Abstract
Ziconotide is a powerful analgesic drug that has a unique mechanism of action involving potent and selective block of N-type calcium channels, which control neurotransmission at many synapses. The analgesic efficacy of ziconotide likely results from its ability to interrupt pain signaling at the level of the spinal cord. Ziconotide is a peptidic drug and has been approved for the treatment of severe chronic pain in patients only when administered by the intrathecal route. Importantly, prolonged administration of ziconotide does not lead to the development of addiction or tolerance. The current review discusses the various studies that have addressed the in vitro biochemical and electrophysiological actions of ziconotide as well as the numerous pre-clinical studies that were conducted to elucidate its antinociceptive mechanism of action in animals. In addition, this review considers the pivotal Phase 3 (and other) clinical trials that were conducted in support of ziconotide’s approval for the treatment of severe chronic pain and tries to offer some insights regarding the future discovery and development of newer analgesic drugs that would act by a similar mechanism to ziconotide but which might offer improved safety, tolerability and ease of use.
Introduction
Ziconotide, which is also known as SNX-111, is a novel non-opioid analgesic drug. It is a synthetic version of ω-conotoxin MVIIA (ω-MVIIA), which is a peptide that is found in the venom of the fish-eating marine snail, Conus magus. Ziconotide has only limited ability to cross the blood–brain barrier and so in order to achieve optimal analgesic efficacy with reduced potential for serious side-effects, it must be administered intrathecally to patients. This spinal route of administration permits ziconotide to reach its maximum local concentration in a short time, which encourages a rapid onset of analgesia. Following the successful completion of three pivotal double-blind, placebo-controlled trials, intrathecal infusion of ziconotide was recently approved by regulatory bodies worldwide as a therapeutic approach for the symptomatic management of severe chronic pain, particularly in patients who are refractory to treatment with morphine and for whom intrathecal therapy is a viable option. The “Ziconotide Intrathecal Infusion” product is marketed by Elan Pharmaceuticals as Prialt® and is intended for continuous delivery via a programmable surgically implanted variable rate infusion device such as the Medtronic SynchroMed® EL, the SynchroMed® II Infusion System, or the CADD-Micro® Ambulatory Infusion Pump. Alternatively, an external microinfusion device can be used temporarily. The use of an infusion pump allows the dose of ziconotide to be titrated incrementally according to patients’ personal needs and comfort in order to achieve an optimal balance of analgesic efficacy and side-effects.
Ziconotide’s pharmacological effects have been investigated extensively in pre-clinical in vivo and in vitro models. Briefly, intrathecal ziconotide is a powerful antinociceptive drug in several animal models of chronic pain and it appears to have a completely novel mechanism of action that involves potent and selective block of pre-synaptic neuronal N-type calcium channels in the spinal cord. In fact, it is the only selective N-type channel blocker that is currently approved for clinical use. Evidence suggests that ziconotide delivers its antinociceptive efficacy by reducing the release of pronociceptive neurotransmitters in the dorsal horn of the spinal cord, thereby inhibiting pain signal transmission. Intrathecal ziconotide’s clinical efficacy is consistent with the hypothesis that spinal N-type calcium channels are key regulators of nociceptive signaling in humans, although it is fair to say that its precise analgesic mechanism in humans remains unconfirmed at this time. There are several recent publications that are relevant to the topics in this review and they will be cited where appropriate.
Acute and chronic pain
Pain has been defined as “an unpleasant sensory and emotional experience that is associated with actual or potential tissue damage” (International Association for the Study of Pain®) and can be classified according to a variety of characteristics including its duration (acute or chronic) and intensity (mild, moderate, or severe). Acute pain is a normal experience that is usually short-lasting and serves to alert the body about ongoing tissue damage so that protective or evasive measures can be taken. Acute pain usually lessens over time as a consequence of the healing process. In contrast, chronic pain represents an abnormal experience that is long-lasting and persists in the absence of any apparent tissue damage. Chronic pain is not equivalent to long-lasting acute pain; it appears to serve no useful purpose and is often associated with diseases involving tissue inflammation (leading to chronic inflammatory pain) or damage to peripheral or central neurons (leading to chronic neuropathic pain). More complex chronic pain syndromes may exhibit signs of both inflammatory and neuropathic pain.
Pain is experienced through a complex neural network that has two anatomically defined and functionally interacting systems that control pain perception and pain modulation (CitationAlmeida et al 2004; CitationApkarian et al 2005). During normal pain sensation, components of the pain perception system are activated first and subsequently the pain modulation system may contribute inhibitory and/or facilitatory input to alter the strength and duration of the pain. During pain perception, the peripheral nerve endings of high-threshold mechanosensitive and polymodal nociceptive neurons, whose cell bodies are located in the dorsal root ganglia (DRG), are excited by noxious stimuli, leading to the generation and propagation of sodium channel-dependent action potentials along small diameter finely myelinated (Aδ fiber) or unmyelinated (C fiber) axons. The Aδ and C fibers project mainly to the superficial laminae of the dorsal horn in the spinal cord, where they make synaptic connections with secondary sensory neurons (CitationLight and Perl 1979a, Citation1979b; CitationLight et al 1979). In contrast, large diameter low-threshold mechanosensitive Aβ fibers, which encode ordinary tactile information, project mainly to the deeper laminae of the dorsal horn. When the action potentials reach the central terminals of the primary afferent neurons, calcium influx through pre-synaptic voltage-gated calcium channels triggers the release of pronociceptive neurotransmitters and neuromodulators such as substance P, calcitonin gene related peptide (CGRP), and glutamate (CitationLevine et al 1993; CitationDickenson et al 1997; CitationBennett 2000). Under conditions of chronic pain, plastic changes in the nervous system may occur, possibly leading to overactivity in the pain perception system and/or an imbalance in the inhibitory and facilitatory components of the pain modulation system. Both peripheral and central maladaptive mechanisms may contribute to the generation of sensory deficits (CitationKatz and Rothenberg 2005). Peripheral mechanisms include sensitization of Aδ and C fibers, phenotypic switching of Aβ fibers, and awakening of silent nociceptors. Central mechanisms include sensitization of secondary and tertiary sensory neurons, as well as spinal and cortical circuit reorganization.
Many medications are available to treat acute and chronic inflammatory pain, but options for treating chronic neuropathic pain are more limited. Mild to moderate acute pain often can be managed effectively by over-the-counter medications, such as acetaminophen, whereas severe acute pain requires stronger analgesics such as opioid drugs. The exact mechanism of action of acetaminophen is unknown and although it is a very safe drug with few side-effects, a recent study suggests that it may increase the serum levels of liver enzymes when taken at high doses (CitationWatkins et al 2006). The opioid drugs are very effective pain relievers and exert their analgesic effects by agonizing μ, δ, and κ opioid receptors located at spinal and supraspinal sites in the central nervous system. Unfortunately, the opioids can produce serious side-effects, are prone to addiction and promote the development of tolerance with prolonged or repeated use. Drugs that have been used to treat pain associated with inflammation include non-steroidal anti-inflammatory drugs (NSAIDs), such as ibuprofen and naproxen. These drugs are non-selective inhibitors of the two major isoforms of cyclo-oxygenase (COX), ie, constitutive COX-1 and inducible COX-2. The COX inhibitors work by decreasing the production of prostaglandins, which are endogenous agents that are known to sensitize peripheral and central sensory neurons (CitationMcMahon et al 2005). However, these non-selective drugs are associated with the development of gastric ulcers, probably as a result of COX-1 inhibition. In contrast, COX-2 selective inhibitors produce fewer gastrointestinal problems and were prescribed widely for several years, but following controversial revelations regarding potential cardiovascular risks, some COX-2 inhibitors have been withdrawn from the market and others now carry warnings about the potential dangers. Drugs that have been approved for the treatment of neuropathic pain include carbamazepine, gabapentin, pregabalin and duloxetine. In addition, several tricyclic antidepressant, antiepileptic, and antiarrhythmic drugs are commonly used off-label for the symptomatic relief of neuropathic pain. The majority of these drugs appear to act by inhibiting non-selectively the activity of neuronal voltage-gated sodium and calcium channels. However, these drugs usually require high doses, have a high incidence of non-responders and deliver suboptimal efficacy. Consequently, there are significant opportunities for the discovery and development of novel drugs for the treatment of severe and chronic pain conditions although it must be remembered that regulatory agencies will insist that drugs are very safe before granting market approval.
