et al 2002; CitationTafani et al 2003). This IC homeostatic acid-base approach opens new possibilities in the strategy to implement new preventive and/or therapeutic measures in HNDDs, hitting as a key and pivotal therapeutic target pHi homeostasis with any drug or biological compound that, alone or in combination (imidazole, chloroquine, neurotrophic or other growth factors, Bcl-2, cytokines, oncogenes, cell proteins, gene products, platelet transfusions) that activate the NHE-1 and/or decrease nitric oxide (NO) production, thus sustaining pHi within normal limits and allowing IC proton homeostasis and cell function as a whole to be kept within a physiological range ().
Figure 1 Relationships among intracellular signaling factors, pHi, and poptosis.
Notes: For further details see CitationRideout et al 2001; CitationReed 2002, Citation2004; CitationWaldeimer 2003; CitationWaldeimer et al 2004;Broker et al 2005; CitationHarguindey et al 2007.
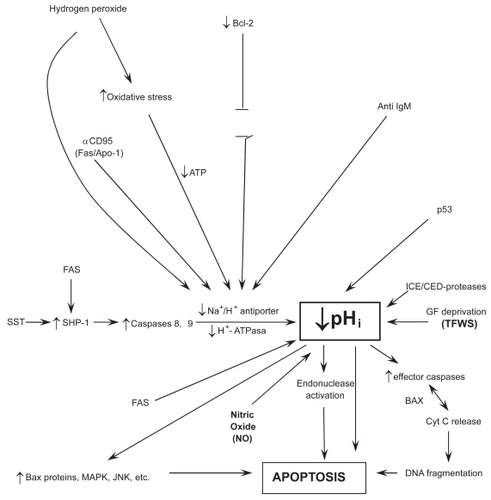
Table 4 pHi, apoptosis and antiapoptosis in HNDDs and cancer
Parallel processes of the pathological apoptosis in HNDDs and the overcoming of the resistance to apoptosis (pathological antiapoptosis) in cancer therapeutics
What spontaneously takes place in HNDDs (pathological apoptosis) (CitationRaina et al 2003), is the same phenomenon that cancer therapeutics attempt to selectively induce through a wide array of different approaches and methods (therapeutic apoptosis) (). Similarly, it has been shown that one of the main mechanisms by which cancer cells are resistant to chemotherapeutic treatment (MDR) is secondary to the fact that most cancer cells posses concerted defensive mechanisms that allow them to maintain their intracellular acid-base situation well above normal levels through all kinds of damaging microenvironmental circumstances. This has been called “the neostrategy of cancer cells and tissues” (CitationHarguindey et al 2005). Otherwise, a successful treatment in cancer is mediated by the activation of a low pHi-mediated apoptotic chain reaction cascade ending in cancer cell death (therapeutic apoptosis) (CitationFamulski et al 1999; CitationPark et al 1999; CitationWilliams et al 1999; CitationHarguindey et al 2005; CitationDi Sario et al 2007; CitationLetai 2008). Some of these mechanisms are mediated by executionary caspases or by the therapeutic activation of a JNK apoptotic pathway in cases of successful chemotherapy (CitationPerona et al 2007). Most interestingly, a parallel phenomenon, namely, an oxidative stress-mediated activation of the c-Jun -N-terminal kinase, takes place in AD (see ). Furthermore, JNK activation is also mediated by directly inducing a low pHi and by amiloride treatment (Lachapelle et al 2007). Other mechanisms of successful cancer treatment include overcoming blockades to cancer cell apoptosis by inhibiting the Bcl-2 antiapoptotic family, whose activity is not surprising that is mediated by IC alkalinization and inhibited by cell acidification (CitationReynolds et al 1996; CitationYang et al 1997; CitationIshaque et al 1998;Shimuzu et al 1998; CitationZanke et al 1998;Zhiuza et al 1998; CitationThangaraju et al 1999; CitationMatsuyama et al 2000; CitationTakashaki et al 2004; CitationWaibel et al 2007). Resistance to chemotherapy (MDR) can also be achieved by similar pH-related methods (CitationRoepe et al 2001; CitationHarguindey et al 2005). A large deal of accumulated data in this area indicates that the study of Bcl-2 as an antiapototic agent seems secondary to an alkalinizing pHi effect, another feature that can be integrated within the H+-mediated model (CitationReynolds et al 1996; CitationThangaraju et al 1999; CitationMatsuyama et al 2000). This makes it highly surprising that even the more recent reviews on cell death mechanisms and resistance to them, either in cancer and HNDDs, either completely ignore the dynamics of the hydrogen ion in these situations or they are granted a meaningless secondary role at the most (CitationRaina et al 2003; CitationWaldeimer 2003; CitationReed 2004; CitationSzakátz et al 2006). Even more, malignant cell apoptosis can be selectively induced by proton pump inhibitors (PPI) like HMA (5-(N, N-hexamethylene)-amiloride) (CitationRich et al 2000), or acid-mediated stimulation of p53 (CitationWilliams et al 1999) ( and ).
Multiple drug resistance to different chemotherapeutic agents (MDR) can be overcome by HMA or other PPI of the amiloride series, another low pHi-mediated phenomena (CitationRoepe et al 2001; CitationHarguindey et al 2005; CitationMiraglia et al 2005, CitationDi Sario et al 2007). In summary, all the available evidence indicates that the more reductionistic and fragmented model represented by the detailed study of any individual factors as well as the intricate abnormalities of the intracellular signaling pathways can be inserted within a more comprehensive and integral homeostatic paradigm of an acid-base nature as represented by the IC–EC dynamics of the hydrogen ion (H+). This acquires further significance when attention is paid to the fact that the dynamics of proton movements in and out of the cell and its transmembrane gradients seem to be the ultimately responsible factor and mechanism for the apoptosis-antiapoptosis machinery in both HNDDs and cancer (CitationGottlieb et al 1996; CitationFurlong et al 1997; CitationFamulski et al 1999; CitationPark et al 1999; CitationVincent el al 1999; CitationWilliams et al 1999; CitationHarguindey et al 2000, Citation2005, Citation2007; CitationMatsuyama et al 2000; CitationRaina et al 2003; CitationTafani et al 2003; CitationWaldeimer 2003; CitationLagadic-Gossmann et al 2004; CitationReed 2004; CitationLetai 2008).
In spite that cancer cells seem able to evade apoptosis by trophic factor deprivation in many cases (CitationTang et al 1998; CitationThompson et al 2005), antihormonal and antigrowth factors are being tried in different therapeutic attempts in the oncology setting (Shegal et al 1994; CitationTaetle et al 1994). These and other parallel considerations relating H+ dynamics and GF activity allow to place the biochemistry and molecular biology of HNDDs and cancer at both ends of an apoptotic-antiapoptotic metabolic spectrum (CitationHarguindey et al 1992b, Citation2007) ( and ). The same unitarian perspective that has made possible to integrate the main areas of basic and clinical oncology research (CitationHarguindey 1992a; CitationHarguindey et al 1995, Citation2005), now allows to advance another new concept: that what is pathological and damaging for some diseases (the spontaneous apoptosis of HNDDs) can be therapeutic and beneficial for their metabolic and homeostatic opposites (the antiapoptosis of malignancy) (). Thus, a primordial approach to IC homeostasis may open new and untrodden ways towards entirely new therapeutic approaches, aiming at the Na+/H+ antiporter, other membrane proton pumps and NO production and control as primordial therapeutic targets, either in the induction of selective low pH-mediated apoptosis in cancer treatment through the utilization of proton transport inhibitors (therapeutic apoptosis) or, in the opposite direction, counteracting IC acidification in HNDDs (therapeutic antiapoptosis) (CitationTroy et al 1996; CitationRich et al 2000; CitationTorigoe et al 2002; CitationIzumi et al 2003; CitationHarguindey et al 2005;Poüysségur et al 2007).
