Abstract
Purpose:
The purpose of this paper is to report 14 new cases of unilateral retinitis pigmentosa and three new cases of cone-rod dystrophy and to compare the similarities and dissimilarities to those found in the bilateral forms of these disorders.
Methods:
A total of 272 cases of retinitis pigmentosa and 167 cases of cone-rod dystrophy were studied by corneal full field electroretinograms and electrooculograms. The student t-test was used to compare categories.
Results:
The percentage of familial and nonfamilial cases was the same for the bilateral and unilateral forms of the disease. In our series, unilateral retinitis pigmentosa makes up approximately 5% of the total population of retinitis pigmentosa, while unilateral cone-rod dystrophy makes up only about 2% of the total. In the familial forms of unilateral retinitis pigmentosa the most common inheritance pattern was autosomal dominant and all affected relatives had bilateral disease.
Conclusion:
Unilateral retinitis pigmentosa and cone-rod dystrophy appear to be directly related to the more common bilateral forms of these disorders. The genetic mechanisms which account for asymmetric disorders are not currently understood. It may be a different unidentified mutation at a single loci or it is possible that nonlinked mutations in multiple loci account for this unusual disorder.
Introduction
Unilateral retinitis pigmentosa (RP) and unilateral cone-rod dystrophy have generally been reported as single case reports or very small series of 2–4 cases. JosephCitation1 reported one case and reviewed the world literature where she found 45 cases. Kolb and GallowayCitation2 reported three cases and found 27 cases reported between 1865 and 1962. Part of this difference in numbers is the acceptance of what constitutes unilateral retinitis pigmentosa. The vast majority of these cases were reported prior to the advent of electroretinography and electrooculography, which means the diagnosis was made based on symptoms and retinal examination alone and that some of the cases may have resulted from other acquired disorders. Since these early reports, an additional 52 cases (obtained by a PubMed search) have been reported in the world literature, with the vast majority having had both electroretinography and electrooculography. The purpose of this manuscript is to report our experience with a series of 14 cases of unilateral retinitis pigmentosa and three cases of unilateral cone-rod dystrophy and to compare these cases with the more typical bilateral cases.
Methods
This clinical neurophysiology laboratory has been providing electroretinography and electrooculography testing since the early 1980s, giving an experience of over 25 years. As such, hundreds of studies have been carried out and all abnormal studies have been maintained in a database. This retrospective study was able to utilize this database for the results presented in this manuscript. The clinical diagnosis of RP by referring ophthalmologist was wrong as 28 cases originally referred as possible RP ended up having cone-rod dystrophy. Only one case was the reverse. The major complaint for patients with RP was defective night vision with decreased peripheral fields (230 cases), this was followed by photophobia (22 cases), and decreased color vision (16 cases). Fundiscopic examination (when data was present) revealed the most common description to be pigmentary changes (71 cases), boney spicule changes (13 cases), compatible with RP (6 cases), and bull’s eye changes (2 cases). The major presenting complaints for cone-rod dystrophy included decreased color vision (56 cases), defective night vision (42 cases), and photophobia (23 cases). Fundiscopic examination showed pigmentary changes (25 cases), maculopathy (23 cases) bull’s eye changes (8 cases), boney spicules (4 cases), and optic atrophy (1 case). The major presenting complaints for cone-rod dystrophy were decreased color vision (38 cases), decreased night vision (32 cases), and photophobia (8 cases). Fundiscopic examination showed pigmentary changes (20 cases), maculopathy (5 cases), Bull’s eye changes (5 cases), and optic atrophy (2 cases). The biggest surprise was the very large number of individuals complaining of decreased night vision loss in patients with cone-rod dystrophy. All of these individuals had normal rod function according to their electroretinogram.