Voltage-gated calcium channels, neurotransmission, and pain signaling
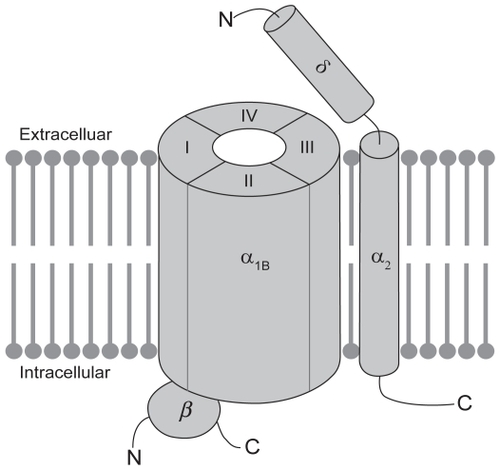
Various subtypes of voltage-activated calcium-permeable ion channels, including L-type, N-type, P/Q-type, and T-type channels have been identified throughout the mammalian nervous system. Most neuronal voltage-activated calcium channels are believed to exist as a complex of proteins (see ), comprising a large α1 subunit, which forms the pore of the channel and is responsible for defining the majority of its biophysical and pharmacological properties, as well as smaller auxiliary disulphide-linked α2δ and cytosolic β subunits, which regulate membrane insertion of the channel complex and modulate its functional properties (CitationArikkath and Campbell, 2003). So far 10 architecturally similar α1 subunits have been identified and structural elements have been identified that correlate with certain functions of the channel (CitationErtel et al 2000; CitationCatterall et al 2003). The α1 subunit is organized into four homologous domains (DI-DIV), each of which contains six membrane-spanning segments (S1–S6). Membrane depolarization is sensed by positively charged amino acids in the so-called voltage-sensors that are located in the S4 transmembrane segment of each domain. The selectivity of the channel for calcium and the process of ion permeation are governed by four critical glutamate residues, one in each of the pore loops (P-loops) that are located between the S5 and S6 segments in each domain of the α1 subunit (CitationSather and McCleskey 2003). Of relevance to the current review, the molecular target of ziconotide appears to be the N-type calcium channel, which is a high-voltage-activated channel that contains the α1B subunit (also known as CaV2.2). The α1B subunit is subject to extensive splice variation (CitationLin et al 1997, Citation1999; CitationMeadows and Benham 1999; CitationPan and Lipscombe 2000; CitationBell et al 2004; CitationCastiglioni et al 2006), which enhances not only the molecular diversity of the N-type calcium channel but also its functional diversity, since there is the potential for altered biophysical and pharmacological properties. Perhaps with the exception of the α2δ subunit, which binds gabapentin and pregabalin, the α1B subunit contains most of the pharmacologically relevant binding sites on the N-type calcium channel. Calcium permeation can be modulated by agents that directly block the pore of the channel, such as divalent cations and peptides derived from venomous species, as well as by small molecule drugs that block the channel in a use-dependent manner, as a result of preferential interactions with activated and/or inactivated states of the channel (CitationWinquist et al 2005).
Figure 1 Schematic representation of the putative structure of the voltage-gated N-type calcium channel. N-type calcium channels are made up of a large pore-forming a1B subunit in association with one or more auxiliary subunits. The a1B subunit contains most determinants of channel function, including its biophysical and pharmacological properties. The proposed membrane topology of the a1B subunit is believed to involve four homologous domains (DI-DIV), each of which contains six transmembrane segments (S1–S6; not shown). The auxiliary subunits include the disulphide-linked a2d subunit, which is anchored in the membrane by a single membrane-spanning segment and the cytosolic b subunit, which interacts with the intracellular loop connecting DI to DII in the a1B subunit.
Voltage-activated calcium channels exhibit subtype-specific cellular and subcellular distributions in the nervous system and play distinct roles in controlling neuronal physiology. For instance, pre-synaptic calcium channels play a critical role in the biochemical cascade of events that leads to the exocytotic release of neurotransmitters via fusion of synaptic vesicles with the plasma membrane (CitationSchneggenburger and Neher 2005). Immunocytochemical approaches have revealed that N-type and P/Q-type calcium channels are localized predominantly on pre-synaptic nerve terminals throughout the nervous system (CitationWestenbroek et al 1998), where they associate with and are regulated by other components of the cellular machinery involved in synaptic transmission (CitationZhong et al 1999; CitationZamponi 2003). Although both subtypes are found pre-synaptically on the terminals of primary sensory neurons in the dorsal horn, only occasionally are they co-localized on the same nerve terminal (CitationWestenbroek et al 1998). The N-type channels are evenly distributed throughout all the laminae of the dorsal horn and are in fact the predominant subtype in the superficial laminae (1 and 2), which is consistent with an involvement in Aδ and C fiber-mediated pain signaling (CitationGohil et al 1994). Furthermore, N-type channels are exclusively co-localized with substance P in presumptive C fiber terminals (CitationWestenbroek et al 1998). In contrast, the P/Q-type channels are not found in lamina 1 of the dorsal horn, although their presence in lamina 2 suggests that they may also play a role in pain signal processing.
In accordance with these distribution data, the use of subtype-selective calcium channel blockers has confirmed that synaptic transmission in the peripheral and central nervous systems is triggered mainly by calcium influx through N-type and P/Q-type channels (CitationGaur et al 1994; CitationWheeler et al 1994; CitationMintz et al 1995), although additional subtypes may also contribute but to a lesser degree (CitationGasparini et al 2001). In the spinal cord, N-type and P/Q-type calcium channels contribute to both excitatory and inhibitory synaptic transmission. Interestingly, the N-type calcium channel is subject to direct regulation by G-protein βγ subunits (CitationDe Waard et al 2005) and a component of the spinal analgesic action of opioid drugs likely involves reduced release of pronociceptive neurotransmitters in the dorsal horn as a consequence of μ-opioid receptor activation and G-protein-dependent inhibition of N-type channels (CitationNorth 1986). The importance of both N-type and P/Q-type calcium channels in the transmission and modulation of nociceptive signaling at the level of the spinal cord is further supported by in vivo pharmacological experiments with subtype-selective blockers, which will be discussed in more depth later (CitationChaplan et al 1994; CitationMalmberg and Yaksh 1994; CitationDiaz and Dickenson 1997; CitationMatthews and Dickenson 2001). In addition, the reader is directed to several recent reviews that have discussed the relative contributions of N-type and other calcium channel subtypes to pain signaling (CitationMcGivern and McDonough 2004; CitationMcGivern 2006; CitationYaksh 2006).
Ziconotide: structural considerations and in vitro biochemical and electrophysiological studies
The ω-conotoxins, such as ω-GVIA, ω-MVIIA, ω-MVIIC, and ω-CVID, constitute a structurally related group of polypeptidic molecules that are found naturally in the venom of certain species of marine snail. In general, the ω-conotoxins bind with high affinity to voltage-gated calcium channels and potently block calcium flux. Despite structural conservation not only among the various ω-conotoxins but also among their binding sites on voltage-activated calcium channels, individual peptides actually exhibit distinguishing specificities for different channels.
ω-MVIIA contains 25 amino acids, 6 of which are cysteine residues that are linked in pairs by 3 disulphide bonds (see ). The disulphide bond linkage pattern is a characteristic feature of ω-conotoxins and serves to ensure correct folding of the peptide and stabilization of its structure in a compact, well-defined, native conformation (CitationChung et al 1995). Disruption of any one of the disulphide bridges greatly destabilizes the structure of ω-MVIIA and renders the remaining disulphide bonds more prone to reduction. Interestingly, the naturally occurring ω-MVIIA is synthesized by Conus magus as a precursor peptide that includes a C-terminally located glycine residue that becomes post-translationally converted to an amide group. This glycine appears to enhance the folding efficiency of the peptide in vivo by promoting molecular interactions that stabilize the native conformation with respect to other disulphide-bonded forms (CitationPrice-Carter et al 1996; CitationPrice-Carter et al 1998). The high resolution three dimensional structure of w-MVIIA has been determined by nuclear magnetic resonance (NMR) spectroscopy. The molecule displays a short triple-stranded anti-parallel β-sheet structure containing four loops, as illustrated in (CitationBasus et al 1995; CitationKohno et al 1995).