Terminal pathogenesis of apoptotic or para-apoptotic cell death in HNDDs. NO, oxidative stress, and cellular pHi
When deprived of trophic factors (TFWS), motor neurons undergo a programmed cell death program, apparently through nitric oxide-dependent apoptosis (CitationHirakura et al 1999; CitationVincent et al 1999; CitationEstévez et al 2002; CitationWilkins et al 2004; CitationEstévez et al 2006), and/or superoxide production (CitationLieberthal et al 1998) and/or caspase activation (CitationTroy et al 1996; CitationMcCarthy et al 1997; CitationChan et al 1999; CitationDeshmukh et al 2000; CitationChoi et al 2002). The induction of NO-related mechanisms share a common final pathway that involves nitric oxide synthase (NOS) and peroxynitrite formation. NO has been linked, partly because to its oxidative role, to the development of several neurodegenerative disorders, seemingly by activating a pHi-related apoptotic cascade (CitationCorzo et al 2007). Furthermore, the formation of NO by astrocytes has been suggested to contribute to the neurodegenerative process, while NO production is significantly elevated in platelets from AD patients (CitationFernandez-Vizarra et al 2004). This very same mechanism proves to be of value in inducing apoptosis, via necrosis, in neoplastic cells (CitationRigas et al 2008) ().
The seminal work of CitationVincent and colleagues (1999) in neurons has shown that lowering pHi from 7.36 to 7.09/7.00 through exposure to NO sets in motion a programmed cell death (PCDP) program, increasing DNA fragmentation and decreasing neuronal survival (low pHi-mediated metabolic collapse). This phenomenon is induced by the activation of three low pHi-dependent endonucleases responsible for neuronal injury (). Furthermore, the formation of pHi-lowering pro-apoptotic NO by astrocytes suggests a potential for NO inhibitors in the treatment of this disease, most likely through the stabilization of IC acid-base homeostasis and the prevention of a deadly drop in pHi (Harguindey et al 2003; CitationArianna et al 2007) (). Other compounds with different anti-NO properties like superoxide dismutase, minocycline, NGF, etc, have also shown neuroprotective effects (CitationKirkland et al 2003; CitationWilkins et al 2004; CitationEstévez et al 2006). The stimulation of oxidative stress by NO has been involved in the pathogenesis of dementia (CitationCorzo et al 2007), while oxidative stress and/or reactive oxygen species (ROS) have been widely considered as a key factor in neuronal degradation (CitationHarguindey et al 2007). Superoxide (SO) formation induced by growth factor deprivation induces apoptosis (CitationLieberthal et al 1998). This phenomenon is further increased after reacting SO with NO to form peroxynitrite (CitationEstévez et al 2006). In contrast, low or physiological concentrations of NO prevents apoptosis (CitationChoi et al 2002). In summary, the dynamics of the killing mechanism (see also ), are:
The study of the molecular biology of cancer cells has shown that the inhibition of the electroneutral membrane exchanger NHE-1 induces intracellular acidification by entrapping hydrogen ions within the cell (CitationLi et al 1995; CitationRebollo et al 1995;Harguindey et al 2003). A severe intracellular acidosis below a certain threshold (pHi:±6.8) is an essential factor in setting up a cascade-like chain reaction ending up in the metabolic collapse preceding apoptosis, either in cancer cells (therapeutic apoptosis) (CitationRich et al 2000, CitationDi Sario et al 2007) or in dying cells of neural origin (pathological or degenerative apoptosis) () (CitationHamilton et al 1993; CitationRoepe et al 1993; CitationGottlieb et al 1996; CitationHarguindey et al 1995; CitationLi et al 1995; CitationRebollo et al 1995; CitationPark et al 1999, Citation2005; CitationVincent et al 1999; CitationThangaraju et al 1999; CitationMatsuyama et al 2000; CitationRich et al 2000; CitationDi Sario et al 2001; CitationSimon 2001; CitationWahl et al 2002; CitationLagadic-Gossmann et al 2004).
Cellular Darwinism. The law of specificity and nonspecificity in biology
From the point of view of biological evolution, the simplest way (and to follow Darwinian rules nature always chooses the simplest possible way in order to solve its biological problems and preserve the stability and continuation of life) is that a wide array of trophic, growth factors and hormones have different kinds of target cells with specific receptors for different cell lineages. This represents a qualitative and specific variation. At the same time, cellular physiology has a very small number of transduction mechanisms from the membrane to the interior of the cell to influence intra-cellular signaling pathways that are shared by all kinds of cells. These mechanisms may only vary from one another in some quantitative, nonspecific aspects. The procedure of choosing the kind of response is through a selective, however variable distribution of specific receptors in the different cell lineages concerted with a few number of membrane-bound signals, as the family of the proton pump transporting systems, and from there on to mostly shared IC signaling pathways. This is the simplest possible way that can be imagined. Much more difficult would be that nature would have chosen that all cells would have the same or similar receptors. Should this have been the case, all cells would be equally sensitive to all different kinds of stimulating or inhibiting hormones and growth factors in order to induce numberless and different effects in each cell or cell lineage. In this case it would have been necessary to select the kind of response through a great number and diversity of signals and/or intracellular transduction mechanisms in the different cells and/or lineages. This complication is so great that it rules itself out. It is much more logical that evolution selects target cells providing them with specific receptors, which are the mediating mechanisms responsible for selecting the kind cellular response, than to have numberless IC signaling factors and pathways for each lineage or cell (CitationMeléndez Hevia 2001; CitationHarguindey et al 2008). In this way, a countless diversification of effects is reduced to the qualitative and quantitative distribution of membrane receptors, which would decrease the information and stimulus-response systems of cells to the smallest number and the least possible degree of complication. All the above-mentioned features can be represented through the following scheme:
| Systemic circulation | Cell membrane | Intracellular signaling |
| Multiplicity of stimulus (hormones, FC, etc.) | Specificityof receptors (qualitative) Same electro- chemical mechanisms | Similar IC pathways (quantitative/nonspecific) |
The fact that all the different kinds of cells of living organisms and a multiplicity of mitogenic stimuli, hormones and growth factors share the same universal transport systems, mainly the Na+/H+ antiporter but also a few other membrane-bound proton pumps at the cell membrane (CitationHarguindey et al 2005), allows to formulate a law that unifies and combines specificity and nonspecificity in biology. Similar examples of this law at the level of intermediary metabolism, are:
Glucose transporters sensitive to insulin (GLUT 4), that are only present in fat, muscle, and heart tissues.
