 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
TP53 is the most commonly mutated gene in human cancers. Approximately 90% of mutations in this gene are localized between domains encoding exons 5 to 8. The aim of this investigation was to examine the ability of the low density DNA microarray with the assistance of double tandem hybridization platform to characterize TP53 mutational hotspots in exons 5, 7, and 8 of the TP53. Nineteen capture probes specific to each potential mutation site were designed to hybridize to specific site. Virtual hybridization was used to predict the stability of hybridization of each capture probe with the target. Thirty-three DNA samples from different sources were analyzed for mutants in these exons. A total of 32 codon substitutions were found by DNA sequencing. 24 of them a showed a perfect correlation with the hybridization pattern system and DNA sequencing analysis of the regions scanned. Although in this work we directed our attention to some of the most representative mutations of the TP53 gene, the results suggest that this microarray system proved to be a rapid, reliable, and effective method for screening all the mutations in TP53 gene.
Introduction
Cancer disease can be attributed to an accumulation of several genetic alterations,Citation1 such as mutations, amplifications, deletions, translocations, virus integration, etc., thus, the identification of these alterations in patient DNA can yield useful molecular markers for diagnosis or prognosis,Citation2 for example, single base mutations in genes such as TP53. This tumor suppressor gene is mapped on the short arm of chromosome 17 and harbors tumor-causing DNA missense mutations in the region that encodes the DNA-binding domain (DBD) (exons 5–8).Citation3 Mutations in this region lead to nuclear accumulation of p53 protein and loss of its normal cellular functions, such as transcriptional transactivation of target genes that regulate the cell cycle and apoptosis.Citation4 Over 24,810 human tumor mutations in the TP53 gene have been registered in the International Agency for Research on Cancer (IARC) TP53 database (R12, November 2007).Citation5 Around 40% of these mutations are localized at the “hotspot” residues R157, R158, R175, G245, R248, R249, R273, and R282.Citation6,Citation7 The detection of such point mutations is useful in screening for cancers,Citation8,Citation9 as well as for typing of cancers to optimize treatment protocols to improve the diagnosis and prognosis.Citation10,Citation11
Microarrays and GeneChips have been of considerable recent interest, due to their remarkable promise for obtaining sequence-specific information in a faster and simpler manner compared to traditional methods. In this context, a considerable effort has been made to develop fast and inexpensive techniques suitable for clinical services and small research laboratories for TP53 mutations detection. Tandem hybridization used in low density DNA microarrays meets the latter requirements. Low-density microarrays certainly have the potential to custom development and the detection of all desired features and could conveniently provide reliable results; decreasing production costs considerably. The low-density DNA microarray used in the present study is a variant of the conventional microarray format employing tandem hybridization, used to detect some point mutations in DNA samples.Citation12 This method offers several advantages over traditional oligonucleotide array mainly due to the annealing of unlabeled target DNA to relatively long (and labeled) stacking oligonucleotides, resulting in an increase of the sequence discrimination, without loss in specificity.Citation13 Previously, we have reported this hybridization strategy to search the most frequently altered codons at exons 7 and 8 of the TP53 gene.Citation14 The microarray designed, has the ability to exclude and confirm the presence of some important mutations on TP53 gene. In the present work, this oligoarray was modified, adding five probes more in order to interrogate frequent point mutations in exon 5. Now, this microarray includes nineteen 7-mer probes to search for 14 mutational hotspots located in codons 157, 158, 248, 249, and 273 of TP53 gene. The method used multiplex-PCR and multiplex-hybridization format that simplifies some steps of the procedure and can be developed using instrumentation available in small laboratories. Besides, this protocol permitted the exact identification of the point mutations coding missense variants, which could have different contributions to the molecular diagnostic and can be used as a screening tool in clinical studies.
Materials and methods
Oligonucleotides
The distribution of nucleotidic changes in the TP53 gene associated with all cancers was retrieved from the IARC TP53 database (see http://www.p53.iarc.fr/p53/database). Both reference (accession #U94788) and mutated TP53 sequences were used to design the probes, stacking oligonucleotides, PCR primers and synthetic targets. All oligonucleotides used for mutations in exons 7 and 8 have been previously described.Citation14 Oligonucleotides used to analyze the mutations in exon 5 were designed in this work ().