Figure 2 Putative structure of ziconotide.

As already mentioned, the molecular target of ziconotide (ω-MVIIA) appears to be the N-type calcium channel. In support of this hypothesis, radioligand binding experiments have demonstrated that ziconotide binds rapidly, reversibly, and with high affinity (see ) to N-type calcium channels in membrane and synaptosome preparations of rat brain (CitationStoehr and Dooley 1993; CitationKristipati et al, 1994). Ziconotide displays a high degree of binding and functional selectivity (>1000-fold) for the N-type calcium channel (CitationOlivera et al 1987; CitationNielsen et al 1999b; CitationLewis et al 2000), whereas in contrast ω-MVIIC is more selective for the P/Q-type calcium channel (CitationHillyard et al 1992). It is believed that the differential potencies of the toxins are determined largely by the relative positions of amino acid side chains on the exposed surface of the toxin peptides (CitationNielsen et al 1996). In the case of ω-MVIIA, it is the non-cysteine amino acids in the loops that determine its binding affinity and calcium-channel-blocking activity. In particular, the second loop located between cysteine-8 and cysteine-15 appears to be most important in directing the selectivity of ω-MVIIA towards N-type channels and away from P/Q-type channels, although the fourth loop also contributes to a lesser degree (CitationNielsen et al 1999b). Alanine substitution experiments have revealed that tyrosine-13 in ω-MVIIA is a critical determinant of binding to N-type calcium channels (CitationKim et al 1995). As one would expect, correct folding of the ω-MVIIA peptide is necessary to ensure appropriate positioning of tyrosine-13 and permit toxin binding to the N-type calcium channel (CitationKohno et al 1995). Furthermore, altering the chirality of tyrosine-13 appears to affect the positions of key residues in the second loop of ω-MVIIA, leading to a reduction in its ability to recognize the N-type calcium channel in a radioligand binding assay (CitationNielsen et al 1999a). In addition, individual amino acid substitutions and chimeric-toxin approaches have revealed the importance of other amino acids such as lysine-2 and arginine-21 as well as those residues in positions 9 through 12 in determining the binding of ω-MVIIA to the N-type calcium channel (CitationNadasdi et al 1995; CitationSato et al 1997, Citation2000).
Table 1 Summary of in vitro studies with ziconotide
Ziconotide appears to bind to the pore region of the α1B subunit and may interfere with calcium permeation by physically occluding the channel. Electrophysiological experiments have demonstrated conclusively that ziconotide inhibits N-type calcium currents in native cells as well as in heterologous expression systems (see ). Most native cells express a variety of different calcium channels and as a result, ziconotide only partially reduces high-voltage-activated calcium currents in differentiated human neuroblastoma IMR32 cells (CitationFox 1995), rat superior cervical ganglion neurons (CitationSanger et al 2000), and rat hippocampal neurons (CitationWen et al 2005). Ziconotide also reduces calcium currents that result from expression of the α1B subunit in HEK cells (CitationBleakman et al 1995; CitationSanger et al 2000), tsa-201 cells (CitationFeng et al 2003), and Xenopus laevis oocytes (CitationNewcomb et al 1995; CitationLewis et al 2000; CitationLuchian 2001). Mechanistically, ziconotide exhibits little or no use-dependence in its inhibitory effect, which probably reflects its equal binding affinity for resting, open, and inactivated states of the channel (CitationFeng et al 2003). Interestingly, the inhibitory potency of ziconotide on α1B-mediated calcium currents may vary depending on whether or not α2d and/or β auxiliary subunits are co-expressed (CitationLewis et al 2000; CitationLuchian 2001; CitationMould et al 2004).
Chimeric-calcium channel α1 subunit approaches along with amino acid substitution experiments have shed light on which regions of the large α1B subunit may define the binding site and determine the characteristics of block by ziconotide. The binding site for ziconotide appears to be overlapping with that of ω-GVIA, which is positioned close to the P-loop of DIII (CitationEllinor et al 1994). Notably, the α1B subunit contains an EF hand-like motif that is located close to the P-loop in DIII. This EF hand-like motif may serve to bind calcium and facilitate its permeation through the pore of the channel. In addition, this motif may also impact ω-conotoxin binding. Indeed, electrophysiological experiments have revealed that mutations at glycine-1326 and glutamate-1332 affect not only calcium permeation but also the channel blocking characteristics of ω-MVIIA. In particular, glycine-1326 appears to restrict access of the toxin to its binding site, thereby decreasing the rate of onset of block and enhancing the reversibility of block (CitationFeng et al 2001). Interestingly, calcium channel block by ziconotide and ω-GVIA are affected differently by amino acid mutations downstream of the EF hand-like motif, supporting the idea that their respective binding sites are overlapping but not identical (CitationFeng et al 2003).
Since it blocks N-type calcium channels so potently, ziconotide has proven to be an effective inhibitor of neurotransmitter release at multiple synapses in the nervous system (see ). In fact due to its subtype-specificity, ziconotide is often used as a tool to define the contribution of N-type calcium channels to synaptic transmission at central and peripheral synapses. Thus ziconotide inhibits norepinephrine release from hippocampal (CitationNewcomb et al 1995; CitationWang et al 1998) and peripheral sympathetic efferent neurons (CitationWang et al 1998). Consistent with the co-localization of N-type calcium channels and substance P in the central nerve terminals of primary afferent neurons (CitationWestenbroek et al 1998), ziconotide potently inhibits the depolarization-evoked release of substance P from spinal cord slices (CitationSmith et al 2002). This result implicates N-type calcium channels in the central processing of pain signals and suggests that this mechanism may contribute to the antinociceptive efficacy of ziconotide. Due to the predominant role of P-type calcium channels in controlling neurotransmitter release at the neuromuscular junction (CitationLlinas et al 1992), ziconotide does not inhibit most nerve-evoked muscle contractions (CitationBowersox et al 1995).
Ziconotide: pre-clinical in vivo studies
Ziconotide has been described as a potent and long-lasting antinociceptive drug when administered by the intrathecal route. Experimental evidence of ziconotide’s antinociceptive properties was first obtained in the early 1990s and since then extensive studies have been conducted to characterize its effects in multiple animal models of pain (see and for details on efficacy and dosing). These studies have revealed that ziconotide is more potent than morphine and is particularly efficacious in models of persistent pain (duration measured in minutes to hours) and chronic pain (duration measured in hours to days). In comparison, it tends to be less effective in tests of acute pain (duration measured in minutes). It is also important to note that ziconotide can be efficacious under a variety of intrathecal dosing regimens, including single bolus injection and acute or chronic continuous infusion. Despite its potent efficacy, the therapeutic index of spinal ziconotide tends to be low and often its antinociceptive effects in animals are accompanied by motor deficits at higher doses. Although ziconotide does not easily cross the blood brain barrier in normal animals, it may cause hypotension if it enters the systemic circulation (CitationBowersox et al 1992; CitationWright et al 2000; CitationTakahara et al 2002). This effect on blood pressure appears to be mediated at least partially by inhibition of sympathetic neurotransmission, probably as a result of N-type calcium channel blockade in sympathetic nerve terminals (CitationWang et al 1998).
Table 2 Summary of experiments conducted with ziconotide in rat behavioral models of acute pain (all dosing was intrathecal)
Table 3 Summary of experiments conducted with ziconotide in rat behavioral models of chronic pain (all dosing was intrathecal)
Formalin is a chemical irritant that when injected subcutaneously into the rat paw evokes a complex behavioral response consisting of an early (acute) phase and a late (persistent) phase of paw flinching and licking. This biphasic behavioral response appears to be correlated temporally with formalin-evoked electrical activity in both primary afferents and secondary sensory neurons in the dorsal horn of the spinal cord (CitationDickenson and Sullivan 1987b). Electrophysiological experiments have revealed an initial period of high intensity C fiber firing, which is followed by a prolonged period of lower intensity C fiber discharging accompanied by facilitation (“wind up”) of dorsal horn neuronal responses. The early phase of neuronal firing appears to be due to the direct excitation of peripheral C fiber nerve endings by formalin (CitationDickenson and Sullivan 1987a), whereas the late phase, although dependent on the early phase for its induction, appears to require additionally the release of pronociceptive neurotransmitters and activation of post-synaptic N-methyl-D-aspartate (NMDA) subtype of glutamate receptors in the spinal cord (CitationHaley et al 1990).