Adrenalin receptors for the stimulation of glucogenolysis, that are only present in muscle and liver.
Glucagon receptors for glucogenolysis, only present in liver.
In all these cases, the mechanism of intracellular stimulation is always the same one: activation of adenylate cyclase to produce cAMP) (CitationMeléndez Hevia 2001). If we apply this law to neuronal neuroprotection, any trophic factors with a clear-cut antiapoptotic activity also stimulate the NHE-1 exchanger (CitationMoolenar et al 1983; CitationL’ Allemain et al 1984). Among the different membrane-bound proton pumps, the NHE1 appears to be the main homeostatic mechanism that protects cells against a pathological decrease in pHi and so, against cell death through cytosolic acidification. Thus, in many cases cellular protection seems to be ultimately mediated by a pHi-sustaining effect responsible for a physiological equilibrium and intracellular homeostasis of an acid-base nature. We have called this phenomena “therapeutic antiapoptosis”, a parallel concept to the “pathological antiapoptosis” which is characteristic to cancer cells, mainly in cases of multiple drug resistance to antichemotherapuetic agents (MDR) (Roepe et al 2003; CitationHarguindey et al 2008) (). In summary, a primordial approach to the dynamics of cellular proton homeostasis gives way to new therapeutic possibilities, strategies and targets both in the treatment of HNDDs and neoplastic diseases (CitationRich et al 2000; CitationHarguindey et al 2005, Citation2007).
The pHi factor and platelet abnormalities in AD. Therapeutic possibilities of human growth factors in the treatment of the trophic factor withdrawal syndrome in HNDDs
Intracellular homeostasis. The pHi factor
The final common pathway mediating cellular responses to different mitogenic stimulus, growth factors and other membrane signals such as hormones, has been repeatedly recognized to be secondary to the induction of an elevation of pHi (CitationBurns et al 1983; CitationMoolenar et al 1983; CitationL’ Allemain et al 1984; CitationHarguindey et al 1995; CitationCardone et al 2005). Contrariwise, apoptosis, either in HNDDs or other settings, is largely mediated by IC acidification (CitationVincent et al 1999; CitationHarguindey et al 2005). Contrariwise, maintaining IC alkaline levels at the level of pHi =7.60 prevents neuronal injury, not only implying pHi as a critical factor in PCD but also indicating a possible role for therapeutic cellular alkalinization in the prevention and treatment of neuronal degeneration, as shown by CitationVincent and colleagues (1999). These authors and others (CitationHarguindey et al 2007) have concluded that counteracting a drop in pHi through trophic and/or peptide growth factors is a most attractive therapeutic target against neuronal degeneration and pathological death. Furthermore, inhibiting NO production by NO inhibitors, like mefenamic acid and thiadiazolidinones, and so IC acidification, has been correctly proposed as a therapeutic option in the treatment of AD (CitationVincent et al 1999; CitationArianna et al 2007; CitationHarguindey et al 2007). Thus, beyond multiple etiological causes and early EC factors that can be involved, a universal mechanism of an acid-base nature appears to be the pivotal and key event in downregulating caspase-mediated or caspase-independent IC signaling pathways leading in many cases to cell death in HNDDs and, similarly, in different lineages of cancer cells (CitationHamilton 1993; CitationLi et al 1995; CitationPérez-Sala et al 1995; CitationRebollo et al 1995; CitationGottlieb et al 1996; CitationReynolds et al 1996; CitationFurlong et al 1997; CitationHirakura 1999; CitationPark et al 1999, Citation2005; CitationVincent et al 1999; CitationThangaraju et al 1999; CitationWilliams et al 1999; CitationLiu et al 2000; CitationMatsuyama et al 2000; CitationRich et al 2000; CitationHirpara 2001; CitationMarches et al 2001; CitationRenz et al 2001; CitationReshkin et al 2003; CitationLagadic-Gossmann et al 2004; CitationRiedl et al 2004;Vaghefiet al 2004; CitationLavrik et al 2005; CitationBredesen et al 2006, CitationDi Sario et al 2007) (). In summary, any therapeutic effort to keep IC acid-base homeostasis within normal or higher than normal levels, is likely to be of significant benefit in the prevention and treatment of HNDDs.
Platelet abnormalities in AD: Interdisciplinary hematology to neurology transversal research
Evidence for a role of platelet dysfunction, structural to functional, in AD patients arises from different studies (CitationZubenko et al 1987a, Citation1987b). CitationArianna and colleagues (2007) and CitationCorzo and colleagues (2007) have reported that abnormal platelet function related to an increase in NO-producing systems play an important role in the pathogenesis of AD and in neuronal degeneration. From the structural side, the presence of abnormal coated-platelets with the capacity to retain amyloid precursor protein in their surface has been shown in early stage AD and MCI (mild cognitive impairment), a feature that also shows a significant relationship with disease progression (CitationProdan et al 2007). Such abnormalities directly relate platelet abnormalities with the pathogenesis of at least some HNDDs, while at the same time they suggest that the supply of healthy exogenous platelets with high contents of PDGF and other platelet-derived GFs may have a significant therapeutic effect in at least certain HNDDs like AD ( and ).
Growth factors in neuronal protection
If one of the main trophic factors that stimulate growth and metabolism of muscle cells is plain exercise, the nervous system is stimulated by certain GFs, neurotrophic, platelet-derived, and beyond. The lack of cell trophism in different degenerative diseases makes it highly advisable to undertake clinical studies to detect the presence, decrease or even absence of either systemic, microenvironmental and/or cellular growth factors and other antiapoptotic substances (PDGF, VEGF, NGF, tyrosine kinases, Bcl-2, Bcl-X, etc.), not only in HNDDs but also in other systemic and nonneural degenerative diseases () (CitationRideout 2001; CitationWaldeimer et al 2003; CitationWaldeimer 2004; CitationAnitua et al 2005, Citation2006, Citation2007a, Citation2007b, Citation2008a, Citation2008b; CitationWallace et al 2006; CitationFarrag et al 2007). Ultrastructural-morphological studies and determinations of the content and activity of α-granules in platelets of patients with HNDDs, as well as measuring the concentration of the different PDGFs in the platelets of patients with AD and other HNDDs represent an unexplored field of research that may lead to improving the understanding of the pathogenesis of HNDDs (CitationSolerte et al 2005). In order to do this, the technology necessary to perform these measurements is now available (CitationAnitua et al 2007a, Citation2007b).
Among the different PDGF, recent evidence points to a pivotal role for VEGF in neuronal protection (CitationJin 2002; CitationCao et al 2004; CitationStorkebaum et al 2004a, Citation2004b; CitationSolerte et al 2005). Decreased VEGF activity has been shown in different HNDDs. Recently, serum VEGF levels of patients with AD were compared with control subjects (CitationMateo et al 2007). The mean concentration of VEGF in the patient group was significantly lower than in the controls. These authors concluded that a decrease in serum levels of VEGF could contribute to the neurodegenerative process in AD. While no differences in serum VEGF levels between AD patients and controls have been found in other studies, low VEGF activity has also been reported in patients with ALS (CitationDel Bo et al 2005). The VEGF deficiency trophic effect can be related to a decrease of VEGF secretion from coated-platelets secondary to already described abnormalities related to the retention of APP protein on their surface (CitationArianna et al 2007; CitationProdan et al 2007). Other authors have found co-accumulation of VEGF with β-amyloid in AD, as well as a strong binding of VEGF to it, suggesting that secondary VEGF deficiency under hypoperfusion may contribute to neurodegeneration and vascular dysfunction in the progression of AD (CitationDel Bo et al 2005).
