Abstract
To examine rhodopsin (Rho) functions in P23H rat, kinetics of Rho regeneration and dephosphorylation were investigated by spectrophotometric analysis and immunofluorescence labeling method using specific antibodies toward phosphorylated 334Ser or 338Ser site. Rho dephosphorylation at both sites was extremely delayed in P23H retina as compared to normal ones. Kinetics of Rho regeneration was not altered between normal and P23H rats under dark adaptation. Next, to study the effects of several Ca2+channel blockers on this model, retinal function and morphology were evaluated. Among them, nilvadipine showed a significant protective effect against P23H retinal degeneration. Neurotrophic factor, fibroblast growth factor-2 and Arc, known to suppress the apoptosis in the central nervous system, were significantly upregulated upon administration of nilvadipine. The present study indicates that misregulation of Rho phosphorylation may be involved as an important step in retinal degeneration of P23H and administration of nilvadipine may be a potential therapeutic agent for the retinal degenerations.
Introduction
Retinitis pigmentosa (RP), a progressive hereditary retinal degeneration, is caused by the result of various genetic mutations (Citationvan Soest et al 1999). Among the most common are mutations in the rhodopsin (Rho) gene, including P23H (CitationDryja 1992). The P23H Rho mutation causes autosomal dominant (ad) retinal degeneration (CitationBerson et al 1991). Retinal dysfunction due to abnormal disc morphology is involved in the molecular pathology causing the retinal degeneration in this mutation (CitationLiu et al 1997). Impaired activation of the phototransduction cascade and recovery from activation are observed in human adRP patients with P23H mutation (CitationBirch et al 1995), and the etiology of these mutations were studied expensively using animal models. P23H mice experiments demonstrated delayed photoresponse recovery by double-flash measurements (CitationGoto et al 1996). Thus, similarly to human adRP patients with P23H mutation, recovery from activation is also slow in the P23H mouse model. These observations suggest that the P23H model animal may be functionally impaired in the recovery state of biochemical reactions such as kinetics of Rho phosphorylation and dephosphorylation, binding of arrestin, and activities of guanylate cyclase and cGMP phosphodiesterase. Interestingly, lower levels of mRNA expressions of α-A crystalline and Rho kinase (RK), which are involved in post-Golgi processing of opsin and Rho phosphorylation, respectively, in Royal College Surgeons (RCS) rats (Maeda et al 2002) in which the retinal pigment epithelium (RPE) cell is affected (CitationMullen and LaVail 1976) by a mutation in the gene encoding the receptor tyrosine kinase Mertk (CitationD’Cruz et al 2000; CitationGal et al 2000). However, expressions of other photoreceptor cell specific proteins including Rho, transducin, arrestin and recoverin were almost comparable between RCS and control rats (Maeda et al 2002). Recently, we demonstrated that dephosphorylation of Rho were extremely delayed in RCS rat retinas during the dark adaptation by a newly developed method to analyze in vivo Rho phosphorylation employing imunohistochemistry with specific antibodies toward phosphorylated Rho at specific sites (CitationOhguro et al 2003). Similarly, we also found significant high levels of Rho phosphorylation in light-induced stress rat retinas (CitationIshikawa et al 2006) and cancer-associated retinopathy (CAR) model (CitationOhguro et al 2001). Therefore taken together, we hypothesized that abnormal kinetics of Rho phosphorylation and dephosphorylation may contribute to persistent misregulation of phototransduction processes in retinal degeneration. If this is the case, photosensitive Rho levels may be insufficient in comparison to deactivated forms of Rho, photolyzed Rho and phosphorylated Rho, and to normalize this imbalance might be a candidate therapy for retinal degeneration. As regards this therapy, we speculated a possible mechanism; modulation of Rho phosphorylation and dephosphorylation kinetics by Ca2+.