Table 1 Sequences of the 5-exon TP53 oligonucleotide microarray
Natural and synthetic DNA samples
To provide confidence in the interpretation of the signals produced by human samples, reference hybridization patterns were prepared using a full set of synthetic DNA targets (group 1). For this purpose, 17 synthetic targets (44–50 nt), representing the wild type and mutant sequences, were used independently and in combination, to represent homozygous and heterozygous conditions. Twenty-four DNA samples from different sources were previously screened for all possible TP53 mutations using our DNA oligonucleotide microarray plus automated sequencing,Citation14 some having known wild type or mutant TP53 sequences and others with unknown TP53 genotype. These included 16 blood samples (six obtained from healthy subjects and 10 obtained from cancer-affected patients), 10 cancer cell lines (A-427, C33 A, Hela, SiHa, Jurkat, T24, SW480, I-37, I-51 and PCS-4), and six recombinant plasmids (pp53, pSCX3, pM47, pC53, Xbal, C1) were obtained from the DNA Bank at the Genomic Oncology Laboratory National Medical Center, Century XXI. From those samples, we selected 11 samples (group 2), with a total of 18 point mutations in TP53, and then were used in a “blind” test for sensitivity and accuracy of hybridization signal. Moreover, for exon 5 analysis, 22 DNA samples from different sources (group 3) with unknown TP53 genotype were analyzed. These included i) 14 blood samples: six obtained from healthy subjects and eight from cancer-affected patients (four obtained from lymphoma-affected patients and four from breast-affected patients); ii) seven bone marrow cells obtained from lymphoma-affected patients; and iii) a Raji cancer cell line. Total genomic DNA was isolated and purified using the Genomic DNA Extraction Kit (Life Technologies Inc., Gaithersburg, MD, USA) and DNA quality and concentration was estimated with Pico Green® dsDNA Quantitation Kit (Molecular Probes Inc., Eugene, OR). Target preparation: as previously described, DNA targets were preannealed with their respective stacking 32P-labeled oligonucleotides.Citation14
Multiplex and asymmetric PCR-based approach analysis and direct sequencing
Exons 5, 7, and 8 of the TP53 gene were amplified from genomic DNA in three amplicons: 129 bp (exon 5: codons 157 and 158); 108 bp (exon 7: codons 248 and 249), and 92 bp (exon 8: codon 273) by multiplex PCR with the same conditions described.Citation14 Briefly: 0.2 mM each dNTP, 50 mM KCl, 10 mM Tris-HCl (pH8.4), 1.5 mM MgCl2, 0.5 μM each primer, 2.5 U of Platinum Taq DNA polymerase (Invitrogen) and purified DNA (50–100 ng) in a final volume of 100 μL. PCR profile consisted of: 94°C for 5 min, 30 cycles for 30 sec at 94°C, 30 sec at 55°C and 72°C for 7 min. The products were run on EtBr-stained agarose gels to verify the success of amplification. PCR-amplified fragments were used to obtain single stranded DNA (ssDNA) by cyclic DNA synthesis. These ssDNA were purified with a QIAEX II kit (Qiagen, Inc., Stanford, USA) according to the manual provided by the manufacturer, and then sequenced directly with the BigDye Terminator Cycle Sequencing FS Ready Reaction kit in a 373 automated DNA sequence (Instruments Core-IMSS) using both the forward and reverse primers.
Target preparation
Target preparation was described previously.Citation14 Briefly, 5 pmol of each dephosphorylated stacking oligonucleotide; 5X Forward Reaction Buffer (5 μl); T4 Polynucleotide Kinase (10 unit); [32P] ATP (10 μCi/μl, 3000 Ci/mmol) 2.5 μl, specific activity 7000 Ci/mmol, diluted in sterile water to 7 μCi/μl, and water HPLC-grade to 25 μl. The mix was incubated 10 min at 37°C and the reaction was stopped by adding 5 mM EDTA (2.5 μl). 32P-labeled stacking oligonucleotides were preannealed with the respective DNA target. The annealing mixture contained 50 μL 20 × SSC, 10 μL 1 M Tris-HCl (pH 8.0), 3 μL 0.5M EDTA, 0.2 pmol of each labeled stacking oligonucleotide, 10 μL of target DNA, and HPLC-grade H2O to a final volume of 90 μL. The mixture was incubated at 95°C for 5 min, 45°C for 5 min, then 6°C for 5 min. Excess [32P] ATP was removed by filtration through an Ultrafree spin filter (3,000 Mr cutoff), and the retained DNA was dissolved in 20 μL of 1 × SSC.