A decreased secretion and release of VEGF in the supernatants of circulating natural killer (NK) immune cells in patients with AD compared to normal controls and patients with other types of senile dementias has also been reported. This down-regulation of VEGF production has been demonstrated in peripheral immune cells of patients suffering from AD (CitationSolerte et al 2005). These findings become even more significant when the pivotal importance of VEGF in brain angiogenesis, neuroprotection, and cerebrovascular exchange of nutrients is considered (CitationZhu et al 2002; CitationStorkebaum et al 2004a; CitationYang et al 2004; CitationDel Bo et al 2005; CitationSolerte et al 2005) From a therapeutic point of view, direct application of VEGF delays the onset of paralysis, improves motor function and increases survival in a rat model of AD (CitationStorkebaum et al 2004b; CitationVende Velde et al 2005). Since the highest known accumulation of VEGF can be found in platelets and leukocytes (CitationKut et al 2007), cyclic transfusions of healthy young platelets and, perhaps, leukocytes too, may represent the easiest and most natural available source of VEGF and other trophic growth factors and a significant new therapeutic approach to certain HNDDs (CitationHarguindey et al 2007).
Similarly, mice lacking epidermal growth factor receptor (EGFR) develop neurodegeneration through Akt-caspase dependent apoptosis of the frontal cortex (CitationWagner et al 2006), this representing a further example of neuronal degeneration as part of the trophic factor withdrawal syndrome (TFWS). Finally, the emerging role of other GFs, like FGF, mainly through its interaction with its receptors, has been reported to present a significant role in brain neuronal trophism, repair and protection, and may also play a role in the treatment of certain mental disorders (CitationRiva et al 2005).
The therapeutic potential of different neurotrophic factors in HNDDs has been studied for more than two decades (Hefty et al 1983; Varon et al 1987; CitationTatton et al 2003, CitationCounts et al 2005). CitationBarbin (1987) initially showed that different neurotrophins induce growth and proliferation of cells of neural origin. The inhibition of NGF has been considered to be involved in the pathogenesis and treatment of the prodromal stages of AD (CitationTatton et al 2003; CitationCounts et al 2005, CitationSchulte-Herbrügen et al 2008). However, more recent clinical trials using NGF have failed to show significant results. Besides, the intracerebral local application of NGF faces many technical problems (CitationBrodski et al 2002; CitationEbert et al 2005; CitationDass et al 2006). However, when glial cell line neurotrophic factor (GDNF) is infused via encapsulated cells in animal models of PD some positive results are obtained (Sajadi et al 2005). Furthermore, it has been reported that the local application of NGF into the putamen has produced sustained benefits in a small series of patients with PD (CitationSlevin et al 2006). Other neurotrophic factors like pigment epithelium-derived factor (PEGF) protects retinal ganglion cells (CitationPang et al 2007). Gene transfer factors and antiapoptotic promoting factors like Bcl-2 or other GFs have also been tried (CitationDass et al 2006; CitationSortwell et al 2007). Finally, hormones like thyrotrophin-releasing hormone (TRH) and its derivatives, as well as testosterone or estrogens, also play the role of GFs in protecting cells from neural injury in a similar manner than certain antioxidants like N-acetylcysteine (CitationYan et al 1995; CitationBialek et al 2004: CitationFaden et al 2004). GFs besides VEGF and EGF, like IGF-1, whose deficiency has also been related to the pathogenesis of AD (CitationAlvarez et al 2007), are able to promote neural survival and differentiation (CitationPoliti et al 2001). Finally, different antiapoptotic therapies for neurodegenerative diseases based upon the application of exogenous products that stimulate cell trophism, such as propargylamines, alpha-2-adrenergic receptor activators, minocycline, etc, are at the present time in preclinical stages of research (CitationTatton et al 2002, Citation2003; CitationLavrik 2005).
Use of PDGFs in clinical medicine and potential utilization of platelet transfusions in the treatment of HNDDs
One possibility of counteracting programmed cell death programs (PCDP) driven by deprivation of trophic and/or growth factors, would be to study the therapeutic potential of PDGF in HNDDs, as it has been recently tried in nonneural pathology (CitationLavrik 2005; CitationAnitua et al 2007b; CitationRay et al 2007) (). Neurons, glial cells, and the pigmentary epithelial cells of the retina all contain platelet-derived growth factor receptors (PDGFR), making them classic therapeutic targets for PDGF. The cyclic administration of PDGFs in HNDDs through the exogenous supply of healthy platelets should be considered in a similar way that a wide variety of human pathologies are nowadays successfully treated through loco-regional application of autologous platelet releseates obtained by simple clinical procedures (CitationFedi et al 1997; CitationAnitua et al 2004). This new research field allows for important and exciting new perspectives in opening untrodden areas of research and treatment of degenerative diseases, either from neural origin or otherwise, where a lack of one or more trophic factors is involved in their pathogenesis, including any kind of degenerative pathology where stimulation of new tissue growth or regrowth is necessary. Nowadays, clot preparations containing high concentrations of PDGF and metabolites that stimulate cell trophism, growth and proliferation, are being used with increasing frequency in a wide range of medical and surgical contexts apart from neurology, from dentistry and oral implantology to orthopaedics, gastric and skin ulcer treatment, eye disorders, etc, successfully inducing new tissue formation and accelerated tissue repair in different tissues and locations (CitationAnitua et al 2004). summarizes some of the therapeutic applications of platelet-based technology in modern clinical medicine. These platelet-releseates contain high concentrations of a wide array of mitogenic and pro-angiogenic factors, mainly platelet-derived growth factor PDGF, TGF-β1, EGF, VEGF, PDEGF, IGF-1, and HGF, as well as cytokines PF4 and CD40L, apart from lower concentrations of other factors like β-FGF (CitationAnitua et al 2007b). Some of these factors show a potent stimulating effect on cell growth, proliferation and viability, to a large extent because of their positive effects on angiogenesis. The possibility of translating these successful clinical results to the prevention and treatment of HNDDs is one of the main medical challenges for the next few years.
Conclusions
The main conclusions of this study are:
The exogenous utilization of human growth factors, platelet-derived or otherwise, present a therapeutic potential in the treatment of certain human neurodegenerative diseases (HNDDs).
There is a large deal of both theoretical and experimental evidence, both at basic and clinical levels, to consider the cyclic and systemic use of healthy platelet transfusions in early stages of certain HNDDs like AD.
The therapeutic failure in the prevention and treatment of HNDDs can be secondary to a certain extent to lack of knowledge about the implications of the different trophic factors in the pathogenesis of HNDDs within the frame of the TFWS.
Every effort to maintain intracellular acid-base homeostasis within the physiological range in HNDDs, thus preventing the metabolic collapse induced by cell acidification and its secondary activation of cell death programs, will help to better understand, prevent and treat HNDDs.
