Abstract
Subcellular distribution of the apoptosis inhibitor survivin and its ability to relocalize as a result of cell cycle phase or therapeutic insult has led to the hypothesis that these subcellular pools may coincide with different survivin functions. The PIK kinases (ATM, ATR and DNA-PK) phosphorylate a variety of effector substrates that propagate DNA damage signals, resulting in various biological outputs. Here we demonstrate that subcellular repartitioning of survivin in MCF-7 cells as a result of UV light-mediated DNA damage is dependent upon DNA damage-sensing proteins as treatment with the pan PIK kinase inhibitor wortmannin repartitioned survivin in the mitochondria and diminished it from the cytosol and nucleus. Mitochondrial redistribution of survivin, such as was recorded after wortmannin treatment, occurred in cells lacking any one of the three DNA damage sensing protein kinases: DNA-PK, ATM or ATR. However, failed survivin redistribution from the mitochondria in response to low-dose UV occurred only in the cells lacking ATM, implying that ATM may be the primary kinase involved in this process. Taken together, this data implicates survivian’s subcellular distribution is a dynamic physiological process that appears responsive to UV light-initiated DNA damage and that its distribution may be responsible for its multifunctionality.
Keywords:
Introduction
Among the regulators of apoptosis, considerable attention has been focused on the inhibitor of apoptosis protein (IAP) survivin.Citation1–Citation3 Survivin is bifunctional as it is implicated in both apoptosis and mitotic regulation and has been shown to reside in the mitochondria,Citation4 nucleus and cytosol of tumors,Citation5 and proliferating normal cells.Citation6 Subcellular distribution and the ability to relocalize as a result of cell cycle phase or therapeutic insult has led to the hypothesis that these subcellular pools may coincide with different survivin functions.
The PIK kinases (ATM, ATR and DNA-PK) phosphorylate a variety of effector substrates that propagate DNA damage signals, ultimately resulting in various biological outputs influencing cell cycle arrest, transcription, DNA repair, and apoptosis.Citation7 ATM is activated by DNA double-strand breaks whereas ATR is recruited to single-stranded DNA regions, which arise at stalled replication forks or during the processing of bulky lesions such as UV photoproducts.Citation8 Both ATM and ATR share overlapping phosphorylation targets but also have distinct substrates. Most notable, ATR specifically phosphorylates and activates the transducer kinase, Chk1, whereas ATM preferentially phosphorylates Chk2.Citation8,Citation9 Tumor cells exposed to DNA damage have been shown to release survivin from their mitochondria in order to counteract cell death.Citation4 This pathway has been shown to be Chk2-dependent as cells depleted of Chk2 are unable to release survivin from the mitochondria and become apoptotic.Citation10
This study reports a survivin compartmentalization in the mitochondria and enhanced cellular apoptosis following treatment of MCF-7 cells with the pan PIK kinase inhibitor wortmannin. Cells also experience a radiation induced decrease in survivin from all three cellular compartments, mitochondria, nucleus and cytosol, following wortmannin treatment. This finding implies that the PIK kinases may be involved in survivin localization to the mitochondria for apoptosis regulation and to the nucleus for cell cycle arrest phenotypes. Mitochondrial redistribution of survivin, as was recorded after wortmannin treatment, occurred in cells lacking the PIK kinases: ATR and DNA-PK. However, failed survivin redistribution to the nucleus in response to UV radiation occurred only in the cells lacking ATM.
Taken together, this data implicates survivin’s subcellular distribution is a dynamic physiological process that is responsive to UV light initiated DNA damage and that its distribution may be responsible for its multifunctionality.
Material and methods
Cell lines and cultures
Osteosarcoma (SaOS2), breast carcinoma (MCF7) cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA) while M059K (DNA-PK+/+), M059J (DNA-PK−/−), GM02184E (ATM+/+), GM03189D (ATM−/−) cells were obtained from the Coriell Institute (Camden, NJ). GM847Kd cells produce a dominant negative ATR kinase inactive mutant (ATRkd) under the control of a tet activator, which can be turned on by the addition of doxycycline in the culture media. These cells were a kind gift from Dr. William A. Cliby, Mayo Clinic, Rochester, MN USA.
For the induction of ATRkd expression in GM847Kd cells, doxycycline (1 μg/ml) was added to the medium for 2–4 days before harvesting. Cells were maintained as defined in Dulbecco’s modified Eagle’s medium (DMEM), McCoy’s, RPMI medium 1640 (RPMI) or Iscove’s modified Eagle’s medium (IMEM) (ATCC) supplemented with 100 units of penicillin, 100 mg/ml streptomycin, 300 mg of L-glutamine and 10%–20% heat inactivated fetal bovine serum (FBS) (ATCC). Cells were grown at 37°C in a humidified atmosphere of 95% air, 5% CO2.