The 14 cases of unilateral retinitis pigmentosa included eight females and six males. The three cases of unilateral cone-rod dystrophy included two females and one male. In none of these cases was there a history of ocular eye trauma, syphilis or other ocular inflammatory disorder, detached retina, or ocular ischemia. No other causes could be found to explain the unilateral nature of these cases. Long term follow-up was impossible to obtain as no personal indentifiers were maintained in our database, in accordance to rules established by the University of Washington Human Research Committee. However, two of the 14 patients were referred for a second study eight and 14 years later and the unaffected eye remained normal.
Electroretinography begins with dark-adapting the patient for 45 minutes to guarantee full dark adaptation. A Ganzfeld stimulator is then used to present a series of different stimuli including single blue flashes (470 nanometers), followed by single red flashes (600 nanometers), then single white flashes. This series provides an initial evaluation of pure rod function, followed by increasing contributions from the cones. Cone oscillations are typically seen with red flash and white flash gives a double a- wave [cone (a1) and rod (a2)] and a very large b-wave. The patient then undergoes a study utilizing 30 Hz white flicker. This test is the first of a series of pure cone measurements. It also light-adapts the patient. The remaining three tests are all done with the patient light-adapted and reflect cone activity.
First, single white flashes are used to stimulate all of the cones. This is followed by stimulation with single red-yellow (>550 nanometers) then single blue-green flashes (<550 nanometers) to measure sub-sets of the cones. All recordings are carried out with a Burien–Allen corneal electrode (Hansen Ophthalmic Laboratories, Iowa City, 1A) or rarely with a gold foil electrode for those who cannot tolerate the Burien–Allen electrode. The amplitude (microvolts) and the implicit time (milliseconds) for each wave are subjected to statistical comparison with a control population. Abnormal results are those that exceed a 99% tolerance limit for 95% of the population (one-tailed).
Electrooculography takes advantage of the electrical potential generated by the retinal pigment epithelial cells. Changes in this electrical potential occur both during the dark stage and following the presentation of light. Measurements of this ocular electrical potential are taken every minute for 15 minutes in the dark followed by measurements taken every minute for 15 minutes after exposing the eye to a bright light. Absolute values are of limited value because of the wide ranges found in the normal population and even between eyes in a given individual, but the ratio of dark trough to light peak (DT/LP) are quite reproducible and reliable. Normal values of DT/LP were >1.72.
The Student’s t-test was used to compare different populations involved in this study.
Results
Table shows the bilateral population of retinitis pigmentosa at 256. There are 14 cases of unilateral retinitis pigmentosa, and two cases of asymmetric retinitis pigmentosa. Unilateral cases make up approximately 5%, and if asymmetric cases are included, 7%. The next most common disorder is cone-rod dystrophy with 164 bilateral cases and three unilateral cases. Unilateral cases make up approximately 2% of the cone-rod dystrophies and this is reflected in the literature where only a couple of case reports exist. Progressive cone dystrophy includes 99 cases with zero cases of unilateral disease and finally 26 cases of Usher disease with zero cases of unilateral disease. Unilateral retinal disease has been previously reported in Usher disease. Table shows the genetic inheritance patterns seen in the bilateral cases and include 52 dominant, 30 recessive (based on affected sibs), and six cases of X-linked retinitis pigmentosa. Unilateral cases of retinitis pigmentosa have a similar pattern, but no examples of X-linked disease were identified in this study, but may be a reflection of the sample size. Unilateral retinitis pigmentosa includes four cases of dominant disease and one case of recessive disease. Bilateral cone-rod dystrophy contained 32 cases of dominant disease and 17 cases of recessive disease. Unilateral cone-rod dystrophy had one case with dominant inheritance. In all groups, sporadic cases exceeded the documented genetically determined cases. In bilateral retinitis pigmentosa, genetic cases make up 34% of the bilateral cases and a similar percentage (36%) is seen in unilateral retinitis pigmentosa. Similar findings are seen in the cone-rod dystrophies where the bilateral form has a genetic makeup of 30% and the unilateral form of 33%.
