Abstract
Purpose:
Describing the ophthalmic findings of an exudative vasculopathy called as Coats-like retinitis pigmentosa on three patients. The etiology of the Coats-like retinitis pigmentosa is obscure. The principal theories have been discussed in this article.
Methods:
Three observational case series have been discussed. Complete ophthalmic examinations and color fundus photos, visual field, and fluorescein angiography have been performed.
Results:
We have identified 3 patients who have some typical clinical features of Coats-like retinitis pigmentosa; peripheral serous retinal detachment, telangiectasia, prominent lipid deposition, pigmentary changes in peripheral retina, and loss of vision. None of the three patients had positive family history. All of the patients have had symptoms of nyctalopia, decreased central vision, and two of them have had constriction of visual field. All of the patients have had cataracts and two of them underwent cataract surgery. Fundus examination and fluorescein angiography of patients revealed typical retinitis pigmentosa with Coats-type changes in bilateral inferiotemporal quadrants.
Conclusion:
A better understanding of clinical features and genetic etiology of Coats-type retinitis pigmentosa will aid diagnosis and development of new therapies. If sufficient conditions arise, genetic factors that influence the expression of CRB1 mutations in Coats-like retinitis pigmentosa should be detected.
Introduction
Retinitis pigmentosa (RP) is a heterogeneous group of disorders that causes degeneration of the photoreceptor cells mainly affecting the rods of the peripheral retina.
Nyctalopia or night blindness is the most common symptom of RP. The classic fundus appearance of RP includes retinal pigment epithelial (RPE) cell changes resulting in retinal hypo or hyperpigmentation, narrowed retinal vessels, pallor of the optic nerve head. Electroretinography demonstrates rod and cone photoreceptor cell disfunction and is a helpful test in the diagnosis and monitoring of patients with RP (CitationHartong et al 2006). The association between RP and exudative retinopathy was first described in 1956 (CitationZamorani 1956) and has been termed a “Coats-like RP” (CitationKhan et al 1988). It affects approximately 1%–4% of cases of RP (CitationKajiwara 1980; CitationPruett 1983).
We present an atypic form of RP in three cases that have bilateral exudative retinopathy diagnosed as Coats-like RP.
Case 1
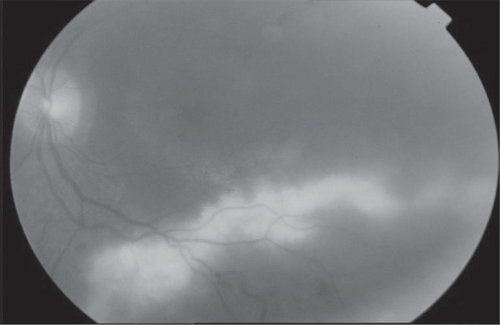
We considered a 35-year old female that presented progressive blurred vision in both eyes with a two years history as our first case. She has subsequently developed progressive nyctalopia, photophobia, and reduced peripheral vision. She did not have any illness in her family history. She had a history of cataract surgery in both eyes. Best corrected visual acuites were 0.2 in the right eye and 0.1 in the left eye. Anterior segment examination showed bilateral pseudophaci. Intraocular pressure values were normal. Fundus examination revealed extensive subretinal exudation, serious retinal detachment and overlying retinal telangiectasia and paled optic discs in both eyes (). Additionally, the mid – periphery of both eyes showed RPE atrophy and bone spicule pigmentation with retinal arteriolar narrowing (). Visual field testing revealed marked constriction in both eyes.
Figure 1 Extensive subretinal exudation, serous retinal detachment, and overlying retinal telangiectasia.

Figure 2 RPE atrophy and bone spicule pigmentation with retinal arteriolar narrowing.
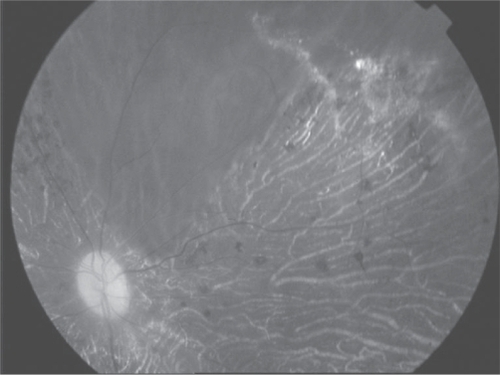
Fluorescein angiogram (FA) showed retinal telangiectasia and serous retinal detachment (). We offered treating the telangiectatic lesions in patient’s both eyes with argon laser photocoagulation but she refused laser photocoagulation treatment.