Ca2+ ions may play a significant role in the cell death by apoptosis, and is a critical factor regulating the recovery of the photoresponse (CitationNicotera and Orrenius 1992). Delayed recovery could therefore result from abnormal Ca2+ ion movement or abnormal levels within cytosol of the photoreceptor cells (CitationKoch and Stryer 1988). In fact, Rho phosphorylation by RK is exclusively regulated in a Ca2+-dependent manner by a photoreceptor specific Ca2+-binding protein called recoverin (CitationKawamura 1991). Therefore, it is plausible that suppression of recoverin-dependent inhibition of RK by the lowering of intracellular Ca2+ levels by some drugs may be efficient for the preservation of photoreceptor cells in retinal degeneration. Indeed, some types of Ca2+ channel blockers have been identified to have preservation effects against retinal degeneration models (CitationFrasson et al 1999; CitationYamazaki et al 2002). As described above, if the kinetics of Rho phosphorylation and dephosphorylation were impaired and photosensitive Rho levels were really insufficient in comparison to phosphorylated Rho in P23H rat model, it would be of great interest to test the effects of Ca2+ channel blockers for therapy.
Therefore, in the present study, to gain new insights into the mechanism of retinal degeneration in P23H rats, photoreceptor functions including Rho regeneration, and Rho phosphorylation and dephosphorylation were systematically studied and the effects of several Ca2+ channel blockers were also investigated.
Materials and methods
All experimental procedures were designed to conform to both the ARVO statement for Use of Animals in Ophthalmic and Vision Research and our own institution’s guidelines.
Animals
Homozygous breeders of transgenic rats carrying Rho mutation of P23H (line 2, albino) were kindly provided by Prof. Matthew LaVail (University of California, San Francisco, CA). Heterozygous P23H rats were produced by mating homozygous breeders with wild-type Sprague Dawley (SD) rats. In the present study, 4- to 8-week-old SD rats (Crea, Tokyo, Japan) and heterozygous P23H rats reared in cyclic light conditions (12 h on/12 h off) were used.
Electroretinogram measurement
Details of preparation, recording technique and measurements of electroretinograms (ERG) have been described elsewhere (CitationMachida et al 2000). Under anesthesia, the rats were laid on its side with its head fixed in place with surgical tape in an electrically shielded room for overnight dark-adaptation. The pupils were dilated with drops of 0.5 % tropicamide. ERGs were recorded with a contact electrode (Kyoto Contact Lens Co., Kyoto, Japan). A grounding electrode was placed on the ear. Responses evoked by white flashes (3.50 × 102 lx, 200 ms duration) using a Ganzfeld dome (SG-2002, Meiyo Co., Japan) were recorded and analyzed with PowerLab Scope version 3.7 (ADI instruments Ltd., Japan).
Light and immunofluorescence microscopy
Enucleated eyes were fixed with methacarn (60% methanol, 30% chloroform, 10% glacial acetic acid) overnight, dehydrated, and embedded in paraffin. Posterior segments of eyes cut from the enucleated eyes were embedded in paraffin. Retinal sections were cut vertically through the optic disc at 2-μm thickness, mounted on subbed slides and dried. The sections were processed with hematoxylineosin staining after deparaffinization. For evaluation of photoreceptor cell survival, the sections including the full length of the retina from optic nerve head through the ora serrata were taken photo and rows of cell nuclei in outer nuclear layer (ONL) were counted at 200-μm intervals along whole horizontal retinal axis.
For immunofluorescence labeling, deparaffinized sections were blocked with phosphate-buffered saline (PBS) containing 3% bovine serum albumin for an hour and then incubated overnight with anti-P-Rho334 antibody, anti-P-Rho338 antibody (1:500), anti-Arc antibody, or anti-FGF2 (1:200) at 4 °C. Sections were washed and incubated with fluorescein-isothiocyanate (FITC) or Cy3-conjugated antibodies to rabbit IgG (Cappel, Durham, NC) for an hour at room temperature. Specificity controls were obtained by omitting the primary antibodies. Sections were observed by a fluorescence microscope (Olympus, model BH-2, Tokyo, Japan) using suitable filters.
Drug administration
Nilvadipine and nifedipine were dissolved in a mixture of ethanol: polyethylene glycol 400:distilled water (2:1:7) at a concentration of 0.1 mg/ml, diluted twice with physiological saline before use, and injected intraperitoneally (1 ml/kg) into anesthetized rats daily for 4 weeks. Nicardipine and D-cis-diltiazem were dissolved in PBS at 0.25 mg/ml and 1 mg/ml, respectively and injected intraperitoneally (1 ml/kg) as above. As control for them, each vehicle solution was administrated as above. The concentrations of these drugs administrated to rats were determined by their concentrations of systemic oral administrations to human patients with hypertension for one day in our clinical practice (CitationTriggle 1981).