Table 1 Case distribution
Table 2 Genetically determined cases
Table compares the age of onset of the various forms of nonfamilial and familial forms of bilateral retinitis pigmentosa, unilateral retinitis pigmentosa, bilateral cone-rod dystrophy, unilateral cone-rod dystrophy and finally the nonfamilial and familial forms of progressive cone dystrophy. Within groups, for example, nonfamilial retinitis pigmentosa and familial retinitis pigmentosa have no statistical differences between ages of onset (Student’s t-test). The same is true for familial and nonfamilial forms of both cone-rod dystrophy and cone dystrophy. There are however, very significant differences in age of onset when nonfamilial retinitis pigmentosa is compared to nonfamilial unilateral retinitis pigmentosa (p = 0.0048) and familial retinitis pigmentosa and familial unilateral retinitis pigmentosa (p = 0.0019). These findings confirm what has been suggested in case reports that unilateral cases tend to have an older age of onset.
Table 3 Age of onset
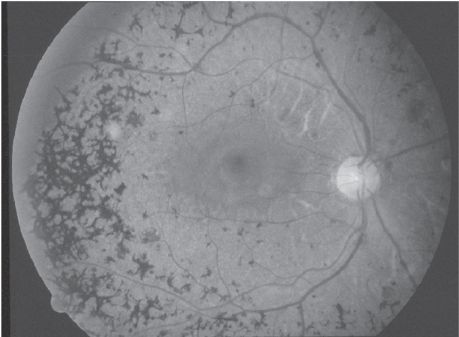
In unilateral retinitis pigmentosa the retina of the affected eye shows the changes characteristic of retinitis pigmentosa including a pigmentary retinopathy (frequently with boney spicule formation), narrowing of the vessels, atrophy of the choroid, and each case shows a reduced peripheral visual field. If the patient has clinical complaints, it is generally that of reduced night vision (see Figure ). The unaffected eye has a perfectly normal retinal examination, including a normal electroretinogram and electrooculogram.
Figure 1 Retinal photograph OS showing widespread pigmentary clumping (boney spicule changes) in the peripheral fields and attenuation of the vessels. OD was perfectly normal.
The electroretinogram and electrooculogram are of great value in establishing the diagnosis of unilateral retinitis pigmentosa and cone-rod dystrophy. Figure is a patient who has normal responses OD, however OS shows no response to blue flash and reduced a-wave and b-wave amplitudes and prolonged implicit times in the dark-adapted and light-adapted states to white flash. The electrooculogram shows the normal response to light OD while OS shows no response to light. This is a typical electroretinogram and electrooculogram seen in a moderately advanced case of retinitis pigmentosa. There are no functioning rods, but continues to have some cone function, albeit abnormal. Figure shows a more advanced case of retinitis pigmentosa where OD is the abnormal eye and there are no responses to any of the test conditions, dark-adapted blue and white flash and light-adapted white flash. Again, the electrooculogram shows a very abnormal OD showing no response to light.
Figure 2 Electroretinogram shows normal responses OD and an absent response OS to blue flash. White flash in both the dark-adapted and light-adapted states have marked reduced a- and b-waves with prolonged implicit times. This pattern is consistent with moderately advanced retinitis pigmentosa with the rod responses being more affected than the cones. The electrooculogram shows a normal light response OD and no response to light OS. These results confirm severe damage to the retinal pigment epithelial layer.
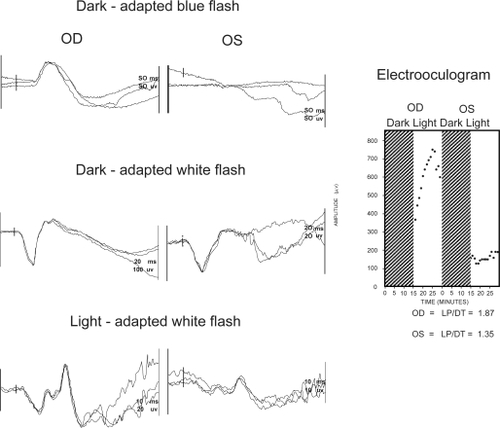
Figure 3 Electroretinogram and electrooculogram in a more advanced case of unilateral retinitis pigmentosa. In this example the affected eye is OD, where no responses to any stimuli elicited. OS has normal responses in both the dark-adapted and light-adapted states. The electrooculogram demonstrates no response to light OD while OS has a normal response to light.