Consistent with their predominant role in controlling release of pronociceptive neurotransmitters in the dorsal horn, N-type calcium channels appear to be involved in defining both the electrophysiological and behavioral responses to peripheral injection of formalin (CitationMalmberg and Yaksh 1994; CitationDiaz and Dickenson 1997). In common with other ω-conotoxins such as ω-GVIA, ziconotide suppresses the formalin-induced hyperexcitability of dorsal horn neurons (CitationDiaz and Dickenson 1997). These electrophysiological effects are consistent with the ability of the ω-conotoxins to exert antinociceptive effects in the formalin model (see ), as assessed during both phases of the behavioral response (CitationMalmberg and Yaksh 1994, Citation1995; CitationBowersox et al 1996; CitationWang et al 2000a; CitationChen et al 2005). The antinociceptive efficacy of ziconotide can be observed under a variety of dosing regimens including administration prior to the injection of 5% formalin, either as single bolus injection (eg, 10 minutes before) or following acute (up to 2 days before) or chronic (up to 7 days before) continuous infusion. In general ziconotide is more efficacious when administered prophylactically, although it does have activity in the late phase of the behavioral response when administered 9 minutes after the injection of formalin. Overall, ziconotide appears to be more effective at inhibiting the late phase rather than the early phase of the behavioral response to formalin. Perhaps consistent with its lesser effects in the early phase of the formalin test, ziconotide is not very effective at increasing pain thresholds in more straightforward models of acute pain (see ), such as the hot plate, radiant heat, and tail immersion tests in rats (CitationMalmberg and Yaksh 1994, Citation1995; CitationWang et al 2000a, Citation2000b; CitationScott et al 2002).
The antinociceptive efficacy of ziconotide was also studied in animal models of inflammatory pain and nerve injury-evoked pain. Typically, animal models of inflammatory pain employ biochemical agents to sensitize or activate primary afferent neurons, leading to spontaneous pain as well as hyperresponsiveness to noxious (hyperalgesia) and innocuous (allodynia) stimuli. On the other hand, animal models of nerve injury-evoked pain usually involve constriction, ligation or partial transection of a peripheral or spinal nerve, leading to the development of behavioral symptoms that mimic some of the sensory abnormalities that are reported by neuropathic pain patients. These sensory phenomena include hyperalgesia and allodynia, both of which can be assessed behaviorally and interpreted as symptoms of neuropathic-like pain. Ziconotide exerts potent antihyperalgesic and antiallodynic effects in models of acute and chronic inflammatory pain (see ) and in models of neuropathic pain (see ). In a model of acute inflammatory pain, intrathecal ziconotide is able to prevent (infusion initiated 1 hour before biochemical challenge) or reverse (continuous infusion initiated 4 hours after biochemical challenge) kaolin and carrageenan-induced secondary heat hyperalgesia in the knee joint (CitationSluka 1998). In a rat incisional model of post-operative pain, a single bolus injection of ziconotide is able to reverse established heat hyperalgesia and mechanical allodynia (CitationWang et al 2000b) and in a model of chronic inflammatory pain, an intrathecal bolus of ziconotide can reverse Freund’s complete adjuvant-induced mechanical hyperalgesia in the hind paw (CitationSmith et al 2002). In models of neuropathic pain, including several that involve surgically-induced damage to the sciatic nerve or spinal nerve roots, ziconotide is effective at reversing established hyperalgesia and allodynia, either when administered by bolus injection or by continuous spinal infusion (CitationChaplan et al 1994, CitationBowersox et al 1996; CitationYamamoto and Sakashita 1998; CitationScott et al 2002; CitationUrban et al 2005). Interestingly, ziconotide can also be efficacious when administered outside the spinal cord. For instance, significant antihyperalgesic and antiallodynic effects have been observed following local application of ziconotide (ID50 <1.0 μg) to the site of sciatic nerve injury in rats (CitationXiao and Bennett 1995). Additionally, antiallodynic effects can be produced following microinjection of ziconotide (ID50 2.8 pmol) into the rostral ventromedial medulla in rats with spinal nerve ligation (CitationUrban et al 2005), presumably by inhibiting neurotransmitter-dependent activation of pain facilitatory neurons that, by way of their descending projections to the dorsal horn of the spinal cord, contribute to the maintenance of hypersensitivity in neuropathic pain states (CitationOssipov et al 2000; CitationPorreca et al 2001).
The anti-nociceptive efficacy of ziconotide in animal models of pain is obviously complex. Nevertheless, the results described above are particularly enlightening with respect to both the mechanism of action of ziconotide and the role of N-type calcium channels in controlling pain signal transmission. The differential efficacy of ziconotide in models of acute versus persistent pain may reflect changes in the expression level of calcium channel subunits under conditions of neuronal hyperexcitability. Both the α1B and α2δ subunits appear to be up-regulated in response to tissue inflammation or nerve injury (CitationLuo et al 2001; CitationNewton et al 2001; CitationAbe et al 2002; CitationCizkova et al 2002; CitationYokoyama et al 2003). Consequently, N-type calcium channels might become functionally more important in hypersensitive states and their role in pain transmission could be greater under conditions of ongoing chronic pain rather than under conditions of acute pain. Alternatively, it is known that the pharmacology of the N-type calcium channel can change depending on its subunit composition, at least in heterologous expression systems (CitationLewis et al 2000; CitationLuchian 2001; CitationMould et al 2004). If this mechanism were to be replicated in native neuronal N-type calcium channels, then tissue inflammation- or nerve injury-induced alterations in the subunit composition of the channels could lead to increased sensitivity to ziconotide’s blocking actions.
The antinociceptive profile of ziconotide in models of acute, persistent and chronic pain is worthy of comparison with that of P/Q-type calcium channel blockers, such as ω-agatoxin-IVA. The potency and efficacy of ziconotide suggests that although N-type calcium channels in the pain pathway contribute to the perception of acute pain (eg, first phase of the formalin test as well as the hot plate test), they play a more significant role in the development and maintenance of multiple hypersensitive painful states (eg, second phase of the formalin test as well as inflammatory and neuropathic pain models). In contrast, the antinociceptive profile of ω-agatoxin-IVA suggests that P/Q-type calcium channels contribute only to inflammation-associated painful conditions (CitationMalmberg and Yaksh 1994; CitationDiaz and Dickenson 1997; CitationNebe et al 1997, Citation1999). They appear to be involved neither in the perception of acute pain nor in the development and maintenance of nerve injury-associated hypersensitive painful states in animal models (CitationChaplan et al 1994; CitationYamamoto and Sakashita 1998). The antinociceptive profiles of subtype-selective calcium channel blockers may reflect the involvement of distinct populations of primary or secondary sensory neurons in the transmission and processing of different types of pain signals. This is a plausible contributory factor because although both N-type and P/Q-type calcium channels are found in the dorsal horn of the spinal cord, they actually display complementary neuronal distributions, with the same nerve terminal rarely containing both subtypes.
In summary, potent antinociceptive effects of ziconotide have been observed in several animal models of pain under a variety of dosing regimens, including acute and chronic administration. The demonstration that ziconotide retains its potent antinociceptive efficacy during chronic administration provides convincing evidence that, unlike the opioids, this drug is not associated with the development of tolerance. This observation has important implications for the long-term therapeutic use of ziconotide in the treatment of pain in patients. The experimental evidence also suggests that N-type calcium channels expressed at multiple sites along the pain pathway are functionally important in the transmission of pain signals. These locations may include the peripheral site of nerve injury, where the N-type calcium channels appear to be involved in the generation of persistent spontaneous neuronal activity under conditions of nerve injury. In addition, N-type calcium channels are important for the transmission of incoming nociceptive signals to secondary sensory neurons in the spinal cord whereas those in the rostral ventromedial medulla may be involved in the activation of descending pain facilitatory systems that have been shown to contribute to the maintenance of neuropathic pain states.