Detection of heterozygous mutation
In order to identify heterozygous mutations, we analyzed a 1:1 mixture of the target DNA from A-427 and pp53 that have wild (AGG) and mutant (AGA) sequence at the codon 249, respectively. The 1:1 mixture (in terms of concentration) of target DNA was prepared of genomic DNA templates that were extracted from both cancer cell lines. Moreover, with one wild type DNA sample and one mutated genomic DNA sample (SW480 cell line) mixed in various ratios, keeping the total DNA constant we determined the lower limit of detection of the mutated p53 sample, to know how sensitive our assay is.
Oligoarray construction and use
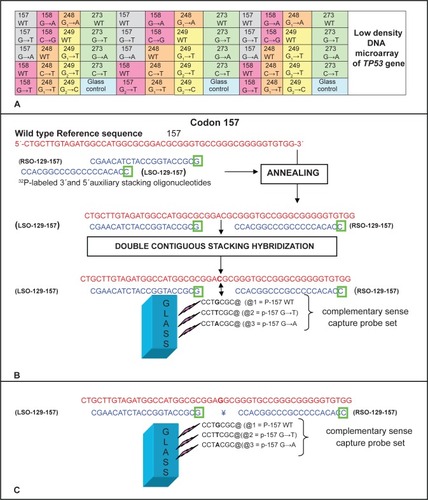
Microarray construction and hybridization were performed as previously describedCitation14 briefly; three replicate spots of each probe were aligned in the same rows of the microarray. Each array contained 19 probes: five wild type sequences and fourteen mutant sequences corresponding to the TP53 targets under study, plus one blank spot without any probe which served as a negative glass control, outlines the array design used in this study. Complementary stacking oligonucleotides were designed to form partially duplex when annealing with single-stranded DNA and . The purified amplicon were diluted in hybridization buffer and hybridized to the TP53 microarray. Finally, the microarray was washed and packed in plastic film and X-ray film exposed for autoradiography, and digitized hybridization images were obtained using a scanner (HP Scanjet 4400c).
Figure 1 Design of the improved TP53 Low density DNA microarray. A) Schematic of the microarray layout (not to scale). B) An example of the alignment of stacking oligonucleotides and probes to their respective synthetic wild type target sequences in 157 and 158 contiguous codons of TP53 gene. As shown a nucleic acid target is first annealed with a molar excess of labeled auxiliary oligonucleotide (the letter inside green square represent the 32P-labeled base of the oligonucleotide). The labeled auxiliary oligonucleotide serves as a reagent to introduce the detection tag into a single specific site within the nucleic acid target. The partially duplex labeled target is then applied to the array of capture probes, typically 7–12 bases long, end-tethered to a glass slide or chip. Each capture probe is designed to hybridize with the target strand in tandem with a labeled stacking oligonucleotide preannealed to the target. Base stacking interactions between the short capture probe and the long stacking probe stabilize the binding of partially duplex labeled target to the glass-tethered capture probe. Hybridization is carried out at an (elevated) temperature at which the short capture probe by itself does not form a stable duplex structure with the target; capture of label to the glass occurs only when the capture probe hybridizes in tandem with the labeled stacking probe. As depicted in C) a single base mismatch between the short capture probe and the target sequence will disrupt the short duplex, preventing binding of label to the glass.
Virtual hybridization (VH)
A VH module of the Genosensor Probe Designer softwareCitation15 was used to predict the stability of hybridization of each capture probe with the DNA target based on calculation of the Gibbs free energy (ΔG°) using thermodynamic parameters compiled by SantaLucia for perfectly matched and mismatched oligonucleotide sequences.Citation16–Citation19 To calculate the ΔG° of the oligonucleotide sequences the VH program can accept thermodynamic calculations for each probe aligned at all positions along the relevant target DNA sequence. The target/probe duplexes having ΔG° values above of a given cut off value (bigger stability) were tabulated. These results were compared with the intensity of signals in order to estimate if the free energy predictions are in agreement with the signals observed. All the described procedures were evaluated and approved by the local ethics committee of the Mexican Institute of Social Security.
Data analysis
ScanAlyze 2.5 software (Stanford University)Citation20 was used to quantify hybridization signals for each probe and subtract background hybridization signals. For every perfect match probe (fully complementary to the target sequence) in each set of probes, another two identical perfect match probes are present elsewhere in the array. This redundancy increases assay sensitivity and specificity by compensating for nonspecific signals that may arise from localized surface imperfections such as microscopic scratches in the array surface. The enhanced signal provides greater sensitivity toward detecting weak hybridization; the specificity was calculated as the ratio of the perfect match probe (PM), fully complementary to the target sequence, hybridization signal of the wild-type target to their single base substitution probe. The raw intensity data for each experiment was normalized calculating the average intensity for each individual dataset; then the average of the averages was calculated. This total average was used as the basis for the normalization. The average of all normalized data thereafter equaled the total average.