Regenerated Rho concentration
Rho regeneration was determined by photospectrometric analysis as described previously (CitationOhguro et al 1995). Briefly, rats were exposed by illumination condition (650 lux) for 6 h and then subjected to dark adaptation. At different times of the dark adaptation, rats were euthanized and eyes were enucleated and cut into two, anterior and posterior segments. The posterior segments were homogenized with 10 mM Hepes buffer, pH 7.5, containing 10 mM dodesyl β-maltoside and 20 mM hydroxylamine by a glass/glass homogenizer. The sample was centrifuged at 20,000 × g and the spectra were recorded before and after complete bleaching. Rho concentrations were determined from difference in A500 before and after bleaching as above by assuming ɛ500 to be 41,000 and the molecular weight to be 38,000.
Results
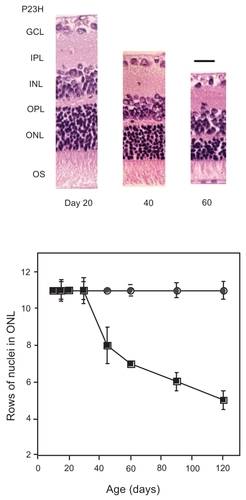
As shown in , retinal degenerative P23H rat strains showed age-dependent thinning of retinal outer layers as described previously (CitationMachida et al 2000), whereas there was no significant difference in the morphology between control rats at Day 20. In the present study, to get an insight into which steps in the phototransduction cascade were affected in this strain, selected biochemical reactions in photoreceptor cells were evaluated, including Rho phosphorylation and dephosphorylation, and Rho regeneration rates that represent the critical step of quenching the photoexcitation, and recovery phase after photoexcitation, respectively.
Figure 1 Retinal function and morphology in P23H rats during their development. Changes in retinal morphology and function by ERG of P23H rat are demonstrated. Hematoxyline-eosine staining of retinal sections at 1 mm from optic disc of P23H rat eyes during different ages were photographed (A), and rows of ONL nuclei were analyzed (B) as described in Materials and methods.
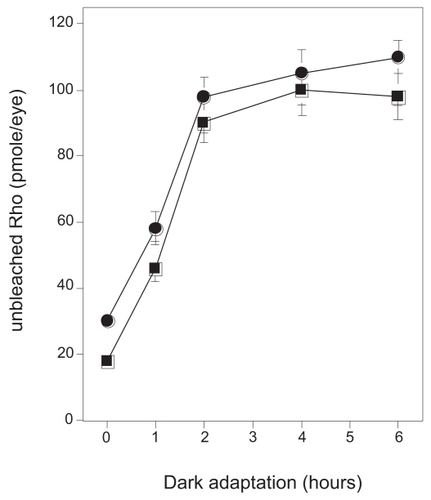
To assess Rho function, regeneration rate after bleach with constant light (650 lux) was firstly examined with P23H rats at the age of Day 20 without evidence of morphological retinal degeneration. Prior bleaching, the amount of Rho was compared and no significant difference was observed between different genetic back grounds. When rats were exposed to constant light for 6 h and then kept in the dark, Rho was completely regenerated in 2 h in both animals employed in the study. Rates of Rho regeneration of P23H strains during dark adaptation from light bleach conditions were almost identical with normal strain, suggesting Rho regeneration was not affected ().
Figure 2 Time course of Rho regeneration during dark adaptation. Control or P23H rats (Day 20) were dark-adapted from regular room light (650 lux). At several time points, 0, 1, 2, 4, and 6 h, rats were euthanized and enucleated eyes were processed to direct Rho concentration analysis as described in the Materials and Methods. For one analysis two eyes from one rat were used. Experiments were performed in triplicate using fresh preparations.
