Abstract
These studies investigated the absorption and metabolic conversion of lisdexamfetamine dimesylate (LDX), a prodrug stimulant that requires conversion to d-amphetamine for activity. Oral absorption of LDX was assessed in rat portal and jugular blood, and perfusion of LDX into isolated intestinal segments of anesthetized rats was used to assess regional absorption. Carrier-mediated transport of LDX was investigated in Caco-2 cells and Chinese hamster ovary (CHO) cells expressing human peptide transporter-1 (PEPT1). LDX metabolism was studied in rat and human tissue homogenates and human blood fractions. LDX was approximately10-fold higher in portal blood versus systemic blood. LDX and d-amphetamine were detected in blood following perfusion of the rat small intestine but not the colon. Transport of LDX in Caco-2 cells had permeability apparently similar to cephalexin and was reduced with concurrent PEPT1 inhibitor. Affinity for PEPT1 was also demonstrated in PEPT1-transfected CHO cells. LDX metabolism occurred primarily in whole blood (rat and human), only with red blood cells. Slow hydrolysis in liver and kidney homogenates was probably due to residual blood. The carrier-mediated absorption of intact LDX, likely by the high-capacity PEPT1 transporter, and subsequent metabolism to d-amphetamine in a high-capacity system in blood (ie, red blood cells) may contribute to the consistent, reproducible pharmacokinetic profile of LDX.
Introduction
Lisdexamfetamine dimesylate (LDX; Vyvanse®, Shire US Inc.) is the first long-acting prodrug stimulant indicated for the treatment of attention-deficit/hyperactivity disorder (ADHD) in children aged six to 12 years and in adults. After oral administration, LDX is converted to l-lysine and d-amphetamine, the latter being the active drug for the treatment of ADHD ().
Figure 1 Enzymatic conversion of LDX to active d-amphetamine.
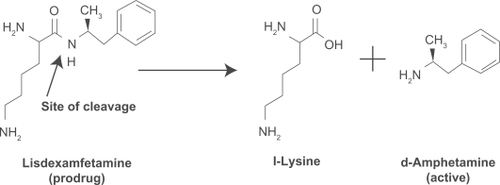
LDX has been shown to provide a long duration of effect, which allows once-daily dosing.Citation1 Low interpatient variability of d-amphetamine plasma levels from LDX has been suggested by consistent delivery of d-amphetamine from patient to patient in a study in children with ADHD and, similarly, within and between subjects in a clinical trial in healthy adults.Citation2,Citation3 Physicochemical and biologic characteristics of LDX, such as high water solubility, lack of effect on absorption/conversion by changes in gastric environment and pH, and gradual conversion of LDX to active d-amphetamine, may impact interpatient and intrapatient variability and consistent delivery of d-amphetamine from patient to patient in these studies.
LDX is the only prodrug available for treatment of ADHD. It is important to understand the absorption and enzymatic conversion of intact LDX to the active moiety, d-amphetamine. The aim of the following studies was to investigate the absorption and enzymatic conversion of LDX in rat and human tissue and cell culture models.
Methods and materials
Male Sprague–Dawley rats weighing approximately 240 to 400 g (Hilltop Lab Animals Inc., Scottdale, PA) were acclimated for at least two days. The rats were maintained on a 12-hour, light–dark cycle, had free access to water, and were fasted overnight before the experiment. For studies conducted in conscious rats, food was returned four hours postdose and the rats had free access to water and food throughout the study. All protocols involving animal experimentation were reviewed and approved by the Institution Animal Care and Use Committee of each test facility. In vivo procedures were in compliance with the National Research Council’s Guide for the Care and Use of Laboratory Animals.Citation4 All protocols involving the use of human tissues were reviewed and approved by the Institutional Review Board for Human Research of each test facility. Unless otherwise noted, all chemicals were obtained from Sigma-Aldrich Corporation (St. Louis, MO). LDX was supplied by Shire (Basingstoke, Hampshire, UK).
Studies of LDX absorption
Oral absorption of LDX in conscious rats
Conscious rats with indwelling cannulae implanted in the jugular and portal veins were administered a single oral dose of LDX by gavage (10 mg/kg of LDX in 5 mL/kg of water). Whole blood was collected with concurrent replacement with donor blood from the portal and jugular veins at predose, 10 and 30 minutes, and 1, 2, 4, 6, 8, and 24 hours postdose into tubes (4°C) containing potassium ethylenediamine tetra-acetic acid (EDTA) as an anticoagulant and centrifuged, and plasma was stored at −20°C prior to analysis.