Results
Reference hybridization patterns (Group 1)
All the synthetic targets, including negative controls, yielded hybridization signals with their respective (“perfectly matched”) probes. Pairs of wild type with each mutant synthetic target sequences (1:1 mixtures) were also hybridized to reproduce heterozygous conditions. All gave the expected signals (data not shown).
Multiplex-PCR and hybridization assay
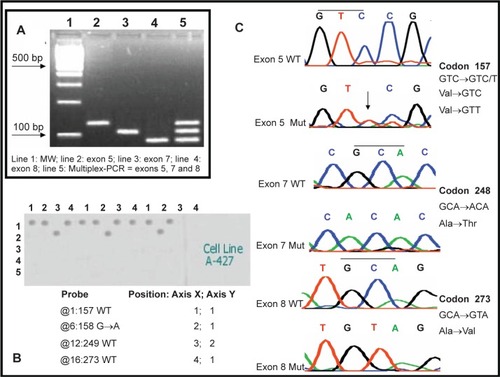
Multiplex-PCR assay from exons 5, 7, and 8 was highly reproducible under the described conditions. These amplicons were run on 4% agarose gels (). Hybridization of gapped duplex, isolated ssDNA target was done for all DNA samples. In general, we observed that all samples hybridized solely with one (homozygous) or two (heterozygous) probes in 33 different samples tested with the hybridization assay. As expected, all probes included in the microarray gave signals and the point mutations were detected by hybridization in all samples. shows a typical hybridization result from this multiplex hybridization assay. DNA sequencing () confirmed all results.
Figure 2 Electrophoretic analysis of PCR products and single-stranded target DNA produced by “cycle sequencing”. A) Electrophoresis in 2% agarose gel from PCR reactions of regions from exons 5, 7 and 8 of the TP53 gene: 100 bp ladder (Line 1), 129 bp PCR product from exon 5 (line 2), 108 bp PCR product from exon 7 (line 3), 92 bp PCR product from exon 8 (line 4), multiplex PCR products of exons 5, 7 and 8. B) Example of the multiplex hybridization of the microarray against a PCR product from the cell line A-427 where wild types 157, 249 and 273 as well as the mutation 158 G→A were detected C) Electropherograms for the sequencing of wild type and mutant PCR products from exons 5, 7 and 8 of the TP53 gene.
The minimum amount of target detected, as estimated by the proportion of equimolar amount of labeled stacking oligonucleotide annealed with the sample, was 5 to 10 femtomoles (data not shown).
Concordance of positive results, blind test with DNA samples (Group 2)
Among the 11 samples that we used to verify reproducibility of the assay, all over again showed the same 18 missense mutations obtained previously by sequencing codons 248, 249, and 273. Besides, from this group of DNA samples, only four samples showed nucleotide changes in exon 5: one of these samples (L1) had two point mutations [(GTC→GAC) Val157Asp; (CGC→CAC) Arg158His]; two cases (L9 and L10) showed the same base change G→T, Val157Phe and another case (P6) showed (GTC→GTT) Val157Val], of which only three were identified by microarray analysis, because in the last case the mutant probe was absent of the microarray. The remaining seven samples did not show any changes in exon 5; therefore they were considered as having only wild type TP53 sequences in the region tested (see ).
Table 2 Mutation detected in exons 5, 7 and 8 by DNA microarray and automated sequencing
Hybridization assay in DNA unknown (Group 3)
Among the 22 DNA samples of the various sources analyzed, 6 point mutations were identified by sequencing. Those included three cases of peripheral blood cells with positive signal for exon 8: B9 Case 11 (CGT→TGT) Arg273Cys; B11 Case 57 (CGT→TGT) Arg273Cys, and B13 Case 80 (CGT→CTT) Arg273Leu missense mutations were identified. Two cases of peripheral blood were positive for exon 5: B10 Case 21 (GTC→GTT) Val157Val; B12 Case 79 (GTC→GTT) Val157Val. One case of bone marrow cell sample showed a mutation in exon 7: BM Case 91 (CGG→CGT) Arg248Arg. Three point mutations were identified by DNA microarray and another three (silent mutations) were detected only for sequencing, since mutant probe was not included on the array. The remaining samples of this group showed only wild type TP53 sequences, see .
