Abstract
Wegener’s granulomatosis (WG) is a necrotizing granulomatous vasculitis characterized by the involvement of the upper or lower airways, lungs, and kidneys, but it can affect almost any organ including the orbit. WG is rare in childhood. This case report describes a 7-year-old girl who presented bilateral idiophatic orbital inflammation and antineutrophil cytoplasmic antibodies-negative titres. Computed tomography scan and magnetic resonance imaging showed enlargement of both lacrimal glands with infiltration. Treatment with corticosteroids achieved remission of the disease. Three years later, she developed a systemic affectation with tracheal stenosis, pulmonary affectation, renal failure, and respiratory tract mucosa inflammation. Lacrimal gland biopsy showed perivascular nonspecific granulomas and ANCA titres remain negative. Treatment with corticosteroids and cyclophosphamide was done. A relapse occurred 2 years later, with complete remission with antitumor necrosis factor-alpha. No other symptoms have appeared after 9 years of follow-up. Early diagnosis and treatment is crucial to increase the survival rate in these patients.
Introduction
Wegener’s granulomatosis (WG) is a chronic inflammatory autoimmune disease of unknown etiology. It is a multisystem necrotizing vasculitis that primarily involves the upper and lower respiratory tract and kidneys but it can affect almost any organ, including the orbit and eye. The involvement of the upper airway is the most common symptom though kidney involvement can be the first one. WG in children is a rare disease.
Only one case has been described previously with cytoplasmic antineutrophil cytoplasmic antibodies (ANCA)-negative titres in the orbit in a child; it was a case of idiophatic orbital inflammation (IOI), also known as orbital pseudotumor (CitationWardyn et al 2003). However, a few cases in childhood with positive ANCA (CitationZiakas et al 2004) have been described.
Case report
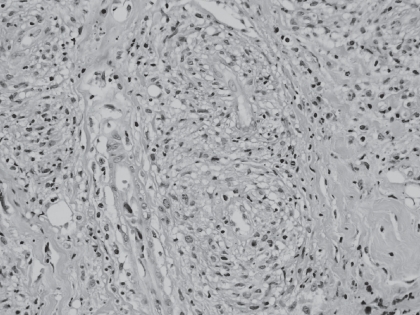
A 7-year-old girl presented with a acute mild proptosis of the left eye pain, along with downwards and medial displacement of the globe and eyelid swelling (), but no diplopia or visual loss. The patient also showed chronic nonspecific constitutional symptoms such as weight loss and height below normal limits. Family history included mother with rheumatoid arthritis. In a few weeks the clinical signs progressed to both orbits. Orbital computed tomography scan () and magnetic resonance imaging showed infiltration and enlargement of both lacrimal glands and sinusitis. Chest X-rays were normal. Immunologic studies showed negative cytoplasmic ANCA and perinuclear ANCA; immunofluorescence and specifities were confirmed by antiproteinase 3, and antimyeloperoxidase. Antinuclear antibodies were positive to a 1/160 dilution (1/20–1/40). C-reactive protein was 0.3 mg/dL (0.0–0.8), rheumatoid factor <20 IU/mL (0–30), erythrocyte sedimentation rate 18 mm/h (0–20), and creatinine clearance was 95 mL/min/1.73 m2 (88–128). Urinanalysis showed normal results. Incisional biopsy of the lacrimal gland showed a nonspecific lymphoplasmacytic infiltrate consistent with IOI. Immunohistochemical studies showed positive CCA, MAC387, lisozyme, and CD68. Special stains for micro-organisms such as fungi and acifast bacteria were negative. Hepatitis B and C infections and human immunodeficiency virus tests were negative. Treatment with corticosteroids, prednisone 1 mg/kg/day for 3 weeks orally and then a tapered dosage to zero over 12 weeks, obtained remission of the disease with disappearance of the orbital symptoms. After 3 years without signs or symptoms, she presented again with the previous ocular symptoms, but also subglottic tracheal stenosis and bilateral basal pulmonary infiltrates, as well as decreased renal function (47 mL/min/1.73 m2), and erithrocyte sedimentation rate of 77. Incisional biopsies of the lacrimal gland, nasal and oral mucosa showed non-specific histiocytic infiltrate with a large compound of plasmacytoid, lymphocytes and eosinophilic cells without anaplasia. The acinar compound of the lacrimal gland was destroyed and the cell infiltrate was located in a perivascular and periductal disposition, with a granulomatous aspect (). The ANCA titres remained within normal limits. At that moment WG was established as a diagnostic, and treatment with oral prednisone 1mg/kg/day, intravenous cyclophosphamide 0.7 gr/m2/month/12 months, 0.5 gr/m2/month/2 months induced remission of the disease. Two years later the patient presented a relapse with new orbital IOI, tracheal stenosis and pulmonary infiltrates, and the treatment applied was an antitumor necrosis factor-alpha (TNF-α), infliximab 5 mg/kg six infusions for 34 weeks, which stabilized the clinical course. After a follow-up of 8 years, the patient is free from any sign of the disease.