Figure 3 Fluorescein angiogram (FA) showed retinal telangiectasia and serous retinal detachment.
Case 2
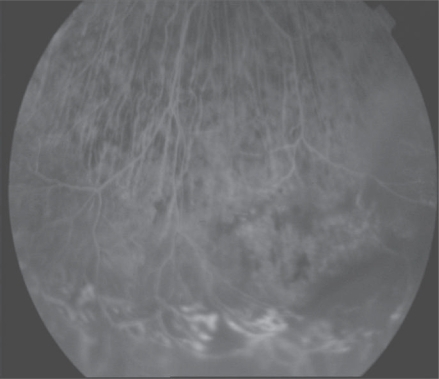
We considered a 26-year old female that presented a progressive blurred vision for 3 years as our second case. She underwent cataract surgery in both eyes. She has also developed progressive nyctalopia also. She did not have any illness in her family history. Best corrected visual acuites were 0.4 in the right eye and 4 counting fingers in the left eye. Anterior segment examination showed bilateral pseudophaci. Intraocular pressure values were normal. Visual field testing showed bilateral constriction in both eyes. Fundus examination revealed waxy pallor of the optic disks, attenuated retinal arterioles, mild paramacular pigmentary changes at the retinal pigment epithelium level, and peripheral bone spicules in both eyes. The inferiotemporal region of her retina showed bilateral massive subretinal exudation with bone spicule pigmentation (). FA showed retinal telangiectasia, serous retinal detachment on inferiotemporal region and pigment epithelial window defects in the macula. Telangiectatic lesions were treated with argon laser photocoagulation. Then she discontinued her regular control visits and treatment and she was dropped from follow-up.
Figure 4 Massive subretinal exudation with bone spicule pigmentation in the inferiotemporal region.
Case 3
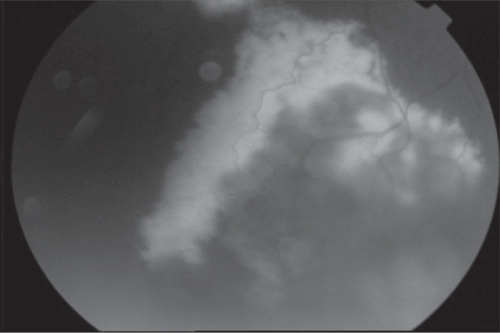
We considered a 26-year old male with congenital nystagmus that presented a one year history of reduced visual acuities in both eyes as our third case. He did not have any illness in his family history and there was no significant medical history. Best corrected visual acuites were 4 counting fingers in the right eye and 3 counting fingers in the left eye. Anterior segment examination showed bilateral mild posterior subcapsuler cataract formation. He had congenital pandular nystagmus. Intraocular pressure values were normal. He had developed progressive nyctalopia. Fundus examination showed bilateral pigmentary changes accompanied by subretinal exudation and exudative retinal detachment in the right eye () and telangiectatic retinal vessels located on inferotemporal region in the left eye. He had bilateral mild macular pigment epithelial changes. Additionally, mottled granularity of the RPE was noted in the mid-periphery of both retinas. FA showed irregular, tortuous and telangiectatic retinal vessels with leakage of the dye (Figure 6). Visual field examination could not be performed because of poor visual acuity and nystagmus. Coats-like RP were diagnosed and telangiectatic lesions were treated with argon laser photocoagulation, nevertheless the retinal telangiectasic lesions could not be resolved, the exudation was slightly less extensive but the visual acuities remained stable even after months.
Figure 5 Pigmentary changes accompanied by subretinal exudation and exudative retinal detachment.
Discussion
Retinitis pigmentosa is an inherited retinal dystrophy caused by the loss of photoreceptors and characterized as retinal pigment deposits on fundus examination. Prevalence of RP is approximately 1/4,000. The most common form of RP is rod-cone dystrophy, in which the first symptom is night blindness, followed by the progressive loss in the peripheral visual field in daylight, and leading to blindness after several decades (CitationHamel 2006). Although RP is usually presented as a nonsyndromic disorder, many syndromic forms may accompany RP; the most common is Usher syndrome. 45 causative genes/loci have been identified in nonsyndromic RP (for the autosomal dominant, autosomal recessive, X-linked, and digenic forms). Molecular diagnosis can be made for some genes; therefore genetic counseling is advised to lay off the underlying syndrome. Currently, there is no therapy that stops or slows down the progression of the disease or restores the vision thus, visual prognosis is poor. The therapeutic approach is restricted to slowing down the degenerative process caused by sunlight protection and vitaminotherapy, treating the complications (cataract and macular edema), and helping patients to survive with the social and psychological impact of blindness. However, new therapeutic strategies are emerging from intensive research (gene therapy, neuroprotection, retinal prosthesis) (CitationBerson et al 1993; CitationWang et al 1997).