Virtual hybridization
Hybridization results are resumed in . In this table probes, targeted sequences and free energy values for the hybridization are indicated for all probes and all target sequences tested. More stable hybridization sites are highlighted in green, medium stable hybridization values are highlighted in red and low stable hybridization values are colored in blue. It is evident in this table that only perfect hybridizations have more stable hybridization values, except two ambiguous hybridizations for exon 5 and two for exon 7. Most part of the ambiguous hybridization has medium or low stable free energy values (more than-9 Kcal/mol). The stability of ambiguous hybridizations is strongly dependent on the identity of the sequences involved.
Table 3 Virtual Hybridization results for the stability of hybridization between probes and PCR products tested
Discussion
Alterations in TP53 gene are probably the most common genetic abnormalities associated with human malignancies. Previous studies using DNA microarrays have delimited a specific pattern of mutational spectrum in some cancers,Citation21–Citation24 making DNA microarray an attractive method in order to define these particular alterations in cancer genetics. A simple method to identify mutations in this gene could improve its usefulness in screening, diagnosis, staging, and treatment of many kinds of cancers. In this work, we evaluated a low density DNA microarray for specific mutation screening in a search for an inexpensive and easy alternative method for small laboratories. This system was developed only to determine some of the most relevant mutations observed in exons 5, 7, and 8 of TP53 gene reported by the IARC (see http://www.p53.iarc.fr/p53/). Besides, one of the aims of this work was to demonstrate the capability of the tandem hybridization approach for the detection of some of the more disease associated relevant mutations in the TP53 gene. And it is critical for us to learn from the previous experience and design a test with the optimal cost/performance relationship with potential application for screening. And it is important to comment that our system was not created to investigate all the potential mutations that could be observed in the TP53 gene. The system was directed only against the hotspot mutations that are more commonly observed in exons 5, 7, and 8 because we think that it is more probable to find mutations in clinical samples based on the frequency of these mutations that represent approximately 20% of the sequence changes affecting the TP53 gene associated to cancer. Moreover, the number of probes can be increased to detect as many mutations as wished. According with the literature available there are two relevant, microarray based, TP53 assays. These are the Arrayed Primer Extension Resequencing APEXCitation25 and the GeneChip array.Citation26 APEX uses an array of 2436, 25-mer oligonucleotides. APEX spans exons 2–9 plus two introns; it covers 95% (1218 bases) of the mutations described in TP53. On average, 97.5% of the arrayed TP53 gene sequence can be analyzed from sense or antisense strands, 81% from both strands. GeneChip has approximately 65000, 18-mer, probes. It covers exons 2–11 and +2/−2 splice site junctures. It searches approximately 1262 bases from TP53 gene. Overall, the oligonucleotide microarray demonstrated a 94% accuracy rate, 92% sensitivity, and 100% specificity. Therefore, neither the APEX array nor the GeneChip array is able to detect all the described TP53 mutations. Besides, they search only part of the TP53 gene. The accuracy in both assays is approximately similar (94%). They cover from 92% to 95% of the published TP53 gene mutations with 2436 and 65000 probes approximately. In comparison the micro-Chip described in this work, which only uses 29 probes and only tests the mutations in the three main hotspots, is able to search on average 20% of the cancer related mutations in the TP53 gene. Our chip is aimed for a primary screen of the somatic mutations. It can be easily made and used at a relatively low cost. And those cases giving negative in this assay can be further tested with other more complex chip, such as the GeneChip. Of course we are in the process of developing a more complex chip using tandem hybridization comprising most of the mutations reported at the IARC database. However, there are several challenges involved in this design. Since additional (stacking) oligonucleotides are included into the hybridization mixture, which add potential interferences in the system. Some important factors should be addressed in this design: (a) such microarray will include a considerable high number of probes and its cost can increase considerably. However in each experiment only a reduced fraction of the probes (if any) will provide useful information. Even including all probes reported at to date some mutations could be missed. This has been raised the idea of the design of a microarray for the detection of the most frequent mutations of this gene as an initial screening. (b) In the tandem hybridization approach stacking oligonucleotides are included, which hybridize contiguously to the site that is recognized by the capture probe in the microarray. When the number of mutations is tested the assays have been working properly even in multiplex assays. However if several mutations (many of the in a close neighborhood) are tested other factors can play an important role as the competition of some of the stacking probes for sites that should be recognized by the capture probe, which can reduce the sensitivity of the approach. Some solutions have proposed has the hybridization in separated experiments of samples which include stacking probes which do not interfere with the capture probes.