Single-pass intestinal perfusion in anesthetized rats
Single-pass intestinal perfusion studies were performed in rats using modifications to established methods.Citation5,Citation6 Briefly, fasted male rats (n = 4 per group) were anesthetized with ketamine/xylazine (1 mL/kg) and placed on a heating pad to maintain body temperature. Intestinal segments were exposed by midline incision and externalized. The proximal ends of intestinal segments were identified as follows: 1 to 2 cm below the pylorus for the duodenum, 10 cm below the pylorus for the jejunum, 15 cm above the ileocecal junction for the ileum, and 1 to 2 cm below the colocecal junction for the colon. A silicone cannula was placed at the proximal ends of each segment, and a small incision was made 10 cm below the proximal cannula. After flushing with warm saline, an intestinal loop was created by cannulation of the distal end of the segment. The lumens of the isolated segments were perfused (0.2 mL/min with a peristaltic pump) with an appropriate perfusion solution (Krebs-Ringer bicarbonate buffer at pH 6.5 for the duodenum and jejunum; pH 7.4 for the ileum and colon) at 37°C.
Experimental perfusions included three reference compounds: fluorescein isothiocyanate-dextran (25 μM FD-4; average molecular mass = 4000 u), an unabsorbed marker; atenolol (100 μM), a low permeability marker; and antipyrine (100 μM), a high permeability marker. Perfusion was initiated with the test solution (LDX 0.125 mg/mL in perfusion solution with markers). Samples of perfusate (3 mL) were collected every 15 minutes for 90 minutes postequilibration with simultaneous collection of blood samples from the femoral vein. Plasma was collected after centrifugation and stored at −20°C.
Permeability of LDX in Caco-2 cells
Caco-2 cell monolayers were grown to confluence on collagen-coated, microporous, polycarbonate membranes in 12-well culture platesCitation7 to establish an intact transporting epithelial layer. The techniques have been previously reported and were adapted with minor modifications. The apical permeability assay buffer (pH 6.0) was Hanks, balanced salt solution (HBSS) containing 10 mM 4 2-(N-morpholino) ethanesulfonic acid and 15 mM glucose. The basolat-eral assay buffer (pH 7.4) was HBSS containing 10 mM N-2-hydroxyethylpiperazine-n-2-ethanesulfonic acid and 15 mM glucose. Cell monolayers were preincubated with appropriate assay buffer at 37°C for 30 minutes to establish a proton gradient. An additional set of monolayers was preincubated with 5 mM glycylsarcosine (Gly-Sar), a known substrate of the peptide transporter-1 (PEPT1) transporter,Citation7 in both chambers for 30 minutes to inhibit PEPT1 activity.
Transport experiments were initiated by the addition of 10 μM LDX or 10 μM cephalexin, a reference substrate for the PEPT1 transporter,Citation8 in assay buffer (pH 6.0) to the apical chambers, and assay buffer (pH 7.4) without agents was added to the basolateral chambers. For inhibition experiments, 5 mM Gly-Sar was also present in both chambers. Incubations were carried out at 37°C with 5% carbon dioxide in a humidified chamber. At one and two hours, 200 μL samples were taken from the basolateral chambers, and fresh assay buffer was added. Prior to the first incubation and at two hours, 100 μL of the apical assay buffer was collected. Each determination was performed in triplicate.
Further experiments assessed the permeability of LDX in Caco-2 cells that had been induced to overexpress the PEPT1 transporter. For these studies, 10 mM Gly-Sar was added to culture media, with daily replacement of the medium to induce expression of the PEPT1 transporter prior to experiments. Expression levels of PEPT1 were confirmed by quantitative polymerase chain reaction (qPCR)Citation9 from total RNA isolated from Caco-2 cells and reverse-transcribed into cDNA. The qPCR was performed with predesigned sense and antisense primers of human PEPT1 and human β-actin as a housekeeping gene. Probes were fluorescein-labeled for fluorescent detection of PCR products. Reactions and detection of PCR products were carried out in the LightCycler® 480 System (Roche, Nutley, NJ).
Inhibition of Gly-Sar transport by LDX in CHO cells
The potential for LDX to modify the uptake of the reporter substrate, Gly-Sar, into Chinese hamster ovary (CHO) cells expressing PEPT transporters was measured using a modification of a previously described method.Citation10 PEPT1-CHO, PEPT2-CHO, and CHO parent cells (100,000 cells/well) were grown for 24 hours in media supplemented with 5 mM sodium butyrate. Before the experiments, the medium was removed and cells were rinsed with Krebs–Henseleit buffer. Uptake experiments were carried out over 10 minutes at 37°C in 50 μL of Krebs–Henseleit buffer containing radiolabeled reporter substrate, Gly-Sar (2 μM) with or without LDX or vehicle, using previously described methods. LDX (10 to 10,000 μM) or vehicle (dimethylsulfoxide [DMSO]) was added to duplicate wells. DMSO concentration did not exceed 1% (vol/vol) in any assays. The reference inhibitors for PEPT1 (tyrosine-phenylalanine) and for PEPT2 (cefadroxil), both at 200 μM, were also assayed in duplicate as a positive control for Gly-Sar uptake inhibition. The amount of reporter substrate taken up by cultured cells was determined by liquid scintillation spectrometry. Uptake activity was reported as % activity: % activity = (A–B)/(C–D) × 100, where A is the amount of reporter substrate in the presence of LDX on transfected cells, B is the amount of reporter substrate in the presence of LDX on parent cells, C is the amount of reporter substrate in the presence of DMSO on transfected cells, and D is the amount of reporter substrate in the presence of DMSO on parent cells. Inhibitory activity of LDX was calculated as IC50 (μM) and efficacy (%) parameters. IC50 (μM) is defined as the concentration of LDX needed to inhibit maximal uptake of reporter substrate by 50%. Efficacy is defined as maximum inhibition achieved by LDX in percent maximal activity. GraphPad Prism 4.03 (GraphPad Software, Inc., La Jolla, CA) was used for curve fitting and determination of reaction parameters.