Coats-like RP is an atypic form of RP and various studies have suggested that 1%–4% of RP cases will show such a response (CitationKajiwara 1980; CitationPruett 1983). This entity can develop in later stages of the disease and is characterized by vascular abnormalities (aneurysmal dilations and telangiectatic retinal veins), yellow extravascular lipid depositions, and retinal detachment, however it differs from classic Coats disease with age (older age), gender (no sex profile), involvement of eye (generally bilateral), progression (more progression), retinal location (inferior quadrans) and shows diffuse pigmented alterations in both fundi (CitationHamel 2006; CitationHartong et al 2006). Lesions are usually found in the inferior and/or temporal quadrants with typical dilated, aneurysmal or telangiectatic veins (CitationHartong et al 2006). Although the age of diagnosis is double that of Coats disease, Coats-type changes were reported as early as age 4-years (CitationKim and Kearney 1997). Patients with RP who develop Coats-type changes show a wide spectrum of disorders, ranging from mild visual difficulties or nyctalopia, as observed in classical RP, to the other extreme in which a proliferative vasculopathy as observed classic Coats disease. The cause is unknown but there are several theories to explain Coats-like RP; one is a vasodilatory response to toxic products of photoreceptor/RPE degeneration. The second one is Coats-type retinopathy may result from chronic microvascular leakage followed by secondary inferior retinal detachment with subsequent retinal hypoxia because of separation from the choroid and later development of telangiectatic abnormalities (CitationPruett 1983).
It has been suggested that genetic factors may be involved in RP with Coats-type exudative vasculopathy (CitationPruett 1983). In a current study, it has been hypothesized that mutations in the human homologue of Drosophila crumbs gene (CRB1) cause a specific form of RP and has been associated with some cases of Coats-type RP (CitationHollander et al 2001). CRB1 is preferentially expressed in the retina, but its function is largely unknown. Development of Coats-type exudative vasculopathy in patients with RP were strongly associated with CRB1 mutations so that CRB1 mutations should be considered as an important risk factor for the Coats-type reaction, and RP patients with CRB1 mutations should be checked regularly for the Coats-type complication in RP (CitationHollander et al 2001). We could not perform genetic study in our patients because of insufficent technical conditions.
Current treatment options include cryotherapy, scleral buckling with subretinal fluid drainage and laser photocoagulation in the telangiectatic and neovascular lesions to induce regression of neovascular tissue and to manage fibrovascular proliferation-induced retinal detachment (CitationHartong et al 2006). Heightened awareness of this potentially treatable complication of RP may facilitate earlier diagnosis of the problem, which may lead to more timely treatment and a better prognosis. That’s why detailed fundus examination should be performed in RP patients not to miss out any exudative and telangiectatic lesions in the periphery of the retina and Coats-like RP should be diagnosed for an earlier treatment that influences the prognosis. Further investigations are needed to determine and to understand the characteristic of Coats-type RP.
References
- BersonELRosnerBSandbergMA1993Vitamin A supplementation for retinitis pigmentosaArch Ophthalmol1111456668240091
- HamelC2006Retinitis pigmentosaOrphanet J Rare Dis14017032466
- HartongDTBersonELDryjaTP2006Retinitis pigmentosaLancet368179580917113430
- HollanderAHeckenlivelyJBornI2001Congenital amaurosis and retinitis pigmentosa with Coat’s-type exudative vasculopathy are associated with mutations in the Crumbs homologue 1 (CRB1) geneAm J Hum Genet6919820311389483
- KajiwaraY1980Ocular complications of retinitis pigmentosa. Association with Coat’s syndromeJpn J Clin Ophthalmol3494755
- KhanJAIdeCHStricklandMP1988Coat’s-type retinitis pigmentosaSurv Ophthalmol32317322457260
- KimRYKearneyJJ1997Coat’s-type retinitis pigmentosa in a 4-year-old childAm J Ophthalmol12484689402836
- PruettRC1983Retinitis pigmentosa: clinical observations and correlationsTrans Am Ophthalmol Soc816937356676982
- WangMLamTTTsoMOM1997Expression of a mutant opsin gene increases the susceptibility of the retina to light damageVis Neurosci1455629057268
- ZamoraniG1956Una rara associazone di retinite di Coat’s con retinite pigmentosaGior Ital Oftalmol942943