In the prevention and treatment of HNDDs, a more effective pharmacological inhibition of the formation of NO by neural cells is needed.
Pathological and spontaneous programmed cell death programs in HNDDs, apoptotic or otherwise, are processes that dwell at opposite ends of an acid-base/homeostatic spectrum when compared with the resistance to the induction of apoptotsis that characterizes malignant cells and tissues (pathological antiapoptosis).
Acknowledgments
This work was supported by a grant of the Castresana Foundation, Vitoria, Spain. We thank Dr. Miriam L. Wahl, for her comments, suggestions and corrections. Moreover, the authors apologize to all investigators who have significantly contributed to the different fields of research reviewed in this contribution, but whose work we were unable to include in our review. The authors report no conflicts of interest in this work.
References
- AlioJLColechaJRPastorS2007Symptomatic dry eye treatment with autologous platelet-rich plasmaOphthalmic Res39124917374962
- AnituaEAndiaIArdanzaB2004Autologous platelets as a source of proteins for healing and tissue regenerationThromb Haemost941514691563
- AnituaEAndiaISanchezM2005Autologous preparations rich in growth factors promote proliferation and induce VEGF and HGF production by human tendon cells in cultureJ Orthop Res23281615779147
- AnituaESanchezMNurdenAT2006Autologous fibrin matrices: a potential source of biological mediators that modulate tendon cell activitiesJ Biomed Mater Res7728593
- AnituaESanchezMNurdenAT2007aReciprocal actions of platelet-secreted TGF-β1 on the production of VEGF and HGF by human tendon cellsPlastic Reconstruct Surg1199509
- AnituaESánchezMOriveGAndíaI2007bThe potential impact of the preparation rich in growth factors (PRGF) in different medical fieldsBiomaterials2845516017659771
- AnituaEAguirreJJAlgortaJ2008aEffectiveness of autologous preparation rich in growth factors for the treatment of chronic cutaneous ulcersJ Biomed Mater Res B8441521
- AnituaESánchezMOriveG2008bDelivering growth factors for therapeuticsTrends Pharmacol Sci29374118037168
- AlvarezACacabelosRSanpedroC2007Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer diseaseNeurobiol Aging28533616569464
- AppelSH1983A unifying hypothesis for the cause of amyotrophic lateral sclerosis, Parkinsonism and Alzehimer’s disease 1981Ann Neurol104995056173010
- ArakiWWurtmanRJ1998Increased expression of amyloid precursor protein and amyloid precursor-like protein 2 during trophic factor withdrawal-induced death of neuronal PC12 cellsMol Brain Res56169779602112
- AriannaVLauraNCinziaM2007Modification of platelet from Alzheimer disease patients: a possible relation between membrane properties and NO metabolitesNeurobiol Aging289879416815594
- BarbinGMaloineSA1987Le viellissement cerebral normal and pathologiqueColloques de la Fondation Nationale de Gerontologie11423
- Belaud-RotureauMALeducqNde GannesFMP2000Early transitory rise in intracellular pH leads to Bax conformation change during ceramide-induced apoptosisApoptosis55516011303914
- BialekMZarembaPBorowiczKK2004Neuroprotective role of testosterone in the nervous system 2004Polish J Pharmacol5650918
- BredesenDERaoRVMehlenP2006Cell death in the nervous systemNature44379680217051206
- BrodskiCVogt WeisenhornMVDechantG2002Therapy of neurodegenerative diseases using neurotrophic factors: cell biological perspectiveExpert Rev Neurotherapeutics28998
- BrökerLEKruytFAEGiacconeG2005Cell death independent of caspases: a reviewClin Cancer Res1131556215867207
- BurnsCPRozengurtE1983Serum, Platelet-derived growth factor, vasopressin and phorbol esters increase intracellular pH in swiss 3T3 cellsBiochem Biophs Res Comm1169318
- CacabelosR2005Pharmacogenomics, nutrigenomics and therapeutic optimization in Alzheimer’s diseaseAging Health130348
- CacabelosRTakedaM2006Pharmacogenomics, nutrigenomics and future therapeutics in Alzheimer’s diseaseDrugs Future31Suppl B5146
- CaoLJiaoXZuzgaDS2004VEGF links hippocampal activity with neurogenesis, learning and memoryNature Genetics368273515258583
- CardoneRACasavolaVReshkinSJ2005The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasisNature Revs Cancer57869516175178
- ConsoloUZaffeDBertoldiC2007Platelet-rich plasma activity on maxillary sinus floor augmentation by autologous boneClin Oral Implants Res182526217348891
- CorzoLZasRRodríguez2007Decreased levels of serum nitric oxide in different forms of dementiaNeurosci Lett420263717556102
- CountsSEMufsonEJ2005The role of nerve growth factor receptors in cholinergic basal forebrain degeneration in prodromal Alzheimer diseaseJ Neuropathol6426372
- ChaoMVRajagopalRLeeFS2006Neurothropin signalling in health and diseaseClin Sci1101677316411893
- ChanSLTammarielloSPEstusS1999Prostate apoptosis response-4 mediates trophic factor withdrawal-induced apoptosis of hippocampal neurones: actions prior to mitochondrial dysfunction and caspase activityJ Neurochem735021210428045
- ChoiB-MPaeH-OJangSIl2002Nitric oxide as pro-apoptotic as well as anti-apoptotic modulatorJ Biochem Mol Biol351162616248976
- DassBWarren OlanovCKordoverJH2006Gene transfer of trophic factors and stem cells grafting as treatments for Parkinson’s diseaseNeurology66S89S10316717256
- Del BoRScarlatoMGhezziS2005Vascular endothelial growth factor gene variability is associated with increased risk for ADAnn Neurol573738015732116
- DeshmukhMJohnsonEM1997Programmed cell death in neurons: focus on the pathway of nerve growth factor deprivation-induced death of sympathetic neuronesMol Pharmacol518979069187255
- DeshmukhMKuidaKJohnsonEM2000Caspase inhibition extends the commitment to neural death beyond cytochrome C release to the point of mitochondrial depolarizationJ Cell Biol1501314310893262
- DiGiammarinoJLeeADSCadwellC2000A novel mechanism of tumorogenesis involving pH-dependent desestabilization of a mutant p53 tetramerNature Struct Biol9121611753428
- Di SarioASvegliati BaroniGBendiaE2001Intracellular pH regulation and Na+/H+ exchange activity in human hepatic stellate cells: effect of platelet-derived growth factor, insulin-like growth factor 1 and insulinJ Hepatology3437885
- Di SarioABendiaEOmenettiA2007Selective inhibition of ion transport mechanisms regulating intracellular pH reduces proliferation and induces apoptosis in cholangiocarcinoma cellsDigestive Liver Dis39609
- EbertADSvendsenCN2005A new tool in the battle against Alzheimer’s disease and aging: ex vivo gene therapyRejuvenation Res8131416144466