In general, oligonucleotide DNA microarrays containing short (15 to 25-mer) oligonucleotide probes provide greater point discrimination than microarrays composed of larger PCR-amplified DNA fragments.Citation13 However, a central challenge to the application of DNA microarrays in mutation detection is achieving the specificity needed to resolve the discriminating between target and no target DNA that differ by a single nucleotide.Citation27 This level of specificity is needed to resolve variants of highly conserved genes (eg, TP53 and K-RAS) which often are point mutated in cancer.
In our understanding this is a critical issue since it will answer the capability of the system to perform early detection in samples commonly containing normal and mutant cells. The proportion of mutant cells grows with the developing of the tumor and the prognostic is generally better for tumors detected in their early stages. Two main factors are involved in the sensitivity and specificity of the detection. The sensitivity depends primarily on the minimum amount of molecules required for fluorescent or radioactive (1012 molecules) detection. PCR technique requires 40 cycles to reach this amount from a single molecule. The PCR in this assay simultaneously amplifies the same DNA fragment from wild type and mutant cells. Therefore, the excessive presence of wild type cells with respect to mutant cells is potentially able to involve the synthesis of mutant fragments by limitation in the reagents available in the PCR mixture. This problem affects all the detection protocols that are depending on PCR reactions. The specificity is based in the capability of the assay to discriminate between the wild type and the mutant sequence. In the hybridization-based approaches this depends on the ΔTm values, which can be accurately determined for single mismatches by Nearest Neighbor (NN) SantaLucia’s Model. We have tested previously our Tandem Hybridization system in the TP53 model. And, as expected a cross hybridization was observed only in a mutation in the N N X context (data not show). This is due to small ΔTm between the GC pair and the GA mismatch in the duplexes formed by annealing of wild type target DNA with the wild type and mutant probes, respectively, in this sequence. To avoid this problem, it is recommended to use as target the other strand, in whose case CG and CT mismatch will be produced in the respective duplexes which show big ΔTm value. The same solution is used in the Resequencing approach. Therefore the Tandem Hybridization approach should include the sets of probes to perform the identification in both strands of the target with independent preannealing of the respective stacking oligonucleotides.
Virtual hybridization is a computer simulation that allows calculating the thermal stability of a DNA duplex even in the presence of single mismatches. For this purpose the nearest-neighbor model for predicting stability of DNA duplexes is used. Allawi and SantaLuciaCitation16–Citation18 have calculated thermodynamic parameters for all the eight possible combinations of mismatches in all sequential contexts. According with their data the general order of stability of these mismatches is as follows:
However, for a precise calculation of the thermal stability is necessary to know the identity of the neighboring bases. The virtual hybridization allows estimating the capacity of discrimination of the possible mismatches, by comparing the difference between the free energy of the perfect hybridization (without mismatches) and the free energy when mismatches are present. Detailed calculations for hybridizations tested in this work by virtual hybridization are showed in , where it can be seen that in general a good discrimination, with low possibility of cross reactions, is predicted, except when the mismatch is located at the ends of the probe because in such cases all the mismatches are more stable. However, the tandem hybridization approach has the effect of accentuate the differences on stability even when mismatches are close to the ends then increasing the specificity of the test, which is an advantage of this test compared with the classical hybridization approach without stacking probes. This increase in specificity is probably due to the additional elimination of base stacking at the contiguous probe and stacking oligonucleotides ends by the production of target/probe mismatches at the center of the probe.
During the hybridization reaction, the guanine form relatively stable mismatches, therefore, the duplexes formed are difficult to discriminate by traditional hybridization.Citation19 Short duplexes are considerably more destabilized by even relatively stable mismatches than longer duplexes, and for this reason short probes have higher discrimination power.Citation28 Short probes when used individually (without tandem hybridization) are not adequate because these sequences can occur by random chance multiple times within relatively long targets, which would limit their specificity in the single-probe approach.Citation29 However, this is not the case in the tandem hybridization approach because the specificity arises from the addition of the sequences contained in the short probes plus the contiguous stacking oligonucleotides (the fact that they must be contiguous in order to see a hybridization signal is an important requirement for this technique). In other words, under stringent hybridization/washing conditions, only 7-mer sites located adjacent to the stacking oligonucleotides are interrogated, since 7-mer duplexes (not stabilized by the base stacking) are unstable under the hybridization conditions used. The stability of 7-mer hybrids stabilized by long stacking probes is similar to that of traditional 14–20-mer single probes, depending on the sequences.Citation30,Citation31
Our system was very specific to indicate the presence of mutations in all cases where a loss of signal was observed, which was separately confirmed by sequencing, so, that the specificity of this assay was high. Other future option to increase the confidence in the results is to perform the analysis with both target strands. This should be especially useful when the base substitution involves a G (which is very frequent), since G forms the most stable mismatches, however the C in the opposite strand produces the most unstable mismatches and therefore are highly discriminatory.Citation32 This approach was successfully used in resequencing by Karaman and colleagues.Citation33
Some mismatches, as AG and TT mismatches, are exceptionally stable, but also the identity of the neighboring bases as well as the mismatch probe position are important stability factors. Exceptionally stable mismatches could be difficult to differentiate using long probes (more than 10-mers), however short probes do not yield enough stable hybridization. This problem is circumvolved by the use of the stacking hybridization were the stability of the probes is significantly increased whereas the mismatch discrimination power is maintained even for the more stable mismatches.