Figure 1 A Patient at presentation showed eyelid swelling with downward and medial displacement of the eye on the left side. B Coronal magnetic resonance imaging showed enlarged lacrimal glands.
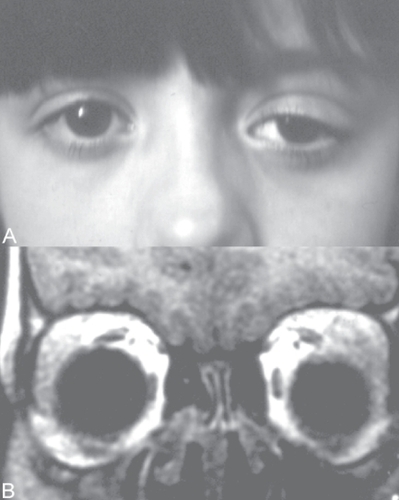
Figure 2 Histopathological examination of lacrimal gland: lymphoplasmacytoid infiltrate with perivascular granulomas (hematoxylineosin, 40×).
Discussion
WG is a systemic granulomatous vasculitis that typically affects the upper airways, lungs, and kidneys. This form of vasculitis is extremely rare in children, the most common form of pediatric systemic vasculitis being Henoch-Schonlein purpura. In the two published case series, the disease was more frequent in females than in males with a proportion (3:1) (CitationReinhold-Keller et al 2001; CitationFechner et al 2002). Ophthalmic involvement is relatively common in the course of the disease and may in fact be the initial presenting feature in some patients. It is therefore imperative to considerer WG in the differential diagnosis when more common disorders are ruled out.
In these patients it is important to rule out the existence of hepatitis B and hepatitis C infections, human immunodefiency virus, immunodeficiency disorders, and malignant diseases, which can produce similar manifestations. Nowadays there are data that suggest a possible toxic exposure, which should be a potentially correctable risk factor (CitationAlbert et al 2005). WG is commonly ANCA-positive but some cases may be ANCA-negative (CitationLangford 1998). WG in the systemic form is commonly cytoplasmic ANCA-positive (95%), while the localized form is usually negative to cytoplasmic ANCA and highly positive to perinuclear ANCA. WG with ANCA has led to its designation as ANCA-associated vasculitis. However, there are patients without ANCA whose clinical and pathological features meet both the American College of Rheumatology 1990 (CitationLeavitt et al 1990) classification criteria for WG and the 1993 (CitationJennette et al 1994) Chapel Hill Consensus Conference on the Nomenclature of Systemic Vasculitis definition of WG. Only 14 cases of active generalized WG without ANCA have been described in the English literature and one in a girl presenting with episcleritis. It is remarkable that most of these patients had cerebral and/or meningeal involvement, but none presented orbital involvement (CitationReinhold-Keller et al 2001).
Histological evaluation usually shows an acute and chronic inflammation, necrotizing vasculitis, poorly formed granulomas, microabscesses, and fibrinoid degeneration of collagen. Nevertheless, the presence of these findings can vary greatly, and small biopsies, especially from the head and neck region, often lack all of them. Therefore, the diagnosis of WG must rest on the finding of a constellation of histological and clinical features (CitationPakrou et al 2006).