Where indicated, cells were exposed to 0, 50, 100, 200, 300 or 400 J/m2 UV irradiation using a Stratalinker 1800 (equipped with standard 254 nm UV bulbs emitting UVC band energy) (Stratagene, La Jolla, CA). After irradiation, cells were returned to the incubator and harvested at the indicated times. Where indicated, cells were preincubated with Wortmannin (Sigma Chemical Co., St. Louis, MO), dissolved in dimethyl sulfoxide (DMSO) and used at a final concentration of 100 mM.
Apoptosis and cell cycle analysis
Subconfluent cultures of the various cell lines were treated as indicated for 24, 48 and 72 hours at 37°C. Cells were harvested, prepared, and analyzed for DNA content as described previously.Citation11,Citation12 DNA content was analyzed using a Becton Dickinson FACScan flow cytometer (Becton Dickinson, San Jose, CA). The distribution of cells in the different phases of the cell cycle was analyzed from DNA histograms using BD CellQuest software (Becton Dickinson and Company, San Jose, CA).
Preparation of nuclear, mitochondrial and cytosolic extracts
Nuclear, mitochondrial and cytosolic fractions were extracted from cells (5 × 107 to 7 × 107) as has been previously described.Citation4 Briefly, extracts were prepared from cells washed in TD buffer (135 mM NaCl, 5 mM KCl, 25 mM Tris-Cl, pH 7.6) and allowed to swell for 10 minutes in ice-cold hypotonic CaRSB buffer (10 mM NaCl, 1.5 mM CaCl2, 10 mM Tris-HCl, pH 7.5, plus protease inhibitors). Cells were Dounce-homogenized with 70 strokes using a type B pestle, with addition of MS buffer (210 mM mannitol, 70 mM sucrose, 5 mM Tris, pH 7.6) to stabilize mitochondria.
The nuclei and cytosol were separated from each other by centrifugation at 600 g for 15 minutes at 4°C. The isolated cytosolic fraction was further centrifuged at 10,000 g for 25 minutes at 4°C, and the supernatant was collected. The purity of mitochondrial fractions was examined by immunoblotting with antibodies to mitochondrial Cox-4.
Western blot analysis
Cells were solubilized, proteins (20–40 μg) separated using 12% and 15% Bis-Tris polyacrylamide gels, proteins transferred onto polyvinlidene difluoride or nitrocellulose membranes (Millipore) and probed using the following antibodies: mouse monoclonal anti-TATA binding protein (AbCam, Cambridge, MA), rabbit polyclonal anti-survivin (Novus, Littleton, CO), rabbit polyclonal antibodies to ATM, ATR, DNA-PK, VDAC-1 (AbCam), rabbit polyclonal antibodies to GAPDH; and Cox IV (Cell Signaling Technologies, Beverly, MA), mouse monoclonal antibodies to β-actin (AbCam, Cambridge, MA).
Secondary antibodies (IR-Dye-conjugated) were goat anti-rabbit and goat anti-mouse immunoglobulin (LICOR, Lincoln, Nebraska).
Immunoreactive bands were detected using the Odyssey imaging system (LICOR) and quantified using ImageQuant software. Protein quantifications presented in this report were normalized with respect to β-actin or GAPDH.
Results and discussion
UV radiation induces survivin protein expression
One of the goals of this work was to investigate the mechanism by which survivin’s intracellular location aids the self-repair response to genotoxic stress, preventing therapy induced apoptosis. One well established feature of the DNA damage response is the slowing or arrest of cell-cycle progression, as a result of what are termed DNA damage “checkpoints”, which delay key cell-cycle transitions until repair has occurred.Citation13–Citation15 Protein phosphorylation has been implicated in the regulation of cell death pathways,Citation12 influencing subcellular localization,Citation16 cytoprotection,Citation12,Citation17,Citation18 and cell cycle transitions.Citation19,Citation20 Survivin phosphorylation on Thr34 by the mitotic kinase p34cdc2-cyclin B1 has been shown to be critical for survivin’s function and stability.Citation12,Citation21,Citation22
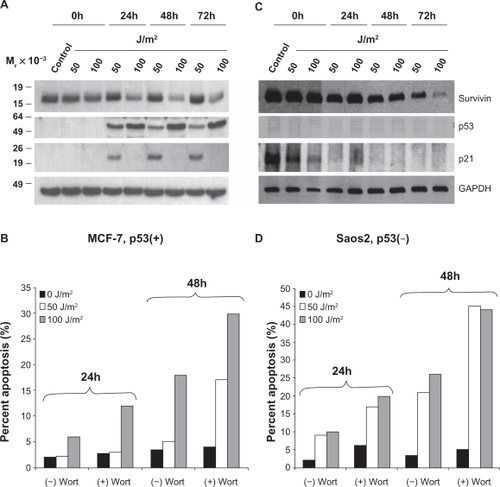