Ziconotide: clinical studies
The development path to regulatory approval of intrathecal ziconotide involved three large randomized, double-blind, placebo-controlled Phase 3 clinical trials that established the safety and analgesic efficacy of this drug in more than 600 patients (see ). All of the patients in these trials were suffering from severe chronic pain of malignant and/or non-malignant origins (CitationStaats et al 2004; CitationRauck et al 2006; CitationWallace et al 2006) and in order for them to be accepted into the trials, it was necessary that their pain was inadequately controlled by other analgesic drugs, including opioids. These clinical trials evaluated the analgesic efficacy of ziconotide under chronic dosing paradigms (up to 3 weeks) in order to determine the potential for this drug to develop tolerance. In addition to these pivotal trials, a smaller placebo-controlled clinical trial demonstrated analgesic efficacy of ziconotide in a post-surgical setting (see ) and a number of open-label studies showed that ziconotide can be an effective therapy in the treatment of neuropathic pain (see ). There are several previously published reviews available that discuss the clinical experiences with ziconotide (CitationJain 2000; CitationHeading 2001; CitationDoggrell 2004; CitationMiljanich 2004; CitationLyseng-Williamson and Perry 2006; CitationStaats 2006).
Table 4 Summary of clinical trials conducted with ziconotide
The first pivotal trial with ziconotide involved patients with chronic pain due to cancer or AIDS (CitationStaats et al 2004). In this trial, 68 patients received ziconotide by continuous intrathecal infusion for an initial 5- to 6-day period, followed by a maintenance period for those who responded to treatment. The starting dose of ziconotide was low (≤0.1 or 0.4 μg/h), although it could be increased frequently (at 12- or 24-hour intervals) either until satisfactory pain relief was achieved, the maximum dose of 2.4 μg/h was reached or adverse events were reported. Moderate to complete pain relief was achieved for most patients during the initial phase, with an average 53% reduction in pain scores, as estimated on a visual analog scale of pain intensity (VASPI). Importantly there was no loss of analgesic efficacy during the maintenance phase, suggesting that humans do not develop tolerance to ziconotide. Adverse events were observed more frequently in the ziconotide group than in the placebo group and in general, their occurrence was reduced either by initiating the drug infusion at lower doses or by using smaller or less frequent dose increments.
The second pivotal trial evaluated the safety and efficacy of ziconotide in patients with chronic non-malignant (mostly neuropathic) pain (CitationWallace et al 2006). In this trial, 169 patients received ziconotide beginning at a low dose (≤0.1 or 0.4 μg/h) and during the subsequent several days, the dose could be doubled at 24-hour intervals either until satisfactory pain relief was achieved, the maximum dose was reached (2.4 or 7.0 μg/h, depending on starting dose) or adverse events were experienced. As in the first trial, patients receiving ziconotide experienced moderate to complete pain relief, although the average reduction in VASPI scores was lower in this second trial. Responders continued to receive drug during the maintenance period, during which the efficacy of ziconotide was maintained. Again, adverse events could be resolved by reducing the dose or frequency of titration or by discontinuation of the drug.
The third pivotal trial evaluated the safety and efficacy of ziconotide in 220 patients with intractable severe chronic pain, the majority of which was neuropathic (CitationRauck et al 2006). This trial was conducted in response to regulatory concerns that were raised about the high incidence and severity of the adverse events as well as the high rate of patient drop-out during the first two trials. Therefore the design of this third trial differed from the earlier trials in several important aspects: (1) a slower titration schedule was employed (increments of 0.1 μg/h no more frequently than every 24 hours); (2) a lower maximum dose was allowed (0.9 μg/h); and (3) the trial length was longer (3 weeks). Significant pain relief was achieved in the majority of patients receiving ziconotide and the average improvement in VASPI scores was estimated to be 15%. The magnitude of this reduction was smaller than what was reported in the previous trials and this seems to be consistent with the lower doses used. In addition, the patients in the ziconotide group consumed 24% less opioid drug compared to the placebo group. Adverse events were experienced at low therapeutic doses of ziconotide in this trial, but most of these were rated as mild or moderate and were slow to develop after the drug infusion was initiated.
A relatively small clinical trial evaluated the ability of ziconotide to reduce post-operative pain arising from major surgery (CitationAtanassoff et al 2000). Low dose (0.7 μg/h) or high dose (7.0 μg/h) ziconotide was given to patients undergoing total abdominal hysterectomy, radical prostatectomy, or total hip replacement. In this trial, ziconotide infusion was initiated before surgical incision and was continued for up to 72 hours post-operatively. For those patients who received ziconotide, significant pain relief was experienced and morphine consumption was reduced. Side-effects, such as dizziness, blurred vision, nystagmus, and sedation, appeared to be more severe in the high-dose drug group and they resolved after discontinuation of the drug.
A number of open-label clinical trials have also been conducted with ziconotide. In one of the earliest reported trials, ziconotide was administered to a patient who had been suffering for more than 20 years from intractable deafferentation pain as a result of brachial plexus avulsion and limb amputation (CitationBrose et al 1997). This patient was given ziconotide by continuous, constant rate, intrathecal infusion and complete pain relief was achieved, even after the dose was lowered to 2 ng/kg hourly to alleviate the patient’s side-effects of dizziness, blurred vision, and nystagmus. Another trial evaluated the analgesic efficacy, safety, and pharmacokinetic properties of intrathecal ziconotide in patients with chronic neuropathic pain (CitationWermeling et al 2003). Ziconotide was infused over a 1-hour period at doses of 1, 5, 7.5, or 10 μg. The analgesic efficacy of ziconotide was dose-related and was correlated with drug exposure in the cerebrospinal fluid. Moreover, efficacy developed rapidly (within 1 hour of initiation of drug infusion) and lasted for up to 48 hours. Most of the adverse events in this study were mild to moderate and serious events were only reported in the highest dose group. Another open-label trial evaluated the ability of ziconotide to relieve the symptoms of long-standing neuropathic pain of various origins in 3 patients (CitationWermeling and Berger 2006). Single dose administration (in 2 patients) or continuous infusion (in 1 patient) of ziconotide alleviated the pain considerably. The patients who received the single dose reported only mild side-effects, whereas the patient who received continuous infusion experienced more severe neurological side-effects. Interestingly, this patient could feel the imminent side-effects and was able to avoid them by reducing the rate of drug infusion. Regarding the pharmacokinetic profile of ziconotide in the cerebrospinal fluid of humans, the measured t½ is around 4½ hours, which is similar to the slow component of drug elimination that has been observed in rats and monkeys (CitationBowersox et al 1997). Ziconotide was not detectable in the plasma of the majority of patients, supporting the idea that this drug cannot easily cross the blood–brain barrier.
In summary, intrathecal ziconotide is a novel, potent and long-lasting analgesic therapy that can be used for the symptomatic relief of severe chronic pain of malignant and non-malignant origins. It is also effective for the prevention of surgically-induced pain. Since ziconotide is administered intrathecally to patients, it is tempting to speculate that its therapeutic mechanism of action primarily involves block of pre-synaptic N-type calcium channels in the spinal cord, leading to a reduction in the release of pronociceptive neurotransmitters from primary afferent nerve terminals and reduced synaptic excitation of secondary sensory neurons in the dorsal horn. Importantly, ziconotide is non-addictive and does not appear to induce the development of tolerance. Therefore it represents an analgesic therapy that is suitable for long-term use and in at least 1 case, a patient continued taking the drug for more than 7 years. Despite the use of an infusion pump to deliver drug directly into the intrathecal space, it is very difficult to predict or control the local concentration of ziconotide that can access the N-type calcium channels located on the central terminals of the primary sensory neurons in the dorsal horn. Therefore the optimal dose of drug needs to be determined empirically. Nevertheless, the therapeutic index of ziconotide tends to be low and adverse events (primarily psychiatric and neurological) may be experienced, particularly if the drug is infused rapidly, a high dose is given or the dose is escalated too frequently. However, the good news is that when adverse events are experienced, they usually resolve when the dose is lowered or the frequency of dose escalation is reduced. In order to minimize the incidence and severity of adverse events in new patients, the manufacturer recommends a “start low, go slow” approach to the use of ziconotide. The current recommendation is to initiate the infusion at a low dose of ≤0.1 μg/h and to titrate the dose upwards no more frequently than 2–3 times/week. This approach has been shown to produce fewer serious adverse events and to reduce the incidence of drug discontinuation by patients.