The disease is usually fatal if untreated, although WG limited to the orbit and adnexal structures without systemic involvement have a good overall prognosis. The prevention of relapses and treatment of refractory cases remain the greatest challenges in the treatment of WG (CitationHellmich et al 2006). Open-label clinical studies suggest a beneficial effect of anti-TNF-α, infliximab, in addition to standard therapy in refractory WG. In contrast, a recent randomized controlled trial showed an increased risk of serious infections and a dose-dependent increased anti-TNF antibody therapy, with higher dosages leading to more remissions (CitationWGET 2005; CitationHellmich et al 2006; CitationBongartz et al 2006). The combination of TNF inhibition and cyclophosphamide may heighten the risk of cancer beyond that observed with cyclosphophamide alone; six cases of solid cancer have been identified with the use of etanercept (CitationStone et al 2006) and the development of a lymphoma with infliximab (CitationBooth et al 2004). Infliximab represents a new therapeutic option and offers better perspectives for a patient group with a previously bad prognosis (CitationBartolucci et al 2002). However, the current data are conflicting and difficult to interpret. As a result, the newer agents cannot be recommended for routine use until a vigorous clinical study confirms their efficacy (CitationWhite and Lynch 2006). While early diagnosis and treatment with immunosuppressives such as cyclophosphamide can induce long term remission, there are also serious toxicities associated (CitationFauci and Wolff 1973). Treatment for maintenance of remission is mandatory.
Our case consisted in a bilateral IOI of the lacrimal gland. WG must be considered in this condition, but the absence of systemic involvement or necrotizing granulomas in the biopsy and the negativity of ANCA made confirmation of the diagnosis impossible. Futhermore, WG rarely affects children and most of the published cases were ANCA-positive. Although there may be ANCA-negative cases in children, the diagnosis is difficult if the histological features are not specific to the disease.
We also point out the lack of aggressiveness of this case, despite its late diagnosis three years after the initial manifestation, and the good response to infliximab, with no relapses after 8 years of follow-up.
Disclosure
The authors do not have any possible financial conflict or interest in this work.
References
- AlbertDAAlbertANVernaceM2005Analysis of a cluster of cases of Wegener granulomatosisJ Clin Rheumatol111889316357755
- BartolucciPRamanoelinaJCohenP2002Efficacy of the anti-TNF-α antibody infliximab against refractory systemic vasculitidies: an open pilot study on 10 patientsRheumatology (Oxford)4111263212364631
- BongartzTSuttonAJSweetingMJ2006Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trialsJAMA29522758516705109
- BoothAHarperLHammadT2004Prospective study of TNFalpha blockade with infliximab in anti-neutrophil cytoplasmic antibody-associated systemic vasculitisJ Am Soc Nephrol157172114978174
- FauciASWolffSM1973Wegener’s granulomatosis: studies in eighteen patients and a review of the literatureMedicine (Baltimore)52535614748591
- FechnerFPFaquinWCPilchBZ2002Wegener’s granulomatosis of the orbit: a clinicopathological study of 15 patientsLaryngoscope11219455012439160
- HellmichBLamprechtPGrossWL2006Advances in the therapy of Wegener’s granulomatosisCurr Opin Rheumatol18253216344616
- JennetteJCFalkRJAndrassyK1994Nomenclature of systemic vasculitides. Proposal of an international consensus conferenceArthritis Rheum37187928129773
- LangfordCA1998The diagnostic utility of c-ANCA in Wegener’s granulomatosisCleve Clin J Med65135409540246
- LeavittRYFauciASBlochDA1990The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosisArthritis Rheum33110172202308
- PakrouNSelvaDLeibovitchI2006Wegener’s granulomatosis: ophthalmic manifestations and managementSemin Arthritis Rheuma3528492
- Reinhold-KellerEde GrootKHoll-UlrichK2001Severe CNS manifestations as the clinical hallmark in generalized Wegener’s granulomatosis consistently negative for antineutrophil cytoplasmic antibodies (ANCA). A report of 3 cases and review of the literatureClin Exp Rheumatol19541911579713
- StoneJHHolbrookJTMarriotMA2006Solid malignancies among patients in the Wegener’s Granulomatosis Etanercept trialArthritis Rheuma54160818
- WardynKAYcinskaKMatuszkiewicz-RowinskaJ2003Pseudotumour orbitae as the initial manifestation in Wegener’s granulomatosis in a 7-year-old girlClin Rheumatol22472414677032
- [WGET] Wegener’s Granulomatosis Etanercept Trial Research Group2005Etanercept plus standard therapy for Wegener’s granulomatosisN Engl J Med3523516115673801
- WhiteESLynchJP2006Pharmacological therapy for Wegener’s granulomatosisDrugs6612092816827598
- ZiakasNGBoboridisKGratsonidisA2004Wegener’s granulomatosis of the orbit in a 5-year-old childEye186586014716328