Studies of LDX hydrolysis
LDX hydrolysis in tissues and blood fractions from rats and human donors
Whole blood, plasma, liver, kidney, upper and lower small intestine, pancreas, and colon tissue samples were collected from three rats and two male human donors (surgical remnant/death from a human tissue procurement center in Philadelphia, PA). Amphetamine abusers were excluded. Tissue samples from rats and human donors were homogenized in 0.1 M phosphate buffer. Because of intersubject variability in the human colon results, colon tissue samples were collected from an additional (third) human donor.
Human blood fractions, including red blood cells (RBCs), peripheral blood mononucleated cells (PBMCs), polymorphonuclear cells (PMNs), and platelets, were collected from two male donors and reconstituted in HBSS to concentrations equivalent to whole blood. Human RBC fractions from fresh blood collected from one male and one female donor were purchased from a commercial human blood cell supplier (Bioreclamation, Inc., New York, NY). Blood from donors with hematocrit outside the normal range were excluded.
Tissue or blood cell samples were incubated at 37°C in HBSS. Incubation reactions were initiated by the addition of LDX (1.0 μg/mL), and samples were collected for assay of LDX and d-amphetamine at 0, 0.25, 0.5, 1, 2, and 4 hours. Additionally, assay conditions included RBC fractions from each donor reconstituted with saline to 10%, 25%, 33%, 50%, 75%, and 90% of the original hematocrit; one RBC fraction from each donor at 90% hematocrit lysed prior to incubation; and one RBC fraction at 100% hematocrit in the presence of additional EDTA (approximately 12.15 mg of potassium EDTA/7.0 mL of blood).
LDX stability in incubations with human liver microsome and rat and human hepatocytes
Human liver microsomes prepared from 15 individuals (male and female) and hepatocytes from rat and human livers prepared according to previously published collagenase perfusion methodsCitation11 were incubated with LDX. Human liver tissue for preparation of hepatocytes was obtained from rejected-for-transplant livers and discarded surgical remnants with appropriate consent from research tissue organizations and participating medical centers. Liver microsomes were incubated with LDX (12.5 or 125 ng/mL) at 37°C for 30 minutes. Hepatocyte suspensions (2 × 106 cells/mL), prepared in Dulbecco’s modified Eagle’s medium with 5% fetal bovine serum, were incubated with LDX (12.5 or 125 ng/mL) at 37°C with samples collected at 0, 1, 2, and 4 hours.
LDX stability in incubations with various peptidases
The stability of LDX was studied in the presence of three hydrolytic peptidase enzymes: human dipeptidyl peptidase IV, cathepsin G, and elastase. Buffer (pH 7.5) containing 25 mM of Tris HCl, 0.130 M of NaCl, and 0.0027 M of KCl was used for incubations with dipeptidyl peptidase IV and elastase and 100 mM HEPES buffer (pH 7.5) for cathepsin G. Enzymes were dissolved in their respective buffer system (approximately 0.25 units/mL of dipeptidyl peptidase IV or 1.0 unit/mL of cathepsin G and elastase). Incubations at 37°C with LDX (1 μg/mL) were for 0, 5, 15, 30, 45, and 60 minutes.
The stability of LDX was studied in the presence of simulated gastric fluid (SGF), simulated intestinal fluid (SIF), and trypsin. LDX (200 μg/mL) was incubated at 37°C with either SGF containing porcine pepsin (pH ∼1.2) or SIF, containing porcine pancreatin (pH ∼6.8) (United States Pharmacopeia-National Formulary, 2005; United States Pharmacopeial Convention, Rockville, MD). Briefly, incubations were at 37°C for 0, 5, 15, 30, 45, and 60 minutes. For incubation with trypsin, LDX (1 μg/mL) was incubated at 37°C in a 0.1 M potassium phosphate buffer (pH 7.4) with trypsin at concentrations of 100, 2000, or 20,000 units/mL. All in vitro reactions were terminated by the addition of 0.5 mL of chilled acetonitrile.
Analytical methods
All samples were extracted by protein precipitation with acetonitrile and analyzed for LDX and d-amphetamine using an electrospray tandem liquid chromatography-mass spectrometry (LC-MS/MS) method. Perfusate samples were also analyzed for the unabsorbed marker, FD-4, using a 96-well plate fluorescence plate reader (FLUOstar, BMG Labtech Inc., Durham, NC) with excitation and emission wavelengths of 492 and 520 nm. Perfusate and plasma samples were also analyzed for atenolol and antipyrine by LC-MS/MS.