Future prospects and concluding remarks
Ziconotide represents a great achievement for current pain therapy but despite its potent analgesic efficacy there remains significant opportunity for improvement. The opportunity derives primarily from the peptidic nature of the drug and its requirement for intrathecal administration in order to yield analgesic efficacy with reduced potential for systemic and central nervous system side-effects. Consequently, drug discovery researchers are considering various approaches to identify and develop novel orally active, N-type calcium channel-selective blockers that have the potential to be superior to ziconotide.
High analgesic efficacy and improved safety and tolerability, relative to both ziconotide and the opioids, are essential properties of a next generation N-type calcium channel blocking drug. This goal could be achieved by exploring approaches to identify compounds that display greater selectivity for sensory neuron-specific splice variants of the N-type calcium channel and/or that possess a use-dependent mechanism of calcium channel block. Regarding the former approach, multiple kinetically distinct splice variants of the calcium channel α1B subunit are known to exist, some of which appear to be exclusive to peripheral neurons (CitationLin et al 1997, Citation1999). In particular, a dorsal root ganglion-specific variant was recently identified in rat (CitationBell et al 2004; CitationCastiglioni et al 2006) and this discovery offers hope that human N-type calcium channels might also exhibit sensory neuron-specific splice variants that could be targeted selectively in the search for safer and more efficacious pain therapeutics. Regarding use-dependent N-type calcium channel blockers, one idea is to identify compounds that bind preferentially to open and/or inactivated states of the channel (CitationWinquist et al 2005). If successful, this approach is likely to lead to the identification of molecules that might inhibit calcium influx more effectively during high-frequency neuronal firing, which occurs in hypersensitive pain states, and less effectively during low-frequency neuronal firing. The hope is that novel molecules displaying a use-dependent mechanism of action will offer both high analgesic efficacy and an improved therapeutic index relative to ziconotide. Neuromed Pharmaceuticals is pioneering efforts in this arena and recently they have partnered with Merck & Co. to develop NMED-160, an orally-available, use-dependent blocker of N-type calcium channels that is in Phase 2 clinical trials for a variety of pain conditions. In pre-clinical testing, this molecule displayed a broad efficacy profile in animal models of neuropathic and inflammatory pain and also had a good safety profile (CitationSnutch et al 2003; CitationSnutch 2004). However, it still remains to be shown that this drug is analgesic in patients with severe chronic pain.
Improved ease of administration is also a desirable property of a novel drug that could negate the requirement for intrathecal therapy that currently hinders widespread testing and use of ziconotide. Indeed, if a novel N-type calcium channel blocker could be delivered systemically then this would not only enable easier delivery to existing patients but could also increase the size of the patient population that stands to benefit from analgesia by this mechanism. An additional benefit of systemic delivery would be to reduce the risk of infection that is associated with a surgically implanted drug delivery device. Although it is theoretically possible to identify peptidic molecules that can cross the blood brain barrier, eg, the ziconotide analog SNX-194 (CitationNewcomb et al 2000) the development hurdles would be difficult to overcome due to the access such a molecule would have to N-type calcium channels throughout the entire nervous system, including the sympathetic neurons that are involved in the control of blood pressure. In addition, due to their widespread distribution in the endocrine system (CitationSher et al 2003; CitationOlsen et al 2005; CitationTakahashi et al 2005), it must be appreciated that drugs targeting N-type calcium channels could have myriad effects on multiple organs that rely on these channels to carry out their normal physiological functions. Therefore based on current knowledge, a structurally novel, orally active small molecule with a use-dependent mechanism of action is considered to be the most desirable target profile for a next generation N-type calcium channel blocking drug for use in the treatment of severe pain.
Acknowledgments
The author is grateful to Barton Manning, Amgen Inc., and George Miljanich, Airmid LLC for useful discussion and constructive feedback on this manuscript.
References
- AbeMKuriharaTHanW2002Changes in expression of voltage-dependent ion channel subunits in dorsal root ganglia of rats with radicular injury and painSpine27151724 discussion 2512131710
- AlmeidaTFRoizenblattSTufikS2004Afferent pain pathways:a neuroanatomical reviewBrain Res1000405615053950
- ApkarianAVBushnellMCTreedeRD2005Human brain mechanisms of pain perception and regulation in health and diseaseEur J Pain94638415979027
- ArikkathJCampbellKP2003Auxiliary subunits:essential components of the voltage-gated calcium channel complexCurr Opin Neurobiol1329830712850214
- AtanassoffPGHartmannsgruberMWThrasherJ2000Ziconotide, a new N-type calcium channel blocker, administered intrathecally for acute postoperative painReg Anesth Pain Med25274810834782
- BasusVJNadasdiLRamachandranJ1995Solution structure of omega-conotoxin MVIIA using 2D NMR spectroscopyFEBS Lett37016397656969
- BellTJThalerCCastiglioniAJ2004Cell-specific alternative splicing increases calcium channel current density in the pain pathwayNeuron411273814715140
- BennettGJ2000Update on the neurophysiology of pain transmission and modulation:focus on the NMDA-receptorJ Pain Symptom Manage19S2610687331
- BleakmanDBowmanDBathCP1995Characteristics of a human N-type calcium channel expressed in HEK293 cellsNeuropharmacology34753658532142
- BowersoxSSGadboisTSinghT1996Selective N-type neuronal voltage-sensitive calcium channel blocker, SNX-111, produces spinal antinociception in rat models of acute, persistent and neuropathic painJ Pharmacol Exp Ther279124398968347
- BowersoxSSMandemaJTarczy-HornochK1997Pharmacokinetics of SNX-111, a selective N-type calcium channel blocker, in rats and cynomolgus monkeysDrug Metab Dispos25379839172958
- BowersoxSSMiljanichGPSugiuraY1995Differential blockade of voltage-sensitive calcium channels at the mouse neuromuscular junction by novel omega-conopeptides and omega-agatoxin-IVAJ Pharmacol Exp Ther273248567714772
- BowersoxSSSinghTNadasdiL1992Cardiovascular effects of omega-conopeptides in conscious rats:mechanisms of actionJ Cardiovasc Pharmacol20756641280738
- BroseWGGutloveDPLutherRR1997Use of intrathecal SNX-111, a novel, N-type, voltage-sensitive, calcium channel blocker, in the management of intractable brachial plexus avulsion painClin J Pain1325699303259
- CastiglioniAJRaingoJLipscombeD2006Alternative splicing in the C-terminus of CaV2.2 controls expression and gating of N-type calcium channelsJ Physiol5761193416857708
- CatterallWAStriessnigJSnutchTP2003International Union of Pharmacology. XL. Compendium of voltage-gated ion channels:calcium channelsPharmacol Rev555798114657414
- ChaplanSRPogrelJWYakshTL1994Role of voltage-dependent calcium channel subtypes in experimental tactile allodyniaJ Pharmacol Exp Ther2691117238014856
- ChenJQZhangYQDaiJ2005Antinociceptive effects of intrathecally administered huwentoxin-I, a selective N-type calcium channel blocker, in the formalin test in conscious ratsToxicon45152015581678