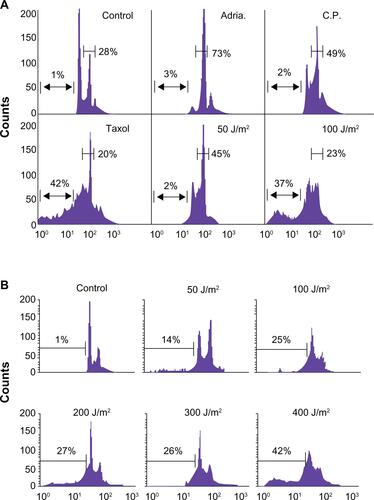
Low dose genotoxic stressors such as Adriamycin (Adria., 100 nM), Cisplatin (C.P., 3 μM) and UV (50 J/m2) arrest MCF-7 cells in the G2/M phase of the cell cycle, while high-dose UV (doses ≥100 J/m2) or the tubulin poison Taxol (2 μM) induce cellular death (Supplemental Figure 1A). Exposure of MCF-7 cells to low-dose (50 J/m2) UV radiation resulted in a sustained to enhanced expression of survivin that was also associated with enhanced p53 and p21-dependent cell cycle arrest (, Supplemental Figure 1B).Citation12 In contrast, high-dose (100 J/m2) UV radiation treatment resulted in an enhanced p53 protein expression, no p21 upregulation and a concomitant decline in survivin expression that was not associated with cell cycle arrest but with modest cellular apoptosis ().
Figure 1 UV radiation induced apoptosis is enhanced by wortmannin’s inhibition of PI3K related kinases. MCF-7 (A) and Saos2 (C) cells exposed to 50 or 100 J/m2 UV for 24, 48, or 72 hours were analyzed by Western blotting. MCF-7 (B) and Saos2 (D) cells were grown in the presence or absence of wortmannin, irradiated as above and analyzed for DNA content after 24 and 48 hours. The percentage of cells with hypodiploid DNA content is indicated.
The p53 stability associated survivin reduction may be the direct effect of p53’s ability to work as a transcriptional repressor.Citation23 However, as survivin’s protein expression is never completely absent and we have not sequenced p53, we cannot be certain. In several studies, the transcriptional repression activity of p53 has been implicated in p53-dependent apoptosis; notable was the finding that deletion of the proline rich domain of p53 renders it competent as a transactivator but unable to induce apoptosis or to repress transcription.Citation24–Citation26
We also investigated the effect of UV irradiation on the p53 deficient cell line Saos2 (). Unlike MCF-7 cells, no p53 protein was produced and p21 levels, which were quite high in control, non-UV treated cells, rapidly reduced in a time- and dose-dependent manner. Survivin levels also gradually declined in a dose- and time-dependent manner () but were not directly associated with p21 as survivin reduction occurred much slower than did p21. Levels of cell death in these cells increased dramatically compared to MCF-7 cells in a time-and dose-dependent manner (). Interestingly, low-dose UV radiation did not markedly induce cell death in the MCF-7 cells but did in Saos2 cells (). This is most likely the result of the p53-dependent, p21-dependent cell cycle arrest and concomitant stable upregulation of survivin.
PI3K related kinases and p53 are involved in survivin redistribution
Tumor cells often become resistant to DNA damage based therapy by releasing survivin from the mitochondria in a p53-independent, but checkpoint kinase 2 (Chk2)-dependent manner.Citation4,Citation10
Several PI3K related kinases, including DNA-PK, ATM, and ATR, participate in DNA damage responsive pathways.Citation7 In order to ascertain their roles in enhanced survivin expression levels after UV radiation induced DNA damage, cells were treated with the pan-PIK kinase inhibitor wortmannin. Wortmannin treatment alone did not alter apoptosis levels in either MCF-7 or Saos2 cells as can be seen in the 0 J/m2 treatment control groups (). However, upon treating both cell lines with 50 or 100 J/m2 UV, a time-dependent increase in cell death was observed in both cell lines. Survivin from MCF-7 cells treated with wortmannin compartmentalized in the mitochondria () and was diminished from the cytosol () and nucleus (). However, UV (50 J/m2) light mediated G2/M growth arrest of MCF-7 cells led to enhanced total cellular survivin () as well as partitioning of this survivin from the cytosol to the mitochondria () and nucleus (). Wortmannin prohibited this repartitioning after low-dose UV treatment.