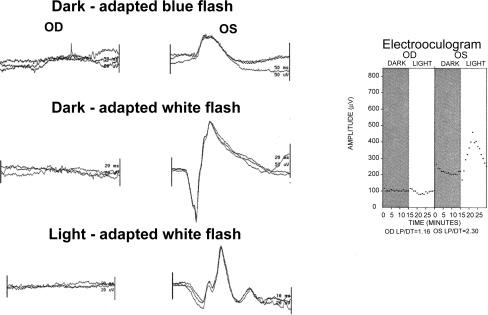
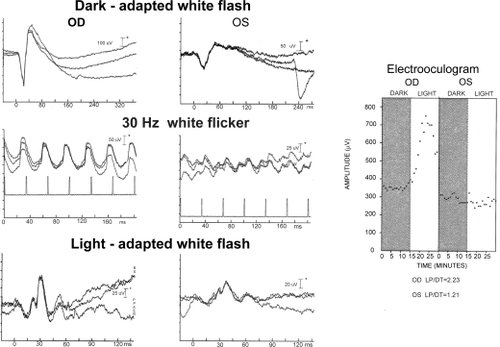
Figure shows the electroretinogram and electrooculogram in a unilateral case of cone-rod dystrophy. OD is the normal eye and OS the abnormal.
Figure 4 Electroretinogram and electrooculogram in one of the individuals with unilateral cone-rod dystrophy. OS is the affected eye and shows residual rod function to white flash in the dark-adapted state. The a- and b-waves are reduced in amplitude and the implicit times prolonged. The b-wave is poorly formed, reduced in amplitude and has a prolonged implicit time to 30 Hz white flicker. White flash in the light-adapted state shows abnormally small a- and b-waves with prolonged implicit times. The electrooculogram demonstrates no response to light OS and a normal response OD.
Note the marked reduction in amplitude and prolonged implicit times in both the a-wave and b-wave, dark-adapted white flash. The b-wave generated by 30 Hz flicker is poorly formed, reduced in amplitude and has a markedly prolonged implicit time. In the light-adapted state, white flash, the a- and b-wave are also reduced in amplitude and the implicit times prolonged. The electrooculogram demonstrates a normal response OD and no response OS.
Discussion
It appears reasonable that unilateral retinitis pigmentosa and cone-rod dystrophy exist as clinical entities. In our series the frequency of unilateral retinitis pigmentosa appears to be 5% of bilateral retinitis pigmentosa. This value is probably on the high side as these studies were performed in a tertiary diagnostic laboratory and there is most likely a bias towards more unusual cases. We report here 14 cases of unilateral retinitis pigmentosa and 256 cases of bilateral retinitis pigmentosa. Kolb and GallowayCitation2 reported four cases of unilateral retinitis pigmentosa and 65 cases of bilateral disease (6%). Their percentage of unilateral cases is virtually identical to our findings (again, this data is from a tertiary center). Many of the single case reports emphasized the lack of familial cases and because of this it was thought that unilateral retinitis pigmentosa was unrelated to bilateral retinitis pigmentosa. JosephCitation1 reported a new case and her review of the world literature identified an additional 45 cases. She eliminated 20 of the cases because there was a possibility that an alternate diagnosis could explain the findings. The other possibilities included trauma, syphilis, inflammatory disease, congenital and defective night vision. This author agrees with all except for those with defective night vision, as patients with congenital stationary night blindness have a normal fundiscopic examination and patients with Oguchi disease have characteristic retinal changes. She also found at least four instances of familial disease in her review, but arbitrarily discounted the familial nature of these cases. Three of the families included only siblings being affected, this being consistent with an autosomal recessive pattern of inheritance, and one instance where a parent was affected. In total, she found four familial cases out of a total of 24 unilateral cases. Our results are not too dissimilar with five familial cases out of 14. This confirms that the ratio of familial cases to nonfamilial cases is about the same as we have found in the bilateral form of the disease. In our series, the familial forms of unilateral disease were found to have two different modes of inheritance, autosomal dominant and recessive. It is of great interest that all of the affected family members suffered from bilateral retinitis pigmentosa. There were no examples of an individual with unilateral retinitis pigmentosa having a relative with unilateral retinitis pigmentosa. In our series and that of the other documented cases of unilateral retinitis pigmentosa there have been no examples of X-linked inheritance, but this may be a reflection of sample size.