- ChungDGaurSBellJR1995Determination of disulfide bridge pattern in omega-conopeptidesInt J Pept Protein Res4632058537186
- CizkovaDMarsalaJLukacovaN2002Localization of N-type Ca2+ channels in the rat spinal cord following chronic constrictive nerve injuryExp Brain Res1474566312444477
- De WaardMHeringJWeissN2005How do G proteins directly control neuronal Ca2+ channel function?Trends Pharmacol Sci264273616009433
- DiazADickensonAH1997Blockade of spinal N- and P-type, but not L-type, calcium channels inhibits the excitability of rat dorsal horn neurones produced by subcutaneous formalin inflammationPain69931009060018
- DickensonAHChapmanVGreenGM1997The pharmacology of excitatory and inhibitory amino acid-mediated events in the transmission and modulation of pain in the spinal cordGen Pharmacol2863389184794
- DickensonAHSullivanAF1987aSubcutaneous formalin-induced activity of dorsal horn neurones in the rat:differential response to an intrathecal opiate administered pre or post formalinPain30349603670880
- DickensonAHSullivanAF1987bPeripheral origins and central modulation of subcutaneous formalin-induced activity of rat dorsal horn neuronesNeurosci Lett83207113441298
- DoggrellSA2004Intrathecal ziconotide for refractory painExpert Opin Investig Drugs138757
- EllinorPTZhangJFHorneWA1994Structural determinants of the blockade of N-type calcium channels by a peptide neurotoxinNature37227257969473
- ErtelEACampbellKPHarpoldMM2000Nomenclature of voltage-gated calcium channelsNeuron25533510774722
- FengZPDoeringCJWinkfeinRJ2003Determinants of inhibition of transiently expressed voltage-gated calcium channels by omega-conotoxins GVIA and MVIIAJ Biol Chem27820171812654924
- FengZPHamidJDoeringC2001Residue Gly1326 of the N-type calcium channel alpha 1B subunit controls reversibility of omega-conotoxin GVIA and MVIIA blockJ Biol Chem276157283511279062
- FoxJA1995Irreversible and reversible blockade of IMR32 calcium channel currents by synthetic MVIIA and iodinated MVIIC omega-conopeptidesPflugers Arch42987357603842
- GaspariniSKasyanovAMPietrobonD2001Presynaptic R-type calcium channels contribute to fast excitatory synaptic transmission in the rat hippocampusJ Neurosci2187152111698583
- GaurSNewcombRRivnayB1994Calcium channel antagonist peptides define several components of transmitter release in the hippocampusNeuropharmacology33121197862257
- GohilKBellJRRamachandranJ1994Neuroanatomical distribution of receptors for a novel voltage-sensitive calcium-channel antagonist, SNX-230 (omega-conopeptide MVIIC)Brain Res653258667982059
- HaleyJESullivanAFDickensonAH1990Evidence for spinal N-methyl-D-aspartate receptor involvement in prolonged chemical nociception in the ratBrain Res518218261975214
- HeadingCE2001Ziconotide (Elan Pharmaceuticals)IDrugs43395016025393
- HillyardDRMonjeVDMintzIM1992A new Conus peptide ligand for mammalian presynaptic Ca2+ channelsNeuron969771352986
- JainKK2000An evaluation of intrathecal ziconotide for the treatment of chronic painExpert Opin Investig Drugs9240310
- KatzWARothenbergR2005Section 3:The nature of pain:pathophysiologyJ Clin Rheumatol11S11516357723
- KimJITakahashiMOhtakeA1995Tyr13 is essential for the activity of omega-conotoxin MVIIA and GVIA, specific N-type calcium channel blockersBiochem Biophys Res Commun206449547826361
- KohnoTKimJIKobayashiK1995Three-dimensional structure in solution of the calcium channel blocker omega-conotoxin MVIIABiochemistry3410256657640281
- KristipatiRNadasdiLTarczy-HornochK1994Characterization of the binding of omega-conopeptides to different classes of non-L-type neuronal calcium channelsMol Cell Neurosci5219288087420
- LevineJDFieldsHLBasbaumAI1993Peptides and the primary afferent nociceptorJ Neurosci132273868501507
- LewisRJNielsenKJCraikDJ2000Novel omega-conotoxins from Conus catus discriminate among neuronal calcium channel subtypesJ Biol Chem275353354410938268
- LightARPerlER1979aSpinal termination of functionally identified primary afferent neurons with slowly conducting myelinated fibersJ Comp Neurol18613350109477
- LightARPerlER1979bReexamination of the dorsal root projection to the spinal dorsal horn including observations on the differential termination of coarse and fine fibersJ Comp Neurol18611731447880
- LightARTrevinoDLPerlER1979Morphological features of functionally defined neurons in the marginal zone and substantia gelatinosa of the spinal dorsal hornJ Comp Neurol18615171447881
- LinZHausSEdgertonJ1997Identification of functionally distinct isoforms of the N-type Ca2+ channel in rat sympathetic ganglia and brainNeuron18153669010213
- LinZLinYSchorgeS1999Alternative splicing of a short cassette exon in alpha1B generates functionally distinct N-type calcium channels in central and peripheral neuronsJ Neurosci1953223110377343
- LlinasRSugimoriMHillmanDE1992Distribution and functional significance of the P-type, voltage-dependent Ca2+ channels in the mammalian central nervous systemTrends Neurosci1535151382335
- LuchianT2001The influence exerted by the beta(3) subunit on MVIIA omega-conotoxin binding to neuronal N-type calcium channelsBiochim Biophys Acta15123293411406110
- LuoZDChaplanSRHigueraES2001Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve-injured ratsJ Neurosci2118687511245671
- Lyseng-WilliamsonKAPerryC2006ZiconotideCNS Drugs20331816599651
- McGivernJG2006Targeting N-type and T-type calcium channels for the treatment of painDrug Discov Today112455316580601
- McGivernJGMcDonoughSI2004Voltage-gated calcium channels as targets for the treatment of chronic painCurr Drug Targets CNS Neurol Disord34577815578964
- McMahonSBCaffertyWBMarchandF2005Immune and glial cell factors as pain mediators and modulatorsExp Neurol1924446215755561
- MalmbergABYakshTL1994Voltage-sensitive calcium channels in spinal nociceptive processing:blockade of N- and P-type channels inhibits formalin-induced nociceptionJ Neurosci144882908046458
- MalmbergABYakshTL1995Effect of continuous intrathecal infusion of omega-conopeptides, N-type calcium-channel blockers, on behavior and antinociception in the formalin and hot-plate tests in ratsPain6083907715945
- MatthewsEADickensonAH2001Effects of spinally delivered N- and P-type voltage-dependent calcium channel antagonists on dorsal horn neuronal responses in a rat model of neuropathyPain922354611323145
- MeadowsHJBenhamCD1999Sensitivity to conotoxin block of splice variants of rat alpha 1B (rbBII) subunit of the N-type calcium channel coexpressed with different beta subunits in Xenopus oocytesAnn N Y Acad Sci868224710414299
- MiljanichGP2004Ziconotide:neuronal calcium channel blocker for treating severe chronic painCurr Med Chem1130294015578997
- MintzIMSabatiniBLRegehrWG1995Calcium control of transmitter release at a cerebellar synapseNeuron15675887546746
- MouldJYasudaTSchroederCI2004The alpha2delta auxiliary subunit reduces affinity of omega-conotoxins for recombinant N-type (Cav2.2) calcium channelsJ Biol Chem279347051415166237
- NadasdiLYamashiroDChungD1995Structure-activity analysis of a Conus peptide blocker of N-type neuronal calcium channelsBiochemistry348076817794920
- NebeJEbersbergerAVanegasH1999Effects of omega-agatoxin IVA, a P-type calcium channel antagonist, on the development of spinal neuronal hyperexcitability caused by knee inflammation in ratsJ Neurophysiol812620610368382
- NebeJVanegasHNeugebauerV1997Omega-agatoxin IVA, a P-type calcium channel antagonist, reduces nociceptive processing in spinal cord neurons with input from the inflamed but not from the normal knee joint—an electrophysiological study in the rat in vivoEur J Neurosci921932019421179
- NewcombRAbbruscatoTJSinghT2000Bioavailability of Ziconotide in brain:influx from blood, stability, and diffusionPeptides2149150110822104