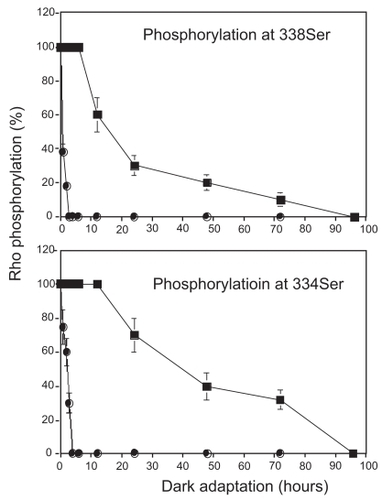
Next, to evaluate quenching function of photoreceptor, Rho phosphorylation and dephosphorylation were assessed by immunohistochemical technique using specific antibodies against phosphorylated 334Ser or 338Ser, both of which have been identified as major sites of phosphorylation in Rho in vivo (CitationOhguro et al 1995). In the control SD rat retina, 334Ser and 338Ser antibody specifically recognized rod outer segments (ROS) of light-adapted but not of dark-adapted retina, and its immunopositivities were then gradually diminished from base to tip of ROS following dark adaptation as described previously (CitationOhguro et al 2003) (data not shown). The kinetics of dephosphorylation of phosphorylated 334Ser and 338Ser were determined by measuring the vertical lengths of immunofluorescence labeled ROS during the dark adaptation. Dephosphorylation of 334Ser and 338 Ser went to completion within 4 h in SD rats. In contrast, in P23H rat dephosphorylation of these sites was extremely prolonged ().
Figure 3 Kinetics of dephosphorylation in phosphorylated 334Ser and 338Ser in control and P23H rats. Four-week-old control and P23H rats were exposed to regular room light (650 lux) for 6 h and then were maintained under dark condition. Rho phosphorylation level was evaluated at three different time points (0, 1, 2, 3, 4, 6, 12, 24, 48, 72, and 96 h, n = 3 per group), with immunofluorescence labeling by anti-P-Rho antibodies. Vertical length of photoreceptor outer segment layers and that of fluorescence labeling was measured at temporal points 1.0 mm apart from optic disc from 6 different points from 3 different eyeballs and their ratios were plotted.
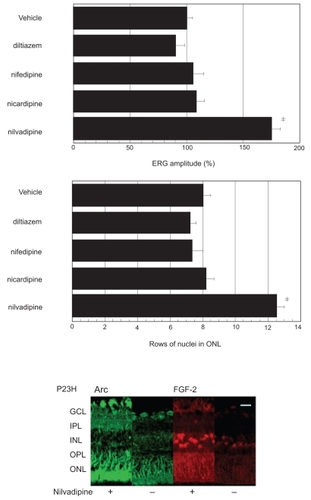
We next studied the effects of several Ca2+ channel blockers, which are candidates as drugs beneficial for retinal degeneration based upon previous studies (CitationFrasson et al 1999; CitationOhguro et al 2001; CitationYamazaki et al 2002; CitationIshikawa et al 2006). Four different Ca2+ channel blockers, D-cis-diltiazem, nifedipine, nicardipine, and nilvadipine, which are used in clinical practice, and their vehicle solutions were systemically administrated to 4-week-old P23H rats daily for four weeks (n = 5 rats, 10 eyes in each condition), and then the retinal function and morphology were compared among these drugs. As shown in , there was no significant difference in rats treated with D-cis-diltiazem, nifedipine or nicardipine and their control rats; however, significant preservation effects in retinal function by ERG and morphology (upper panel) were observed in P23H rats administrated with nilvadipine. To estimate the nilvadipine-dependent preservation effects against P23H retinal degenerations, retinal expressions of FGF2 and Arc, which were significantly enhanced in nilvadipine-treated RCS rat as compared to the control (CitationSato et al 2003), were immunohistochemically investigated. As shown in , lower panel, marked enhancement of the immunoreactivities of FGF2 and Arc was observed in nilvadipine-treated P23H rats in contrast to their controls.
Figure 4 Effects of several Ca2+ channel blockers on retinal function and morphology in P23H rats. P23H rat were treated with Ca2+ channel blockers, D-cis-diltiazem, nifedipine, nicaridipine nilvadipine or their vehicle solutions and thereafter retinal function by ERG and morphological analysis (upper panel) were performed. ERG measurements were performed in 10 eyes (5 rats) in each condition, and b-wave amplitudes of drug treated rats were compared with those of vehicle treated rats. Hematoxyline-eosine stained retinal sections at 1mm from optic disc of P23H rat eyes treated as above were photographed, and rows of ONL nuclei were analyzed as described in Materials and methods.
Abbreviations: GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer, OPL, outer plexiform layer; ONL, outer nuclear layer; OS, outer segment.