The older literature emphasized the later age of onset of unilateral retinitis pigmentosa than bilateral disease. We have confirmed this and the age of onset differences between both the nonfamilial forms and the familial forms of unilateral retinitis pigmentosa are statistically significant (Student’s t-test: p = 0.0048 and 0.0019, respectively). However, this finding may be artifactual in that unilateral disease may take longer for the patient to recognize symptoms than the bilateral form of the disease because the good eye covers up the symptoms of night blindness. It has also become clear that unilateral retinitis pigmentosa remains unilateral. Many cases have now been followed for up to 30 or more years without involvement of the normal eye.
Unilateral cone-rod dystrophy is less common than unilateral retinitis pigmentosa. We found three cases of unilateral disease and 164 cases of bilateral disease. Unilateral disease makes up about 2% of the bilateral cases. This may explain why this condition is so rarely reported in the literature with only two documented cases having been previously reported.Citation3
We found no instances of unilateral disease in the progressive cone dystrophies or in our population of the different forms of Usher disease. However, there appear to be examples of unilateral retinitis in Usher disease in the older literature, including the first case of unilateral retinitis pigmentosa reported in 1865.Citation4 This patient had bilateral deafness. Two possible additional cases were reviewed by Joseph.Citation1
Unilateral retinitis pigmentosa without pigmentary changes has also been reported.Citation5,Citation6 Unilateral retinitis pigmentosa has been reported, associated with a number of other conditions including, heterochromia iridis,Citation7 exfoliation syndrome,Citation8 pit of optic disc,Citation9 temporal arteritis,Citation10 and glaucoma.Citation11 It is not clear whether these conditions are related to the unilateral retinal disease or if they are incidental findings, as only one case each has been reported it is more likely to be the latter.
Great advances have been made in the understanding of the molecular genetic mutations that lead to the various retinal disorders. Over 132 different mutations have now been identified with these disorders.Citation12,Citation13 To date, none of the familial unilateral cases of retinitis pigmentosa or cone-rod dystrophy have been subjected to this type of analysis. In the future familial unilateral cases and their bilaterally affected relatives will need to be studied to determine whether they share a common mutation.
It is clear that the number of familial cases is under-represented in this current study. The number of recessive cases of retinitis pigmentosa and cone-rod dystrophy is on the low side as only families with affected sibs are included in our familial cases. If only one case was present in the family they were included in the nonfamilial form. Even if 2–3 times the reported number of recessive cases were indeed recessive in nature, that still leaves a very high percentage of what are called nonfamilial cases to explain. Are the vast majority of these “nonfamilial cases” genetically determined disorders, but result from mutations that have yet to be identified? Other genetically determined nervous system disorders may provide a clue, to the mechanism that causes unilateral disease and bilateral disease with the same or closely related genetic mutation. For example, disorders with different mutations affecting the micro-tubule associated protein tau may lead to a number of different clinical disorders including progressive nonfluent aphasia, a unilateral degenerative disorder affecting the temporal lobe of the brain or frontotemporal dementia a symmetric disorder affecting both the frontal and temporal lobes of the brain.Citation14–Citation16 Certain forms of autosomal dominant retinitis pigmentosa have been shown to result from unlinked mutations of peripherin/RDS and ROM1 loci.Citation17 It is likely that an unusual mutation or combination of mutations at different loci account for the dominant genetic mechanism responsible for unilateral retinal disorders. The recessive form of the disorders is likely to represent compound heterozygote where each parent provides a different allelic mutation to the offspring. One or the other of these mutations determines the nature of the disorder. Earlier we reported a familyCitation18,Citation19 with two different ages of onset forms of GM1 gangliosidosis. The two affected individuals inherited a common mutation from their grandfather. This man had two different wives, each producing an offspring, one individual born to each wife, married an unrelated carrier of the trait and on one side of the family a child with infantile onset GM1 gangliosidosis was born and on the other side a child with juvenile onset GM1 gangliosidosis was born. Each case progressed as expected for the age of onset.