Results
Studies of LDX absorption
Oral absorption of LDX in conscious rats
illustrates the plasma concentration-versus-time curves for systemic and portal circulation of LDX and d-amphetamine administered orally to rats. Pharmacokinetic studies in portal and jugular vein-cannulated rats showed that after administration of a single oral dose of LDX, intact LDX was rapidly absorbed from the gastrointestinal (GI) tract (). Plasma concentrations of LDX in both portal and systemic circulation peaked by 10 minutes postdose (time to maximum plasma concentration [Tmax] 0.17 hours). d-Amphetamine was measurable in both portal and systemic plasma shortly after dosing, suggesting rapid conversion of LDX to d-amphetamine after absorption, and plasma d-amphetamine in both portal and systemic circulation peaked between one and two hours postdosing. LDX and d-amphetamine had similar area under the plasma concentration time curve from time 0 hour to last time point (AUClast) in portal plasma, but the AUClast of d-amphetamine was approximately 3.2-fold higher than the AUClast for LDX in systemic plasma, suggesting there is presystemic (ie, in portal blood before entry into the systemic circulation) conversion of LDX to d-amphetamine (). In addition, portal blood levels of LDX were approximately 10-fold higher than systemic levels (AUClast for LDX in systemic and portal blood was 158.0 and 1501.1 ng•h/mL, respectively, ), indicating presystemic conversion of LDX to d-amphetamine in the rat model.
Figure 2 Mean (SD) of systemic (A) and portal (B) plasma concentration versus time for LDX and d-amphetamine in rats.
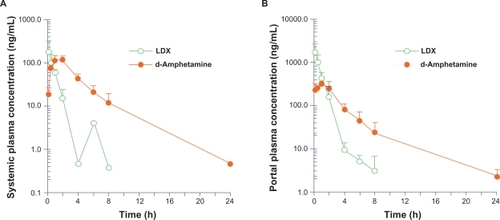
Table 1 Mean (SD) systemic and portal pharmacokinetic parameters in Sprague–Dawley rats
Single-pass intestinal perfusion in anesthetized rats
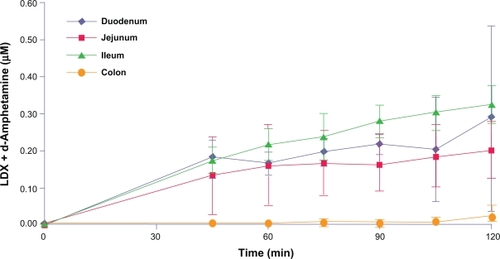
LDX absorption was detected from the duodenum, jejunum, and ileum during regional perfusions of isolated intestinal segments in rats. LDX and d-amphetamine plasma concentrations rose initially during perfusion of the jejunal segments and then remained stable, whereas LDX and d-amphetamine plasma concentrations during duodenal and ileal perfusion increased steadily throughout the experiments (). Very little plasma LDX and d-amphetamine was seen during perfusion of the colon (). In addition, plasma d-amphetamine concentrations rose steadily throughout the experiment after perfusion of all segments except the colon. Mean (SD) d-amphetamine concentrations in plasma were 24.2 (16.4), 19.0 (7.0), and 29.5 (6.0) ng/mL at 120 minutes after perfusion of the duodenum, jejunum, and ileum, respectively, and 0.9 (1.2) ng/mL in the colon.
Figure 3 Absorption of total LDX (intact LDX and d-amphetamine) from rat intestinal segments.
Carrier-mediated transport of LDX in noninduced and PEPT1-overexpressing Caco-2 cells
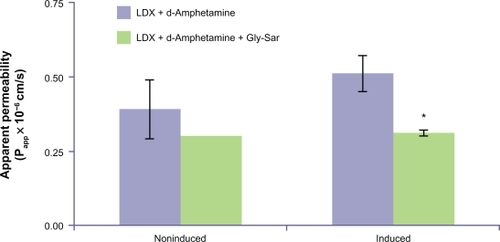
Carrier-mediated directional LDX transport was demonstrated in Caco-2 cells that endogenously express PEPT1. Approximately half the LDX and d-amphetamine transported across Caco-2 cells was converted to d-amphetamine, presumably by endogenous peptidases, so total LDX transport was measured by assay of LDX and d-amphetamine in incubates. Total LDX was transported across the epithelial layer of Caco-2 cells, although the apparent permeability (Papp) rate was low. The mean [SD] Papp of total LDX transport in Caco-2 cells (0.67 [0.17] × 10−6 cm/s) was similar to that seen for cephalexin (0.64 [0.08] × 10−6 cm/s). The mean [SD] Papp of total LDX decreased (P < 0.05) by approximately 50% in the presence of Gly-Sar (0.30 [0.04] × 10−6 cm/s), an inhibitor of PEPT1, suggesting LDX transport was mediated by PEPT1.