- EstévezAGSampsonJBZhuangYX2000Liposome-derived superoxide dismutase prevents nitric oxide-dependent motor neuron death induced by trophic factor withdrawalFree Rad Biol Med284374610699756
- EstévezAJordánJ2002Nitric acid and superoxide, a deadly cocktailAnn NYAS96220711
- EstévezASahawnehMALangePS2006Arginase 1 regulation of nitric oxide production is key to survival of trophic factor-deprived motor neuronsJ Neurosci268512616914676
- FadenAIKnoblachSMMovsesyanVA2004Novel small peptides with neuroprotective and nootropic propertiesJ Alzheimer Dis6S93S97
- FamulskiKEMacDonaldDPatersonMC1999Activation of low pH-dependent nuclease by apoptotic agentsCell Death Differ6281910200579
- FarragTYLeharMVerhaegenP2007Effect of platelet rich plasma and fibrin sealant on facial nerve regeneration in a rat modelLaryngoscope1171576517202946
- FediPTronickSRAaronsonSAHollandJFBastRCMortonDL1997Growth factorsCancer MedicineBaltimoreWilliams and Wilkins4774
- Fernández-VizarraPFernándezAPCastro-BlancoS2004Expression of nitric oxide system in clinically evaluated cases of Alzheimer diseaseNeurobiol Dis1528730515006699
- FurlongIJAscasoRLopez RivasA1997Intracellular acidification induces apoptosis by stimulating ICE-like protease activityJ Cell Sci110653619092947
- GottliebRAGiesingHAZhuJY1995Cell acidification and apoptosis: granulocyte colony-stimulating factor delays programmed cell death by up-regulating the vacuolar ATPaseProc Natl Acad Sci U S A92596587541139
- GottliebRANordbergJSkowronskiE1996Apoptosis induced in Jurkat cells by several agents is preceded by intracellular acidificationProc Natl Acad Sci U S A9365488570610
- GrinsteinSRotinDMasonMJ1989Na+/H+ exchange and growth factor-induced cytosolic changes. Role in cellular proliferationBiochem Biophys Acta98873972535787
- GuoQSopherBJFurukawaK1997Alzheimer presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and β-amiloid peptide: involvement of calcium and oxyradicalsJ Neurosci174212229151738
- HamiltonGCosentiniEPTelekyB1993The multidrug-resistance modifiers verapamil, cyclosporine A and tamoxifen induce an intracellular acidification in colon carcinoma cell lines in vitroAnticancer Res136A2059637905252
- HanahanDWeinbergRA2000The hallmarks of cancerCell100577010647931
- HarguindeySCragoeEJJrKleymanThRSimchowitzL1992aUse of Na+/H+ antiporter inhibitors as a novel approach to cancer treatmentAmiloride and its analogs: unique cation transport inhibitorsNew YorkVCH Publishers Inc31734
- HarguindeyS2002Integrating fields of cancer research through pivotal mechanisms and synthetic final pathways: A unifying and creative overviewMed Hypotheses5844452 Erratum. 2003. Med Hypotheses, 61:318–1912323109
- HarguindeySCragoeEJJr1992bThe Na+/H+ antiporter in oncology in the light of the spontaneous regression of cancer and cell metabolismMed Hypotheses39229371335544
- HarguindeySPedrazJLGarcía CañeroR1995Hydrogen ion-dependent oncogenesis and parallel new avenues to cancer prevention and treatment using a H+-mediated unifying approach: pH-related and pH-unrelated mechanismsCritical Rev Oncog61338573605
- HarguindeySPedrazJLGarcía CañeroR2000Edelfosine, apoptosis, MDR and Na+/H+ exchanger: induction mechanisms and treatment implicationsApoptosis587911227496
- HarguindeySOriveGPedrazJL2005The role of pH dynamics and the Na+/H+ antiporter in the etiopathogenesis and treatment of cancer. Two faces of the same coin-one single natureBioch Biopyhs Acta Revs Cancer1756124
- HarguindeySReshkinSJOriveG2007Growth and trophic factors and the pH, Na+/H+ exchanger in Alzheimer’s disease, other neurodegenerative diseases and cancer: new therapeutic possibilities and potential dangersCurr Alzheimer Res4536517316166
- HarguindeySOriveGAnituaE2008Hacia un nuevo enfoque integrado del tratamiento de las enfermedades neurodegenerativas (ENDs): de etiopatogenia a tratamiento. El síndrome de deficiencia de factores tróficos y de crecimiento (SDFC)Gen T3829
- HeftiF1983Alzheimer’s disease caused by a lack of nerve growth factor?Ann Neurol13109106830157
- HirakuraYLinM-ChKaganBL1999Alzheimer amyloid aβ1-42 channels: effect of solvent, pH, and congo redJ Neurosci Res574586610440895
- HirparaJClémentM-VPervaizS2001Intracellular acidification triggered by mitochondrial-derived hydrogen peroxide is an effector mechanism for drug-induced apoptosis in tumor cellsJ Biol Chem2765142111016925
- IshaqueAAl-RubeaiM1998Use of intracellular pH and annexin V flow cytometric assays to monitor apoptosis and its suppression by bcl-2 over-expression in hybridoma cell cultureJ Immunol Methods22143579894897
- IzumiHTTorigoeTIshiguchiH2003Cellular pH regulators: potentially promising molecular targets of cancer chemotherapyCancer Treat Rev29541914585264
- JinKZhuYSunY2002Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivoProc Natl Acad Sci U S A99119465012181492
- KerrJFWyllieAHCurrieAR1972Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kineticsBr J Cancer26239574561027
- KhaledARMoorANLiA2001Thropic factor withdrawal: p38 mitogen-activated protein kinase activates NHE1, which induces intracellular alkalinizationMol Cell Biol2175455711604491
- KirklandRAFranklinJI2003Prooxidant effects of NGF withdrawal and MEK inhibition in sympathetic neuronesAntioxid Redox Signal5635914580320
- KutCMac GabhannFPopelS2007Where is VEGf in the body? A meta-analysis of EGF distribution in cancerBr J Cancer979788517912242
- Lagadic-GossmannDHucLLecureurV2004Alterations of intracellular pH homeostasis in apoptosis: origins and rolesCell Death Differ119536115195071
- L’AllemainGFranchiACragoeEJ1984Blockade of the Na+/H+ antiport abolishes growth factor-induced DNA synthesis in fibroblastsJ Biol Chem259431396323465
- LavrikINGolksAKrammerPH2005Caspases: pharmacological manipulation of cell deathJ Clin Invest11526657216200200
- LambrechtsDStorkebaumEMorimotoM2003VEGF is a modifier of amyothropic lateral sclerosis in mice and humans and protects motoneurons against ischemic deathNat Genet34357812923539
- LetaiAG2008Diagnosing and exploiting cancer’s addition to blocks in apoptosisNature Revs Cancer81213218202696
- LiJEastmanA1995Apoptosis in an interleukin-2-dependent cytotoxic T lympohcyte cell line is associated with intracellular acidification. Role of the Na(+)/H(+)-antiportJ Biol Chem2703203117852405
- LieberthalWTriacaVKohJS1998Role of superoxide in apoptosis induced by growth factor withdrawalAm J Physiol275F691F7029815127