Discussion
It has been hypothesized that quenching mechanisms during the phototransduction pathway are functionally affected in RCS rat, based upon the experimental evidence of several alterations in expressions of opsin (CitationNir et al 1987), and ROS protein phosphorylation levels (CitationHeth and Schmidt 1992). Recently, to test this hypothesis, we developed specific antibodies toward Rho phosphorylated at 334Ser or 338Ser sites (CitationOhguro et al 2003), major phosphorylation sites in Rho in vivo (CitationOhguro et al 1995), and studied the kinetics of dephosphorylation of phosphorylated photolyzed Rho in living retina. During dark adaptation of normal control rats, dephosphorylated at 338Ser and 334Ser sites were completed within several hours (0–4 h). In contrast, the same immunoreactivities directed toward phosphorylated-338Ser and -334Ser sites resulted in a diminution at between 4 to 7 days in RCS rat retinas under dark adaptation (CitationOhguro et al 2003). Such prolonged kinetics of the Rho dephosphorylation during dark adaptation was also evident in light-stress-induced retinal degeneration (CitationIshikawa et al 2006), and significant enhancement of the phosphorylated form of Rho as well in a model of cancer-associated retinopathy (CAR), which is produced by intravitreous administration of anti-recoverin antibody to rats (CitationOhguro et al 2001). In addition, P23H rats in this study showed changes in Rho dephosphorylation kinetics during phototransduction cascade reactions. Consideration of the above suggests that deficits in regulation of phototransduction cascade by Rho phosphorylation and dephosphorylation is presumably a common mechanism responsible for most retinal photoreceptor degeneration. In fact, this hypothesis is supported by the previous observation that retinitis pigmentosa patients (CitationBirch et al 1995) and mouse model (CitationGoto et al 1996) with Rho mutations already known to show delayed photo-response recovery, suggesting that the quenching and adaptation process by Rho phosphorylation is impaired, in addition to a suggested disease mechanism of mislocalization of Rho within the photoreceptor cells (CitationGreen et al 2000). The status of Rho phosphorylation and dephosphorylation of the P23H rats after nilvadipine administration is very critical issue to prove our speculation that misregulation of Rho phosphorylation and dephosphorylation kinetics is the mechanism causing retinal degeneration and this may be normalized by administration of the suitable Ca channel blocker. Nevertheless, analysis of Rho dephosphorylation is very difficult at present because the retinal degeneration is also progressive during the drug administration period (4 weeks) and degenerative outer segments is not suitable for the proper analysis using immunohistochemical assay as above. Thus, we have to develop a more precise and reliable assay system for evaluating Rho phosphorylation and dephosphorylation at real-time in vivo. Therefore this is our next project.
Since the reactions responsible for quenching and adaptation of the phototransduction are Ca2+-dependent (CitationKoch and Stryer 1988), it is reasonable to speculate that Ca2+ channel blockers may have a potentially beneficial effect on the retinal degeneration. CitationFrasson and colleagues (1999) first described the rescue of rod photoreceptor cells by a Ca2+ channel blocker, D-cis-diltiazem, in rd mouse, in which the gene encoding cGMP phosphodiesterase is affected. However CitationBush and colleagues (2000) claimed that D-cis-diltiazem had no effects on the photoreceptor degeneration in the Rho P23H rat. Therefore, the protective effect of Ca2+ channel blocker against retinal degeneration is controversial. Recently, to clarify this ambiguity regarding the effect of Ca2+ channel blocker, we systematically studied the effects of several kinds of Ca2+ channel blockers, including D-cis-diltiazem, nicardipine, nilvadipine and nifedipine against several models with retinal degeneration. From among these, only nilvadipine had a preservative effect on photoreceptor cells during retinal degeneration of RCS rat (CitationYamazaki et al 2002), light-stress-induced rat (CitationIshikawa et al 2006) and CAR model rat (CitationOhguro et al 2001), whereas, other Ca2+ channel blockers had no effects. Surprisingly, the present study consistently showed that nilvadipine also had protective effects against P23H retinal degeneration. Among four Ca2+ channel blockers used in the present study, three dihydropyridine (DHP) (nilvadipine, nicaridipine, and nifedipine) and diltiazem, it was revealed that nilvadipine is a much higher hydrophobic chemical and well distributed in various types of tissue, including brain, after the systemic administration than others (CitationTokuma et al 1987; CitationSuzuki et al 1988). Thus, as to the reason why only nilvadipine was effective for the retinal degeneration we speculated the occurrence of preferable transmission of nilvadipine to the central nervous system, including retina as compared with other Ca2+ channel blockers. It was also revealed that nilvadipine had also a low-voltage-activated Ca2+ channel blocking action in addition to the L-type high-voltage-activated Ca2+ channel blocking action (CitationIshibashi et al 1998). However, in contrast, much less effects on a LVA Ca2+ channel blocking action of nifedipine, nicaridipine and diltiazem have been reported (CitationAkaike et al 1989). Thus this may be another possibility since presence of retinal LVA Ca2+ channels has been reported (CitationGuenther et al 1994).