Incubation of Caco-2 cells with Gly-Sar resulted in induction of PEPT1 mRNA and functional activity, as well as induction of Gly-Sar-inhibitable total LDX transport. Preliminary experiments found 2.15- and 1.40-fold PEPT1 mRNA induction after 48 hours of incubation with 10 mM Gly-Sar. This induction time was used in all subsequent experiments. Functional induction of PEPT1 by 48-hour incubation in Gly-Sar was demonstrated by an increased Papp for cephalexin from 0.81 × 10−6 cm/s to 1.47 × 10−6 cm/s (P < 0.05). As shown in , induction of Caco-2 cells with Gly-Sar for 48 hours also resulted in a 1.3-fold increased Papp for total LDX from 0.39 × 10−6 cm/s to 0.51 × 10−6 cm/s, although this increase was not statistically significant. Incubation in the presence of Gly-Sar and LDX decreased total LDX transport in these Caco-2 cells by 39% (Papp decreased from 0.51 × 10−6 cm/s to 0.31 × 10−6 cm/s, P < 0.05) ().
Figure 4 Transport of total LDX (intact LDX and d-amphetamine) across Caco-2 cell monolayers with and without induction of PEPT1 expression. *Indicates P < 0.05 versus corresponding group without Gly-Sar added. Mean data from n = 2 or n = 3 determinations. Error bars illustrate SD except where n = 2.
Inhibition of Gly-Sar transport by LDX in CHO cells
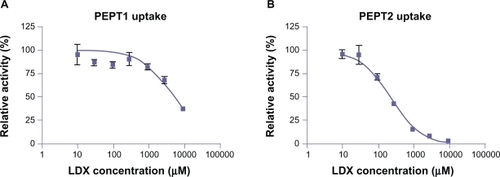
LDX inhibited uptake of radiolabeled Gly-Sar by CHO cells stably expressing either human PEPT1 or PEPT2 (dipeptide transporters), in a concentration-dependent manner (). For PEPT1 uptake inhibition, the maximum efficacy was 63% at 10 mM LDX concentration, with an IC50 of approximately 4 mM. For PEPT2 uptake inhibition, maximum efficacy was 100% at 10 mM LDX, with an IC50 of 246 μM.
Figure 5 Uptake of radiolabeled Gly-Sar in the presence of increasing concentration of LDX by CHO cells expressing PEPT1 (A) or PEPT2 (B).
Hydrolysis of LDX
LDX was stable and no d-amphetamine formation was measured in all negative controls for tissue and enzyme incubations.
LDX stability in rat tissues
LDX was metabolized in rat whole blood and liver tissue. There was a rapid decrease in LDX concentrations and a corresponding increase in d-amphetamine appearance in incubations with liver homogenates and in whole blood. The half-life (t½) of disappearance of LDX was 2.5 and 1.0 hours in rat liver and whole blood, respectively. In contrast, LDX was stable, and the formation of d-amphetamine was negligible in incubations with the other rat tissue homogenates investigated (upper and lower intestines, pancreas, and plasma). There was a slight decline in LDX levels over time, with a corresponding slight increase in d-amphetamine in incubations with kidney and colon homogenates.
LDX stability in human tissues
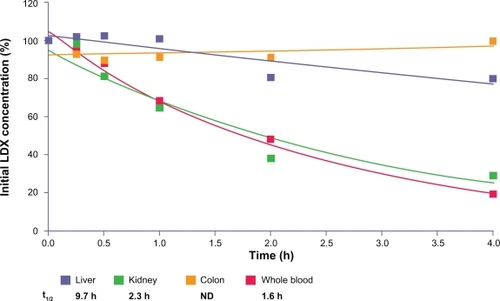
LDX was metabolized in human whole blood and kidney homogenate and to a lesser extent in liver homogenate. LDX levels rapidly decreased over time with concomitant formation of d-amphetamine () in whole blood and kidney homogenates. The t½ of disappearance of LDX averaged 1.6, 2.3, and 9.7 hours for whole blood, kidney homogenate, and liver homogenate, respectively. LDX concentrations were stable and d-amphetamine formation was negligible in incubations with homogenates of the upper and lower intestines, pancreas, and plasma from human donors (data not shown). In incubations of colon homogenates, there was intersubject variability. In the incubation of colon tissue from donor 1, LDX concentrations decreased (t1/2 was approximately 3.7 hours), whereas in the incubation of colon tissue from donor 2, LDX concentrations were stable. Colon tissue from a third donor was incubated and LDX concentrations were stable ().
Figure 6 LDX remaining (percentage of initial concentration) after incubation with human tissue homogenates from donors.
In complementary experiments, LDX was stable in incubations over 30 minutes, with human liver microsomes and in incubations over four hours with freshly isolated rat and human hepatocytes. No d-amphetamine formation was detected in these incubations.