- LiuDMartinoGThangarajuM2000Caspase-8-mediated intracellular acidification preceds mitochondrial dysfunction in somatostatin-induced apoptosisJ Biol Chem27592445010734062
- MarchesRVitettaESUhrJW2001A role for intracellular pH in membrane IgM-mediated cell death of human B lymphomasProc Natl Acad Sci U S A983434911248096
- MateoILlorcaIInfanteJ2007Low serum VEGF levels are associated with Alzheimer’s diseaseActa Neurol Scand11656817587256
- MatsuyamaSLlopisJDeverauxQL2000Changes in intramitchondrial and cytosolic pH: early events that modulate caspase activation during apoptosisNature Cell Biol23182510854321
- MatsuyamaSReedJC2000Mitochondria-dependent apoptosis and cellular pH regulation 2000Cell Death Differ711556511175252
- McCarthyMJRubinLLPhilottKL1997Involvement of caspases in sympathetic neuron apoptosisJ Cell Sci1102165739378766
- Meléndez HeviaE2001Natural selection and thermodynamics in biological evolution: from the origin of life to cancerServicio de Publicaciones de la Universidad de La LagunaTenerife, Spain188
- MiragliaEViarisioDRigantiCH2005Na+/H+ exchanger activity is increased in doxorubicin-resistant human colon cancer cells and its modulation modifies the sensitivity of the cells to doxorubicinInt J Cancer115924915729714
- MoolenarWHMummeryCLvan der Saag1983Na+/H+ exchange and cytoplasmatic pH in the action of growth factors in human fibroblastsNature30464586410286
- OriveGReshkinSJHarguindeyS2003Hydrogen ion dynamics and the Na+/H+ antiporter in cancer angiogenesis and antiangiogenesisBr J Cancer893959
- ParkHSLeeBKParkS2005Effects of sabiporide, a specific Na+/H+ exchange inhibitor on neuronal cell death and brain ischemiaBrain Res1061677116225853
- Pérez-SalaDCollado-EscobarDMollinedoF1995Intracellular alkalinization supresses lovastatin-induced apoptosis in HL60 cells through the inactivation of a pH-dependent endonucleaseJ Biol Chem2706235427890761
- MishraAPavelkoT2007Treatment of chronic elbow tendinosis with buffered platelet-rich plasmaAm J Sports Med341774816735582
- MurrayMMSpindlerKPAbreuE2007Collagen-platelet rich plasma hydrogel enhances primary repair of the porcine anterior cruciate ligamentJ Orthop Res25819117031861
- PangI-HZengHFleenorD2007Pigment epithelium-derived factor protects retinal ganglion cellsBMC Neurosci811117199885
- ParkHJLyonsJCOhtsubo1999Acidic environment causes apoptosis by increasing caspase activityBr J Cancer801892710471036
- PeronaRSánchez-PérezI2007Signalling pathways involved in clinical responses to chemotherapyClin Transl Oncol96253317974523
- PolitiLERotsteinNPSalvadorG2001Insulin-like growth factor- I is a potential trophic factor for amacrine cellsJ Neurochem76119921111181839
- PoserSImpeySXiaZ2003Brain-derived neurotrophic factor protection of cortical neurones from serum withdrawal-induced apoptosis is inhibited by cAMPJ Neurosci234420712805282
- PoüysségurJDayanFMazureNM2006Hypoxia signalling in cancer and approaches to enforce tumor regressionNature4414374316724055
- ProdanCIRossEDVincentAS2007Coated-platelets correlate with disease progression in Alzheimer’s diseaseJ Neurol254548917380246
- RainaAKZhuXRottkampCA2000Cyclin’ towards dementia: cell cycle abnormalities and abortive oncogenesis in Alzheimer’s diseaseJ Neurosci Res611283310878584
- RainaAKHochmanAIckesH2003Apoptotic promoters and inhibitors in Alzheimer’s disease: who wins out?Prog Neuropsychopharmacol Biol Psych272514
- RaySBritschgiMHerbertCH2007Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteinsNature Med1313596217934472
- RebolloAGómezJde AragónAM1995Apoptosis induced by IL-2 withdrawal is associated with an intracellular acidificationExp Cell Res21858157796894
- ReedJC2002Apoptosis-based therapiesNat Revs Drug Discov11112112120092
- ReedJC2004Apoptosis mechanisms: Implications for cancer drug discoveryOncology18112015651172
- RenzABerdelWEKreuterM2001Rapid extracellular release of cytochrome c is specific for apoptosis and marks cell death in vivoBlood981542811520805
- ReshkinSJBellizziACaldeiraS2000Na+/H+ exchanger-dependent intracellular alkalinization is an early event in malignant transformation and plays an essential role in the development of subsequent transformation-associated phenotypesFASEB J1421859711053239
- ReshkinSJBellizziACardoneM2003Paclitaxel induces apoptosis via Protein Kinase A- and p38 Mitogen-activated Protein-dependent inhibition of the Na+/H+ Exchanger (NHE) Isoform 1 in Human Breast cancer cellsClinical Cancer Res923667312796407
- ResnickLFennellM2004Targeting JNK3 for the treatment of neurodegenerative disordersDrug Disc Today99329
- ReynoldsJELiJFCraigRW1996BCL-2 and MCL-1 expression in chinese hamster ovary cells inhibits intracellular acidification and apoptosis induced by staurosporineExp Cell Res22543068660932
- RichIRWorthington-WhiteDGardenOA2000Apoptosis of leukemic cells accompanies reduction in intracellular pH after targeted inhibition of the Na+/H+ exchangerBlood9514273410666221
- RideoutHJStefanisL2001Caspase inhibition: a potential strategy in neurological diseasesHistol Histopathol689590811510981
- RiedlAJShiY2004Molecular mechanisms of caspase regulation during apoptosisNature Revs Mol Cell Biol589790715520809
- RigasBSunY2008Induction of oxidative stress as a mechanism of action of chemopreventive agents against cancerBr J Cancer9811576018253125
- RivaMMolteniRBedogniF2005Emerging role of the FGF system in psychiatric disordersTrends Pharmacol Sci262283115860368
- RoepePDWeiLYCruzJ1993Lower electrical membrane and altered pHi homeostasis in multidrug-resistant (MDR) cells: further characterization of a series of MDR cell lines expressing different levels of P-glycoproteinBiochemistry3211042568105888
- RoepePDGilliesRG2001pH and multidrug resistanceThe tumor microenvironment: causes and consequences of hypoxia and acidity Novartis Foundation Symposium, No. 240Chichester, New YorkJohn Wiley and Sons Ltd23247
- SajadiABensadounJ-ChSchneiderBL2006Transient striatal delivery of GNDF via encapsulated cells to sustained behavioral improvement in a bilateral model of Parkinson’s diseaseNeurobiol Dis221192916300956
- Schulte-HerbrüggenOJockers-ScherübiMCHellwegR2008Neurotropins: from physiopathology to treatment of Alzheimers’s diseaseCurr Alz Res53844
- ShackaJJRothKA2005Regulation of neural cell death and neurodegeneration by members of the Bcl-2 family: therapeutic implicationsCurr Drug Targets CNS Neurol Dis42539
- SehgalIPowersSHuntleyB1994Neurotensin is an autocrine trophic factor stimulated by androgen withdrawal in human prostate cancerProc Natl Acad Sci U S A91467378197117