In our previous study, to elucidate the molecular mechanism responsible for the drug effects of Ca2+ channel blocker nilvadipine on the retinal degeneration, we analyzed altered gene expression by mRNA profiling assay and found that neurotrophic factor, fibroblast growth factor-2 (FGF-2) and Arc, known to suppress the apoptosis in the central nervous system were remarkably up-regulated among 1101 genes commonly expressed in rodent in nilvadipine-treated RCS rat (CitationSato et al 2003). Thus, we suggested that systemic administration of nilvadipine to RCS rats increases the expression of endogenous FGF-2 and Arc in retina, and potentially have a protective effect against retinal degeneration. In the present study, nilvadipine-induced enhancement of the endogenous FGF-2 and Arc expressions were also recognized in P23H model. FGFs constitute a large family of polypeptides that are important in the regulation of cell growth and differentiation and play a key role in oncogenesis and developmental processes including limb formation, mesoderm induction and neuronal development (CitationWagner 1991). Among FGFs, in vivo and in vitro studies have revealed that basic FGF (FGF-2) has been recognized as an important neuro-survival factor. Several in vitro and in vivo studies have revealed that FGF-2 prevented retinal degeneration (CitationFaktorovich et al 1990; CitationSteward and Worley 2001). Arc (activity-regulated cytoskeleton-associated protein) was first identified as one of the immediate-early genes in neurons (CitationSteward and Worley 2001). It was shown that Arc mRNA is constitutively expressed within the cell body, but is delivered into dendrites and accumulated at synapses upon an appropriate stimulus, such as a single electroconvulsive seizure (CitationLyford et al 1995). In addition, this specific localization of Arc mRNA was shown to be dependent on local signaling through the NMDA receptor (CitationSteward and Worley 2001). Thus if these factors indeed cause beneficial effects against retinal degenerations, nilvadipine-induced enhancement of endogenous these factors is a promising candidate therapeutic strategy for retinal degeneration.
Disclosure
The authors report no conflicts of interest in this work.
References
- AkaikeNKanaideHKugaT1989Low-voltage-activated calcium current in rat aorta smooth muscle cells in primary cultureJ Physiol416141602558173
- BersonELRosnerBSandbergMA1991Ocular findings in patients with autosomal retinitis pigmentosa and rhodopsin gene defect (Pro-23-His)Arch Ophthalmol109921011987956
- BirchDGHoodDCNusinowitzS1995Abnormal activation and inactivation mechanisms of rod transduction in patients with autosomal dominant retinitis pigmentosa and the pro-23his mutationInvest Ophthalmol Vis Sci361603147601641
- BushRAKononenLMachidaS2000The effect of calcium blocker D-cis-diltiazem on photoreceptor degeneration in the rhodopsin Pro23His ratInvest Ophthalmol Vis Sci41269770110937585
- D’CruzPMYasumuraDWeirJ2000Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS ratHum Mol Genet96455110699188
- DryjaTP1992Rhodopsin and autosomal dominant retinitis pigmentosaEye61101358680
- FaktorovichEGSteinbergRHYasumuraD1992Basic fibroblast growth factor and local injury protect photoreceptors from light damage in the ratJ Neurosci123554671527595
- FaktorovichEGSteinbergRHYasumuraD1990Photoreceptor degeneration in inherited retinal dystrophy delayed by basic fibroblast growth factorNature3478362168521
- FrassonMSahelJAFabreM1999Retinitis pigmentosa: rod photoreceptor rescue by a calcium-channel blocker in the rd mouseNature Med51183710502823
- GalALiYThompsonDA2000Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosaNat Genet26270111062461
- GotoYPeacheyNSZiroliNE1996Rod phototransduction in transgenic mice expressing a mutant opsin geneJ Opt Soc Am1357785