LDX stability in human blood cell fractions
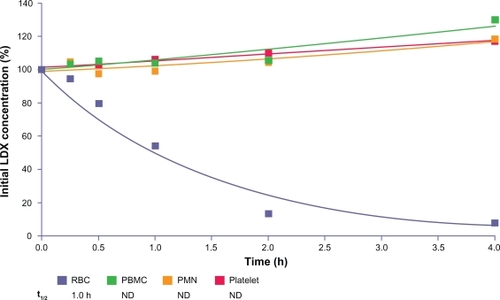
LDX was metabolized by human RBCs. In incubations with RBCs, LDX was converted rapidly and the formation of d-amphetamine increased concurrently (). The t½ of disappearance of LDX in incubations with RBCs averaged 1.0 hours (0.87 and 1.10 hours for RBC fractions from two human donors). LDX concentrations were stable and d-amphetamine formation was negligible in incubations with PBMCs, PMNs, and platelets ().
Figure 7 LDX remaining (percentage of initial concentration) after incubation with blood fractions from donors.
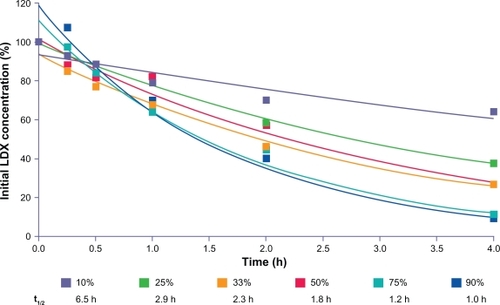
The concentration of LDX decreased over time, and d-amphetamine levels increased in the incubations with reconstituted RBC fractions at various hematocrits, with the highest hematocrit (90%) showing a conversion rate most like that of fresh whole RBCs ( and ). The rate of hydrolysis of LDX tended to decrease as the RBC fractions were diluted, although there was still substantial conversion at the lowest concentrations of RBC fractions (10% and 25% of normal hematocrit). Lysis of RBCs did not affect the rate of LDX conversion. The addition of EDTA to reconstituted RBCs did not inhibit the rate of LDX hydrolysis.
Figure 8 LDX remaining (percentage of initial concentration) after incubation with varying hematocrits of RBCs from a male and female donor.
LDX stability in in vitro assays
LDX was stable and d-amphetamine formation was negligible in incubations with dipeptidyl/peptidase IV, cathepsin G, and elastase. LDX was also stable in SGF and SIF up to 60 minutes. LDX was stable in incubations with trypsin, even when very high trypsin concentrations were used.
Discussion
The intact prodrug, LDX, was readily absorbed from the GI tract after oral administration, with relatively high levels of LDX detected in the portal blood of rats. When LDX was perfused into isolated segments of rat duodenum, jejunum, and ileum, it was absorbed into the systemic circulation; however, LDX was not absorbed during perfusion into colonic segments. These findings indicate that absorption of LDX occurs via carrier-mediated transport in the small intestines and are consistent with the physicochemical properties of LDX (ie, high aqueous solubility of greater than 0.85 g/mL within the physiologically relevant pH range of 1 to 8Citation12 and low lipophilicity [logP–1.75, unpublished data]), which would predict poor passive diffusion across biologic membranes. The prodrug LDX can be considered to be a peptidomimetic, in which the naturally occurring amino acid, l-lysine, is covalently linked to d-amphetamine. Therefore, it is likely that LDX will be a substrate for peptide transport proteins such as PEPT1 and PEPT2. Because PEPT1 is highly expressed in the small intestines, but not in the colon of adult rats,Citation13 the profile of LDX absorption observed in our studies is consistent with PEPT1-mediated transport.
To further investigate the involvement of the PEPT1 transporter in absorption of LDX, permeability experiments in Caco-2 cells were conducted, as PEPT1 has been shown to be expressed in this cell line.Citation14–Citation16 LDX flux across Caco-2 cells was similar in magnitude to the measured flux of cephalexin, a known substrate for the PEPT1 transporterCitation15,Citation17,Citation18 and was partially inhibited by Gly-Sar, an established competitive substrate for PEPT1. Additionally, LDX transport in Caco-2 cells correlated well with the expression level of PEPT1, in that higher transport was demonstrated in cells in which PEPT1 had been induced (both in terms of mRNA and functionality). The affinity of LDX for human PEPT1 and PEPT2 transporters was confirmed in transfected CHO cells. Based on inhibition of Gly-Sar uptake, LDX had a low affinity for PEPT1 (∼4 mM) and a relatively high affinity for PEPT2 (∼200 μM), values that reflected the low affinity/high capacity and high affinity/low capacity of these two transporters, respectively. LDX is, therefore, a potential substrate for both transporters; however, the localization of PEPT1 in the small intestines suggests that LDX absorption is likely to be mediated by PEPT1 rather than PEPT2. Of course, the involvement of other peptide and amino acid transporters in the absorption of LDX in man cannot be ruled out. PEPT1 transport is characterized as a high-capacity uptake system for amino acids and drug ligands that mediates fast and efficient intestinal uptake, even in settings of impaired mucosal function.Citation19 These findings are consistent with the apparent lack of effect of concomitant food intake on absorption of PEPT1 substrates such as cephalexinCitation20 and, similarly, with data demonstrating that food does not impair LDX bioavailability.Citation21
d-Amphetamine was detected in portal blood shortly after oral dosing, suggesting rapid metabolism of LDX to d-amphetamine, either during or immediately after absorption. Previous evidence suggested that this enzymatic conversion likely occurs by first-pass intestinal and/or hepatic metabolism.Citation22 The current findings suggest that LDX is primarily absorbed intact in the small intestines into the portal circulation and that enzymatic hydrolysis of LDX occurs primarily in the blood.