- NewtonRABinghamSCasePC2001Dorsal root ganglion neurons show increased expression of the calcium channel alpha2delta-1 subunit following partial sciatic nerve injuryBrain Res Mol Brain Res951811687271
- NewcombRPalmaAFoxJ1995SNX-325, a novel calcium antagonist from the spider Segestria florentinaBiochemistry34834177541240
- NielsenKJAdamsDAAlewoodPF1999aEffects of chirality at Tyr13 on the structure-activity relationships of omega-conotoxins from Conus magusBiochemistry3867415110346894
- NielsenKJAdamsDThomasL1999bStructure-activity relationships of omega-conotoxins MVIIA, MVIIC and 14 loop splice hybrids at N and P/Q-type calcium channelsJ Mol Biol28914052110373375
- NielsenKJThomasLLewisRJ1996A consensus structure for omega-conotoxins with different selectivities for voltage-sensitive calcium channel subtypes:comparison of MVIIA, SVIB and SNX-202J Mol Biol2632973108913308
- NorthRA1986Opioid receptor types and membrane ion channelsTrends Neurosci91147
- OliveraBMCruzLJde SantosV1987Neuronal calcium channel antagonists. Discrimination between calcium channel subtypes using omega-conotoxin from Conus magus venomBiochemistry262086902441741
- OlsenHLTheanderSBokvistK2005Glucose stimulates glucagon release in single rat alpha-cells by mechanisms that mirror the stimulus-secretion coupling in beta-cellsEndocrinology14648617016081632
- OssipovMHLaiJMalanTPJr2000Spinal and supraspinal mechanisms of neuropathic painAnn N Y Acad Sci909122410911921
- PanJQLipscombeD2000Alternative splicing in the cytoplasmic II-III loop of the N-type Ca channel alpha 1B subunit:functional differences are beta subunit-specificJ Neurosci2047697510864934
- PorrecaFBurgessSEGardellLR2001Inhibition of neuropathic pain by selective ablation of brainstem medullary cells expressing the mu-opioid receptorJ Neurosci215281811438603
- Price-CarterMGrayWRGoldenbergDP1996Folding of omega-conotoxins. 2. Influence of precursor sequences and protein disulfide isomeraseBiochemistry3515547578952509
- Price-CarterMHullMSGoldenbergDP1998Roles of individual disulfide bonds in the stability and folding of an omega-conotoxinBiochemistry379851619657699
- RauckRLWallaceMSLeongMS2006A Randomized, Double-Blind, Placebo-Controlled Study of Intrathecal Ziconotide in Adults with Severe Chronic PainJ Pain Symptom Manage3139340616716870
- SangerGJEllisESHarriesMH2000Rank-order inhibition by omega-conotoxins in human and animal autonomic nerve preparationsEur J Pharmacol388899510657551
- SatherWAMcCleskeyEW2003Permeation and selectivity in calcium channelsAnnu Rev Physiol651335912471162
- SatoKRaymondCMartin-MoutotN1997Binding of chimeric analogs of omega-conotoxin MVIIA and MVIIC to the N- and P/Q-type calcium channelsFEBS Lett41448049315745
- SatoKRaymondCMartin-MoutotN2000Binding of six chimeric analogs of omega-conotoxin MVIIA and MVIIC to N- and P/Q-type calcium channelsBiochem Biophys Res Commun269254610694509
- SchneggenburgerRNeherE2005Presynaptic calcium and control of vesicle fusionCurr Opin Neurobiol152667415919191
- ScottDAWrightCEAngusJA2002Actions of intrathecal omega-conotoxins CVID, GVIA, MVIIA, and morphine in acute and neuropathic pain in the ratEur J Pharmacol4512798612242089
- SherEGiovanniniFCodignolaA2003Voltage-operated calcium channel heterogeneity in pancreatic beta cells:physiopathological implicationsJ Bioenerg Biomembr356879615000528
- SlukaKA1998Blockade of N- and P/Q-type calcium channels reduces the secondary heat hyperalgesia induced by acute inflammationJ Pharmacol Exp Ther28723279765342
- SmithMTCabotPJRossFB2002The novel N-type calcium channel blocker, AM336, produces potent dose-dependent antinociception after intrathecal dosing in rats and inhibits substance P release in rat spinal cord slicesPain961192711932068
- SnutchTP2004N-type channels, small organic molecules and painSpring Pain Research ConferenceGrand Cayman, BWI
- SnutchTPFengZPBelardettiF2003Novel N-type calcium channel blockers efficacious in animal models of chronic pain226th American Chemical Society National MeetingNew York, NY
- StaatsP2006Ziconotide:a viewpoint by peter staatsCNS Drugs2033941
- StaatsPSYearwoodTCharapataSG2004Intrathecal ziconotide in the treatment of refractory pain in patients with cancer or AIDS:a randomized controlled trialJAMA291637014709577
- StoehrSJDooleyDJ1993Characteristics of [125I]omega-conotoxin MVIIA binding to rat neocortical membranesNeurosci Lett16111368255536
- TakaharaAKoganeiHTakedaT2002Antisympathetic and hemodynamic property of a dual L/N-type Ca(2+) channel blocker cilnidipine in ratsEur J Pharmacol43443711755164
- TakahashiEItoMMiyamotoN2005Increased glucose tolerance in N-type Ca2+ channel alpha(1B)-subunit gene-deficient miceInt J Mol Med159374415870896
- UrbanMORenKSabladM2005Medullary N-type and P/Q-type calcium channels contribute to neuropathy-induced allodyniaNeuroreport16563615812308
- WallaceMSCharapataSGFisherR2006Intrathecal Ziconotide in the Treatment of Chronic Nonmalignant Pain:A Randomized, Double-Blind, Placebo-Controlled Clinical TrialNeuromodulation97586
- WangYXBezprozvannayaSBowersoxSS1998Peripheral versus central potencies of N-type voltage-sensitive calcium channel blockersNaunyn Schmiedebergs Arch Pharmacol357159689521489
- WangYXGaoDPettusM2000aInteractions of intrathecally administered ziconotide, a selective blocker of neuronal N-type voltage-sensitive calcium channels, with morphine on nociception in ratsPain842718110666532
- WangYXPettusMGaoD2000bEffects of intrathecal administration of ziconotide, a selective neuronal N-type calcium channel blocker, on mechanical allodynia and heat hyperalgesia in a rat model of postoperative painPain84151810666519
- WatkinsPBKaplowitzNSlatteryJT2006Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily:a randomized controlled trialJAMA296879316820551
- WenLYangSQiaoH2005SO-3, a new O-superfamily conopeptide derived from Conus striatus, selectively inhibits N-type calcium currents in cultured hippocampal neuronsBr J Pharmacol1457283915880145
- WermelingDPBergerJR2006Ziconotide infusion for severe chronic pain:case series of patients with neuropathic painPharmacotherapy2639540216503720
- WermelingDDrassMEllisD2003Pharmacokinetics and pharmacodynamics of intrathecal ziconotide in chronic pain patientsJ Clin Pharmacol436243612817525
- WestenbroekREHoskinsLCatterallWA1998Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminalsJ Neurosci186319309698323
- WheelerDBRandallATsienRW1994Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmissionScience264107117832825
- WinquistRJPanJQGribkoffVK2005Use-dependent blockade of Cav2.2 voltage-gated calcium channels for neuropathic painBiochem Pharmacol704899915950195
- WrightCERobertsonADWhorlowSL2000Cardiovascular and autonomic effects of omega-conotoxins MVIIA and CVID in conscious rabbits and isolated tissue assaysBr J Pharmacol13113253611090104
- XiaoWHBennettGJ1995Synthetic omega-conopeptides applied to the site of nerve injury suppress neuropathic pains in ratsJ Pharmacol Exp Ther274666727636726
- YakshTL2006Calcium channels as therapeutic targets in neuropathic painJ Pain7S133016426997
- YamamotoTSakashitaY1998Differential effects of intrathecally administered N- and P-type voltage-sensitive calcium channel blockers upon two models of experimental mononeuropathy in the ratBrain Res794329329622667
- YokoyamaKKuriharaTMakitaK2003Plastic change of N-type Ca channel expression after preconditioning is responsible for prostaglandin E2-induced long-lasting allodyniaAnesthesiology9913647014639150
- ZamponiGW2003Regulation of presynaptic calcium channels by synaptic proteinsJ Pharmacol Sci92798312832834
- ZhongHYokoyamaCTScheuerT1999Reciprocal regulation of P/Q-type Ca2+ channels by SNAP-25, syntaxin and synaptotagminNat Neurosci29394110526329