- GreenESMenzMDLaVailMM2000Chacterization of rhodopsin mis-sorting and constitutive activation in a transgenic rat model of retinitis pigmentosaInvest Ophthalmol Vis Sci4115465310798675
- GuentherERotheTTaschenbergerH1994Separation of calcium currents in retinal ganglion cells from postnatal ratBrain Res633223357907933
- HethCASchmidtSY1992Protein phosphorylation in retinal pigment epithelium of Long-Evans and Royal College of Surgeons ratsInvest Ophthalmol Vis Sci332839471526733
- IshibashiHMuraiYAkaikeN1998Effect of nilvadipine on the voltage-dependent Ca2+ channels in rat hippocampal CA1 pyramidal neuronsBrain Res81312179824683
- IshikawaFOhguroHOhguroI2006Prolonged rhodopsin phosphorylation in light-induced retinal degeneration in rat modelsInvest Ophthalmol Vis Sci4752041117122104
- KawamuraS1991Rhodopsin phosphorylation as a mechanism of cyclic GMP phosphodiesterase regulation by S-modulinNature36285578386803
- KochKWStryerL1988Highly cooperative feedback control of retinal rod guanylate cyclase by calciumNature3346462455233
- LiuXWuT-HStoweS1997Defective phototransductive disk membrane morphogenesis in transgenic mice expressing opsin with a mutated N-terminal domainJ Cell Sci1102589979372448
- LyfordGLYamagataKKaufmannWE1995Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendritesNeuron14433457857651
- MachidaSKondoMJamisonJA2000P23H rhodopsin transgenic rat: correlation of retinal function with histopathologyInvest Ophthalmol Vis Sci413200910967084
- MaedaAOhguroHMaedaT1999Low expression of alphaA-crystallins and rhodopsin kinase of photoreceptors in retinal dystrophy ratInvest Ophthalmol Vis Sci4027889410549637
- MullenRJLaVailMM1976Inherited retinal dystrophy: Primary defect in pigment epithelium determined with experimental rat chimerasScience20110235
- NicoteraPOrreniusS1992Ca2+ and cell deathAnn Ny Acd Sci6481727
- NirISagieGPapermasterDS1987Opsin accumulation in photoreceptor inner segment plasma membranes of dystrophic RCS ratsInvest Ophthalmol Vis Sci286292948935
- OhguroHOgawaKMaedaT2001Retinal dysfunction in cancer-associated retinopathy is improved by Ca2+ antagonist administration and dark adaptationInvest Ophthalmol Vis Sci4225899511581204
- OhguroHOhguroIMamiyaK2003Prolonged survival of the phosphorylated form of rhodopsin during dark adaptation of royal college surgeons ratFEBS Lett5511283212965217
- OhguroHVan HooserJPMilamAH1995Rhodopsin phosphorylation and dephosphorylation in vivoJ Biol Chem27014259627782279
- SatoMOhguroHOhguroI2003Study of pharmacological effects of nilvadipine on RCS rat retinal degeneration by microarray analysisBiochem Biophys Res Commun3068263112821116
- StewardOWorleyPF2001Selective targeting of newly synthesized Arc mRNA to active synapses requires NMDA receptor activationNeuron302274011343657
- SuzukiHOnoEUenoH1988Physico-chemical properties and stabilities of the highly potent calcium antagonist benidipine hydrochlorideArzneim Forsch38167163219139
- TokumaYFujiwaraTNoguchiH1987Absorption, distribution and excretion of nilvadipine, a new dihydropyridine calcium antagonist, in rats and dogsXenobiotica17134193433803
- TriggleDHWeissGB1981Calcium antagonist: basic chemical and pharmacological aspectsNew Perspectives on calcium antagonistsBethesda, MAAmerican Physiological Society1981121
- van SoestSWesterveldADejongPTVM1999Retinitis pigmentosa: defined from a molecular point of viewSurv Ophthalmol433213410025514
- WagnerJA1991The fibroblast growth factors: an emerging family of neural growth factorsCurr Top Microbiol Immunol165951182032466
- YamazakiHOhguroHMaedaT2002Nilvadipine, a Ca2+ antagonist, effectively preserves retinal morphology and functions in Royal College Surgeons ratInvest Ophthalmol Vis Sci439192611923229