Although these rat studies were not intended to establish a detailed pharmacokinetic profile in this setting, the results are consistent with previous LDX studies in healthy human volunteers. After intravenous administration, the pharmacokinetic profile of d-amphetamine from intact LDX (50 mg) was delayed compared with injected d-amphetamine (20 mg).Citation23 However, mean (SD) total exposure (AUC0–24 hours) to d-amphetamine from LDX was 562.7 (115.5) versus 681.4 (96.69) for injected d-amphetamine.Citation23 Interestingly, while the pharmacokinetic profiles of intact LDX following oral and intranasal administration differ in that plasma LDX reaches an earlier higher peak after intranasal versus oral LDX, the pharmacokinetic profiles of d-amphetamine from LDX after intranasal and oral administration are virtually identical.Citation24 Pharmacokinetic parameters of d-amphetamine from LDX were dose-proportional over a wide range of oral doses, ranging from therapeutic doses to doses three to eight times higher than therapeutic range.Citation3 The consistent findings that administration of LDX results in an extended-release profile of d-amphetamine across multiple routes of administration and an extended dose range provides substantial support that systemic delivery of d-amphetamine is dependent on the rate-limiting conversion of LDX.
LDX was not metabolized in vitro by SGF, SIF, or trypsin, supporting our view that any conversion of LDX to d-amphetamine in the GI tract in vivo is likely to be minimal. Taken together, these findings are consistent with a model of absorption of predominantly intact LDX with subsequent enzymatic conversion to active d-amphetamine and l-lysine.
LDX was metabolized to d-amphetamine in rat and human whole blood but not in plasma. Although metabolism was also observed in rat and human liver homogenates and in human kidney homogenate, it is possible that the conversion of LDX in these tissues was related to residual blood that was not removed when these tissues, which have very rich blood supply, were excised from the animal or human donor. The lack of conversion of LDX in assays with liver microsomes and assays with freshly isolated hepatocytes provides further support that RBCs in the portal circulation, rather than liver tissue, are responsible for the presystemic conversion of LDX to d-amphetamine.
LDX was metabolized to d-amphetamine by human RBCs but not by other blood fractions, such as white blood cells or platelets. Moreover, RBCs had a very high capacity for the metabolism of LDX. The rate of metabolism was not substantially reduced until concentrations of RBCs were reduced to 10% of normal hematocrit. Lysis of the RBCs also had little impact on the rate of metabolism of LDX, and nor did EDTA (a calcium chelator that inhibits the activity of calcium-dependent enzymes).
In conclusion, LDX is absorbed as the intact prodrug and converted to inactive l-lysine and active d-amphetamine primarily in the blood by RBCs. The absorption of LDX likely occurs via a high-capacity carrier-mediated transport system involving PEPT1 in the small intestine, although the involvement of other peptide and amino acid transporters cannot be ruled out. The subsequent metabolism of LDX to d-amphetamine by RBCs also appears to be via a high-capacity system. Thus, saturation of enzymatic conversion of LDX in the blood is unlikely to occur at therapeutic doses. The dual findings of high-capacity absorption and enzymatic conversion may contribute to the consistent and reproducible pharmacokinetic profile of LDX in humans.Citation2,Citation3 That LDX is subject to enzymatic hydrolysis after absorption, but is not subject to variations in absorption related to changes in factors such as gastric pH and variations in normal GI transit timeCitation12,Citation21,Citation25 may result in a consistent rate of hydrolysis and delivery of active d-amphetamine, compared with formulation-based delivery systems of d-amphetamine. Thus, the biotransformation process, and not the dissolution of intact LDX, appears to control the rate of delivery of active d-amphetamine.
Acknowledgements
This research was supported by funding from Shire Development Inc. Writing and editorial assistance was provided by Susan Kralian, PhD, a former employee of Health Learning Systems and Micheal Pucci, PhD, an employee of Health Learning Systems. Editorial assistance in the form of proofreading, copy editing, and fact checking was provided by Health Learning Systems. Assays were performed at QPS, LLC, Newark, DE; Absorption Systems LP, Exton, PA; CellzDirect Inc, Austin, TX; and SOLVO Biotechnology, Budaörs, Hungary. These data were presented at the 49th Annual New Clinical Drug Evaluation Unit Meeting, June 29–July 2, 2009, Hollywood, FL.
Disclosure
The author is a full-time employee and stockholder of Shire Pharmaceutical Development Ltd.