VH hybridization allows predicting stability values for hybridization of probes against the targets studies and then is an important tool for designing and evaluating microarray performance. The minimum amount of target detected, as estimated by the proportion of equimolar amount of labeled stacking oligonucleotide annealed with the sample, was 5 to 10 femtomoles. It is important to notice that the intensity of signals seen with the same sample, using 7-mer probes, under traditional, single tandem and double tandem hybridizations increases from 3 to 6 times in Single Tandem Hybridization (which uses only a single stacking oligonucleotide) and 6 to 12 times in the Double Tandem Hybridization (which uses both stacking oligonucleotides) when compared with traditional hybridization (data not published). In this work we are using a radioactive label with high specific activity (6000 Ci/mmol) due to its high sensitivity, besides is cheaper and easy to reveal, yet it is becoming less attractive for routine clinical tests and therefore in future studies we will need to incorporate nonradioactive labeling methods.
In the present study, we showed that the TP53 point mutations obtained in the DNA of different cancer samples, using our DNA microarray, are similar to the frequent point mutations reported in the IARC TP53 Mutation Database. In general, of the 165 codons studied (5 codons in the 33 samples tested), only 28 codons (17%) harbored point mutations. The remaining codons studied with the oligoarray only gave hybridization signal with the probes corresponding to wild type sequences. Selecting specimens from a series of different DNA sources generated the sequencing data of . Because the type of the mutation was not preselected in this study, it was remarkable to observe that in some cases the affected codon encodes for Arginine. These mutations will probably affect the biological activity of p53, because many basic amino acids, such as Arginine, are necessary for DNA binding of p53.39 It is important to notice that the limited number of capture probes on the oligoarray did not allowed us to detect some of the mutations, this can be easily solved by adding the needed probes in future arrays. We cannot exclude the chance of a mutation occurring outside of exons 5, 7 and 8 in these samples. However, previous studies have shown that mutations outside of these exons are infrequent (less than 5%) in the TP53 gene.Citation34,Citation35
Conclusions
In conclusion, low density DNA microarray allowed the detection of some frequent point mutations present in the samples tested. Further studies using arrays with a higher number of probes and the possibility to combine with labeled fluorescent, might be useful for the detection of specific mutations that are relevant for the screening diagnosis of the genesis and progression of cancer disease.
Acknowledgments
The authors with to thank Dr H. Mayani, Head of Oncology Research Unit, and Dr M. Madngal for providing the bone marrow cells, and P. Pina for critical review of the manuscript. During this work AR was an IMSS fellowship recipient. This work was partially supported by grants from CONACYT Mexico (Fondos Sectoriales 7114) and FOFOI-IMSS.
Disclosure
The authors report no conflicts of interest in this work.