Since there are no examples of X-linked inheritance in the reported cases of unilateral retinitis pigmentosa, asymmetric inactivation of the X-chromosome in carriers of X-linked retinitis pigmentosa cannot be considered a common mechanism to explain the unilateral nature of the disorder.
To date, all evidence points to a close relationship between unilateral retinitis pigmentosa and bilateral retinitis pigmentosa. The genetic mechanisms to explain this rare disorder remain to be identified.
Disclosure
The author does not have a proprietary interest in this study.
References
- JosephRUnilateral retinitis pigmentosaBr J Ophthalmol1950359811314821283
- KolbHGallowayNRThree cases of unilateral pigmentary degenerationBr J Ophthalmol19644847147914218207
- SievingPA‘Unilateral cone dystrophy’: ERG changes implicate abnormal signaling by hyperpolarizing bipolar and/or horizontal cellsTrans Am Ophthalmol Soc199492471474
- PedragliaCKlinische beobachtungen. Retinitis pigmentosaKlin Mbl Augenheilk18653114117
- JacobsonJHStephensGUnilateral retinitis pimentosa sine pigmentoArch Ophthalmol19626745645814036508
- PearlmanJTSaxtonJHoffmanGUnilateral retinitis pigmentosa sine pigmentoBr J Ophthalmol197660354360952804
- GrisantiSDiestelhorstMLebekJWalterPHeimannKUnilateral pigmentary degeneration of the retina associated with heterochromia iridisGraefes Arch Clin Exp Ophthalmol19982369409449865626
- Paolo de FeliceGBottoniFOrzalesiNUnilateral retinitis pigmentosa associated with exfoliation syndromeInt Ophthalmol1988112192263182175
- GodelVRegenbogenLUnilateral retinitis pigmentosa and pit of optic nerveArch Ophthalmol19769414171418949285
- BankHPascoMGodelVUnilateral retinitis pigmentosa and temporal arteritisArch Ophthalmol197232213216
- KrillAEIserGUnilateral retinitis pigmentosa with glaucomaAMA Arch. Ophthalmol19596162663013636552
- BokDContributions of genetics to our understanding of inherited monogenic retinal diseases and age-related macular degenerationsArch Ophthalmol200712516016417296891
- KoenekoopRKLopezIden HollanderAIAllikmetsRCremersFPGenetic testing for retinal dystrophies and dysfunctions: benefits, dilemmas and solutionsClin Exp Ophthalmol200735473485
- KnibbJAKippsCMJodgesJRFrontotemporal dementiaCurr Opin Neurol20061956557117102695
- BoctiCRockelCRoyPGaoFBlackSETopographical patterns of lobar atrophy in frontotemporal dementia and Alzheimer’s diseaseDement Geriatr Cogn Disord20062136437216534206
- CaffreyTMWade-MartinsRFunctional MAPT haplotypes: Bridging the gap between genotype and neuropathologyNeuro Biol Dis200727110
- KajiwaraKBersonELDryjaTPDigenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 lociScience1994264160416088202715
- FarrellDFOchsUGM1 gangliosidosis: phenotypic variation in a single familyAnn Neurol198192252316784662
- FarrellDFMacMartinMPGM1 gangliosidosis: Enzymatic variation in a single familyAnn Neurol198192322366784663