References
- CowlesBJLisdexamfetamine for treatment of attention-deficit/hyperactivity disorderAnn Pharmacother200943466967619318601
- BiedermanJBoellnerSWChildressALopezFAKrishnanSZhangYLisdexamfetamine dimesylate and mixed amphetamine salts extended-release in children with ADHD: A double-blind, placebo-controlled, crossover analog classroom studyBiol Psychiatry200762997097617631866
- ErmerJHomolkaRMartinPBuckwalterMPurkayasthaJRoeschBLisdexamfetamine dimesylate: Linear dose-proportionality, low intersubject and intrasubject variability, and safety in an open-label single-dose pharmacokinetic study in healthy adult volunteersJ Clin Pharmacol2192010 [Epub ahead of print]
- Institute of Laboratory Animal ResourcesAnimal environment, housing, and managementGuide for the Care and Use of Laboratory AnimalsWashington, DCNational Academies Press19962155
- UsanskyHHHuPSinkoPJDifferential roles of P-glycoprotein, multidrug resistance-associated protein 2, and CYP3A on saquinavir oral absorption in Sprague-Dawley ratsDrug Metab Dispos200836586386918256206
- SalphatiLChildersKPanLTsutsuiKTakahashiLEvaluation of a single-pass intestinal-perfusion method in rat for the prediction of absorption in manJ Pharm Pharmacol20015371007101311480535
- BretschneiderBBrandschMNeubertRIntestinal transport of ß-lactam antibiotics: Analysis of the affinity at the H+/peptide symporter (PEPT1), the uptake into Caco-2 cell monolayers and the transepithelial fluxPharm Res199916155619950279
- ShimizuRSukegawaTTsudaYItohTQuantitative prediction of oral absorption of PEPT1 substrates based on in vitro uptake into Caco-2 cellsInt J Pharm20083541–210411018276092
- HayeshiRHilgendorfCArturssonPComparison of drug transporter gene expression and functionality in Caco-2 cells from 10 different laboratoriesEur J Pharm Sci200835538339618782614
- HanH-KRhieJKOhD-MCHO/hPEPT1 cells overexpressing the human peptide transporter (hPEPT1) as an alternative in vitro model for peptidomimetic drugsJ Pharm Sci199988334735010052994
- MacDonaldJMXuASLHiroshiKEx vivo maintenance of cells from the liver lineageLanzaWLLangerRVacantiJMethods of Tissue EngineeringSan Diego, CAAcademic Press2001
- ShojaeiAErmerJCKrishnanSLisdexamfetamine dimesylate as a treatment for ADHD: Dosage formulation and pH effectsPoster presented at: 160th Annual Meeting of the American Psychiatric AssociationMay 19–24, 2007San Diego, CA Poster NR 740.
- ShenHSmithDEBrosiusFCIIIDevelopmental expression of PEPT1 and PEPT2 in rat small intestine, colon, and kidneyPediatr Res200149678979511385139
- YamashitaSHattoriEShimadaANew methods to evaluate intestinal drug absorption mediated by oligopeptide transporter from in vitro study using Caco-2 cellsDrug Metab Pharmacokinet200217540841515618692
- ChuX-YSánchez-CastañoGPHigakiKOhD-MHsuC-PAmidonGLCorrelation between epithelial cell permeability of cephalexin and expression of intestinal oligopeptide transporterJ Pharmacol Exp Ther2001299257558211602669
- HilgendorfCAhlinGSeithelAArturssonPUngellA-LKarlssonJExpression of thirty-six drug transporter genes in human intestine, liver, kidney, and organotypic cell linesDrug Metab Dispos20073581333134017496207
- MitsuokaKKatoYKuboYTsujiAFunctional expression of stereoselective metabolism of cephalexin by exogenous transfection of oligopeptide transporter PEPT1Drug Metab Dispos200735335636217142565
- BrandschMKnütterILeibachFHThe intestinal H+/peptide symporter PEPT1: Structure-affinity relationshipsEur J Pharm Sci2004211536014706811
- DanielHMolecular and integrative physiology of intestinal peptide transportAnnu Rev Physiol20046636138414977407
- Keflex® (Package insert)Germantown, MDMiddlebrook Pharmaceuticals, Inc.2006
- KrishnanSZhangYRelative bioavailability of lisdexamfetamine 70-mg capsules in fasted and fed healthy adult volunteers and in solution: A single-dose, crossover pharmacokinetic studyJ Clin Phar-macol2008483293302
- Vyvanse® (Package insert)Wayne, PAShire US Inc.2009
- JasinskiDRKrishnanSHuman pharmacology of intravenous lisdexamfetamine dimesylate: Abuse liability in adult stimulant abusersJ Psychopharmacol200923441041818635707
- ErmerJDennisKHaffeyMPharmacokinetics of intranasal versus oral administration of lisdexamfetamine dimesylate in healthy adultsPoster presented at: American Psychiatric Association ’s 162nd annual meetingMay 16–21, 2009San Francisco, CA
- HaffeyMBBuckwalterMZhangPEffects of omeprazole on the pharmacokinetic profiles of lisdexamfetamine dimesylate and mixed amphetamine salts extended release in adultsPostgrad Med20091215111919820270