References
- LodishHBerkAZipurskyLMatsudairaPBaltimoreDDarnellJMolecular cell biology4th edNew YorkWH Freeman2000
- KirkBWFeinsodMFavisRKlimanRMBaranyFSingle nucleotide polymorphism seeking long term association with complex diseaseNucleic Acids Res2002303295331112140314
- VelculescuVEEl DeiryWSBiological and clinical importance of the p53 tumor suppressor geneClin Chem1996428588688665676
- VogelsteinBLaneDLevineAJNature200040830731011099028
- PetitjeanAMatheEKatoSImpact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 databaseHum Mutat20072862262917311302
- ChoYGorinaSJeffreyPDPavletichNPCrystal structure of a p53 tumor suppressor–DNA complex: understanding tumorigenic mutationsScience19942653463558023157
- MartinACRFacchianoAMCuffALIntegrating mutation data and structural analysis of TP53 tumor-suppressor proteinHum Mutat20021914916411793474
- KirkGDLesiOAMendyM249 (ser) TP53 mutation in plasma DNA, hepatitis B viral infection, and risk of hepatocellular carcinomaOncogene2005245858586716007211
- LeroyKHaiounCLepageEP53 gene mutations are associated with poor survival in low and low-intermediate risk diffuse large B-cell lymphomasAnn Oncol2002131108111512176791
- BerghJTorbjornNSjogrenSLindgrenAHolmsbergLComplete sequencing of the p53 gene provides prognostic information in breast cancer patients, particularly in adjuvant systemic therapy and radiotherapyNat Med19951102910347489358
- MillerLDSmedsJGeorgeJAn expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survivalPNAS20051021355113555
- Maldonado-RodríguezRBeattieKLAnalysis of nucleic acids by tandem hybridization on oligonucleotide microarraysMethods Mol Biol200117015717111357679
- RelógioASchwagerCRichterAAnsorgeWValcárcelJOptimization of oligonucleotide-based DNA microarraysNucleic Acids Res200230e5112034852
- Rangel-LópezAMaldonado-RodríguezRSalcedo-VargasMEspinosa-LaraJMMéndez-TenorioABeattieKLLow density microarray for the detection of most frequent TP53 missense point mutationsBMC Biotechnol20055815713227
- Reyes-LópezMAMéndez-TenorioAMaldonado-RodríguezRDoktyczMJFlemingJTBeattieKLFingerprinting of prokaryotic 16S rRNA genes using oligodeoxiribonucleotide microarrays and virtual hybridizationNucleic Acids Res20033177978912527788
- AllawiHTSantaLuciaJNearest neighbor thermodynamic parameters for internal G: A mismatches in DNABiochemistry199837217021799485363
- AllawiHTSantaLuciaJThermodynamics of internal C: T mismatches in DNANucleic Acids Res199826269427019592156
- AllawiHTSantaLuciaJNearest-neighbor thermodynamics of internal A-C mismatches in DNA. Sequence dependence and pH effectsBiochemistry199837943594449649326
- PeyretNAnandaSPAllawiHTSantaLuciaJNearest neighbor thermodynamics and NMR of DNA sequences with internal A.A, C.C, G.G and T.T mismatchesBiochemistry1999383468347710090733
- SherlockGHernandez-BoussardTKasarskisAThe Stanford Microarray DatabaseNucleic Acids Res20012915215511125075
- AhrendtSAHalachmiSChowJTRapid p53 sequence analysis in primary lung cancer using an oligonucleotide probe arrayProc Natl Acad Sci U S A1999967382738710377423
- MillerLDSmedsJGeorgeJAn expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survivalPNAS2005102135501355516141321
- PrixLUciechowskiPBöckmannBGiesingMSchuetzAJDiagnostic biochip array for fast and sensitive detection of K-ras mutations in stoolClin Chem20024842843511861435
- Sánchez-CarbayoMUse of high-throughput DNA microarrays to identify biomarkers for bladder cancerClin Chem200349233112507957
- TonissonNZernantJKurgAEvaluating the arrayed primer extension resequencing assay of TP53 tumor suppressor geneProc Natl Acad Sci U S A2002995503550811960007
- WenWHBernsteinLLescallettJComparison of TP53 Mutations Identified by Oligonucleotide Microarray and Conventional DNA Sequence AnalysisCancer Res2000602716272210825146
- BinderHKirstenTLoefflerMStadlerPThe sensitivity of microarray oligonucleotide probes – variability and the effect of base compositionJ Phys Chem B20041081800318014
- NaefFLimDAPatilNMagnascoMDNA hybridization to mismatched templates: a chip studyPhys Rev E20026540924096
- BinderHPreibischSSpecific and nonspecific hybridization of oligonucleotide probes on microarraysBiophys J20058933735215834006
- WuCHCartaRZhangLSequence dependence of cross-hybridization on short oligo microarraysNucleic Acid Res200533e8415914663
- HalperinABuhotAZhulinaEBSensitivity, specificity, and the hybridization isotherms of DNA ChipsBiophys J20048671873014747310
- LeeIDombkowskiAAAtheyBDGuidelines for incorporating non-perfectly matched oligonucleotides into target-specific hybridization probes for a DNA microarrayNucleic Acids Res20043268169014757833
- KaramanMWGroshenSLeeCCPikeBLHaciaJGComparison of substitutions, insertions and deletion probesNucleic Acids Res200533e3315722479
- SzymanskaKHainautPTP53 and mutations in human cancerActa B Pol200350231238
- MontesanoRHollsteinMHainautPGenetic alterations in esophageal cancer and their relevance to etiology and pathogenesis: a reviewInt J Cancer1996692252358682592