- SimonSGilliesR2001The multiple mechanisms of drug resistance and cellular pHThe tumor microenvironment: causes and consequences of hypoxia and acidity Novartis Foundation Symposium No. 240Chichester, New YorkJohn Wiley and Sons, Ltd26981
- SlevinJTGashDMSmithCD2006Unilateral intraputaminal glial cell line-derived neurotrophic factor in patients with Parkinson’s disease: response to 1 year each of treatment and withdrawalNeurosurg Focus20E116711657
- SolerteSBFerrariECuzzoniG2005Decrease release of the angiogenic peptide vascular endothelial growth factor in Alzheimer’s disease: recovering effect with insulin and DHEA sulphateDement Geriatr Cogn Disord1911015383738
- SortwellCEBowersWJCountsSE2007Effect of ex vivo transduction of mesencephalic reagggregates with bcl-2 on grafted dopamine neuron survivalBrain Res113433417196186
- SperandioSde BelleIBredesenDE2000An alternative, nonapoptotic form of programmed cell deathProc Natl Acad Sci U S A97143768111121041
- StorkebaumECarmelietP2004aVEGF: a critical player in neurodegenerationJ Clin Invest113141814702101
- StorkebaumELambrechtsDDewerchinM2004bTreatment of motoneuron degeneration by intraventricular delivery of VEGF in a rat model of ALSNature Neurosci8859215568021
- SzakácsGPatersonJKLudwigJA2006Targeting multidrug resistance in cancerNature Revs521931
- TafaniMCohnJAKarpinichNO2003Regulation of intracellular pH mediates Bax activation in HeLa cells treated with staurosporine on tumor necrosis factor-alphaJ Biol Chem277495697612393866
- TaetleRDos SantosBOhsuigiY1994Effect of combined antigrowth factor receptor treatment on in vitro growth of multiple myelomaJ Natl Cancer Inst8645058120920
- TakashakiAMasudaSunM2004Oxidative stress-induced apoptosis is associated with alterations in mitochondrial caspase activity and Bcl-2-dependent alterations in mitochondrial pH (pHm)Brain Res Bull6249750415036564
- TangDGLiLChopraDP1998Extended survivability of prostate cancer cells in the absence of trophic factors: increased proliferation, evasion of apoptosis, and the role of apoptosis proteinsCancer Res583466799699682
- TattonWGChamblers-RedmanRJuWJH2002Propargylamines induce antiapoptotic new protein synthesis in serum- and nerve growth factor (NGF)-withdrawn, NGF-differentiated PC-12 cellsJ Pharm Exp Therap30175364
- TattonWChenDChamblers-RedmanR2003Hypotheses for a common basis for neuroprotection in glaucoma and Alzheimer’s disease: antiapoptosis by alpha-2-adrenergic receptor activationSurvey Ophtalmol48Suppl 1s25s37
- ThangarajuMSharmaKLeberB1999Regulation of acidification and apoptosis by SHP-1 and BCL-2J Biol Chem274295495710506221
- ThompsonCBBauerDELumJJ2005How do cancer cells acquire the fuel needed to support cell growth?Cold Spring Harbor Symp Quant Biol703576216869772
- TorigoeTUIzumiHIseT2002Vacuolar H+-ATPase: functional mechanisms and potential as a target for cancer chemotherapyAnticancer Drugs132374311984067
- TroyCMStefanisLProchiantzA1996The contrasting roles of ICE family proteases and inteleukin-1β in apoptosis induced by trophic factor withdrawal and by copper/zinc superoxide dismutase down-regulationProc Natl Acad Sci U S A935635408643629
- VaghefiHHughesAlNeetKE2004Nerve growth factor withdrawal-mediated apoptosis in naïve and differentiated PC12 cells through p53/caspase-3-dependent and -independent pathwaysJ Biol Chem279156041414739302
- VaronSManthorpeMWilliamsLR1983Neuronotrophic and neurite-promoting factors and their clinical potentialsDev Neurosci673866088207
- Vende VeldeChClevelandDW2005VEGF: multitasking in ALSNature Neurosci85715622408
- VincentAMTenBroeckeMMaieseK1999Neuronal intracellular pH directly mediates nitric oxide-induced programmed cell deathJ Neurobiol401718410413448
- WagnerBNatajaranAGrünaugS2006Neuronal survival depends on EGFR signalling in cortical but not in midbrain astrocytesEMBO J257526216467848
- WahlMLOwenJABurdR2002Regulation of intracellular pH in human melanoma: potential therapeutic implicationsMol Cancer Therap16172812479222
- WaibelMKramerSLauderK2007Mitochondria are not required for death-receptor mediated cytosolic acidificaction during apoptosisApoptosis126233017195091
- WaldeimerPCTattonW2004Interrupting apoptosis in neurodegenerative disease: potential for effective therapy?Drug Discover Today92108
- WaldeimerPC2003Prospects for antiapoptotic drug therapy of neurodegenerative diseasesProg Neuropsychopharm Biol Psychiatry2730321
- WallaceJLDicayMMcKnightW2006Platelets accelerate gastric ulcer healing through presentation of vascular endothelial growth factorBr J Pharmacol148274816565732
- WilkinsANikodemovaMCompstonA2004Minocycline attenuates nitric oxide-mediated neuronal axonal destruction in vitroNeuron Glia Biol129730518634603
- WilliamsACTCollardJParaskevaC1999An acidic environment leads to p53 dependent induction of apoptosis in human adenoma and carcinoma cell lines: implications for clonal selection during colorectal carcinogenesisOncogene18319920410359525
- YanCYFerrariGGreeneLA1995N-acetylcysteine-promotes survival of PC12 cells is gluthation-independent but transcription-dependentAm Soc Biochem Mol Biol2702682732
- YangSPBaeAGKangHJ2004Co-accumulation of vascular endothelial growth factor with β-amyloid in the brain of patients with Alzheimer’s diseaseNeurobiol Aging252839015123332
- YangJXuesongLiuKapilBhalla1997Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blockedScience751129329027314
- YangS-PDong-GooBaeKangHJ2004Co-accumulation of vascular endothelial growth factor with β-amyloid in the brain of patients with Alzheimer’s diseaseNeurobiol Aging252839015123332
- ZankeBWLeeCArabS1998Death of tumor cells after intracellular acidification is dependent on stress-activated protein kinases (SAPK/JNK) pathway activation and can not be inhibited by Bcl-2 expression or inteleukin 1β-converting enzyme inhibitionCancer Res58280189661894
- ZhiuhaXSchendelSMatsuyamaS1998Acidic pH promotes dimerization of Bcl-2 family proteinsBiochemistry376410189572858
- ZhuJKSunYMaoXO2002Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivoProc Natl Acad Sci U S A99119465012181492
- ZhuXOgawaOWangY2003JKK1, an upstream activator of JNK/SAPK, is activated in Alzheimer’s diseaseJ Neurochem85879312641730
- ZubenkoGSCohenBMReynoldsCF1987aPlatelet membrane fluidity in Alzheimer’s disease and major depressionAm J Psychiatry14486083605398
- ZubenkoGSWusylkoMCohenBM1987bFamily study of platelet membrane fluidity in Alzheimer’s diseaseScience238539423659926