Figure 2 UV radiation’s repartitioning of survivin from the cytosol to the mitochondria and nucleus is reduced after wortmannin treatment in p53 expressing MCF-7 cells. MCF-7 and Saos2 cells, exposed to 50 or 100 J/m2 UV for 24 hours were subcellularly fractionated into cytosol (A), mitochondria (B) and nuclear (C) fractions and analyzed by Western blotting.
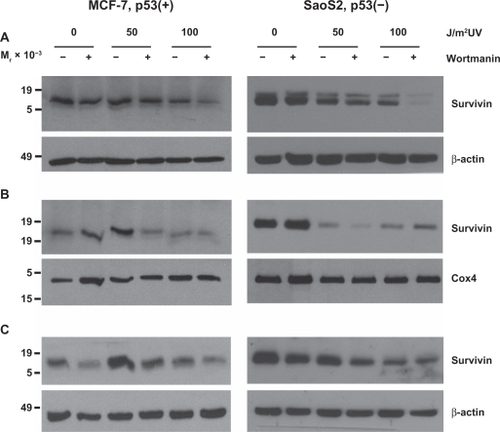
In Saos2 cells, a UV dose-dependent reduction in survivin protein () was associated with survivin reduction from all three compartments, cytosol, mitochondria and nucleus (). In both cell lines, 100 J/m2 UV combined with wortmannin resulted in survivin reduction from all three compartments () and enhanced apoptosis in both cell lines (). Wortmannin’s inability to enhance the apoptotic killing of 100 J/m2 over that of 50 J/m2 UV in Saos2 cells () did not surprise us as survivin was not repartitioned to nucleus, cytosol or mitochondria (). It has been shown that ionizing radiation modulates survivin independently of gene expression or the G2/M checkpoint,Citation10 and that the treating of MCF-7 cells with ionizing radiation induced a discharge of mitochondrial survivin that coincided with increased cytosolic survivin.
It has already been shown that survivin’s cytoprotective function involved the discharge of mitochondrial survivin to the cytosol where it prevents caspase activation and inhibits apoptosis.Citation4 Interestingly and contrary to these two reports,Citation4,Citation10 UV treated Saos2 cells experienced a notable mitochondrial release () of survivin that did not result in a stable cytosolic accumulation. Instead, survivin depletion was recorded from all three subcellular pools.
These findings imply that the PIK kinase-dependent p53 pathway may be involved in survivin relocalization to the mitochondria for apoptosis regulation and the nucleus for cell cycle arrest phenotypes.
Loss of PI3K related kinases increases cellular apoptotic response to UV irradiation
As shown in , wortmannin blocked survivin’s redistribution in MCF-7 cells, suggesting that one or more PI3K related kinases are involved in these UV irradiation redistribution events.
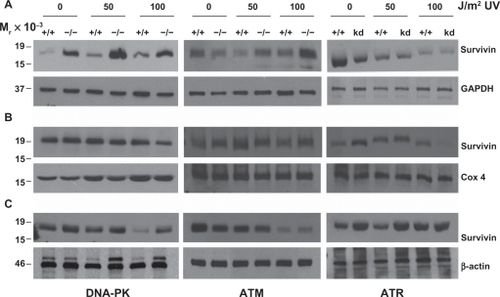
To explore which PI3K related kinase(s) is/are required for Survivin redistribution within the UV treated cell, we used a panel of cell lines that were either proficient or deficient in DNA-PK, ATM or ATR. DNA-PK and ATM deficiency rendered the cells noticeably more sensitive to UV induced apoptosis () in both a time- and dose-dependent manner. Cells that had been made kinase dead for ATRCitation27 exhibited little more than a modest enhanced cell death.
Figure 3 Loss of DNA-PK, ATM and expression of a kinase dead (kd) ATR renders cells sensitive to UV radiation. M059K (DNA-PK+/+), M059J (DNA-PK−/−), GM02184E (ATM+/+), GM03189D (ATM−/−), GM847 (ATR+/+) and GM847Kd (ATR kinase dead) were exposed to 50 or 100 J/m2 UV and analyzed for DNA content after (A) 24 and (B) 48 hours. The percentage of cells with hypodiploid DNA content is indicated.
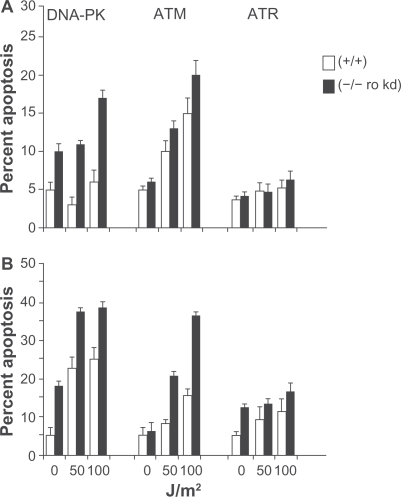
These are consisten with the known role of these proteins in sensing DNA damage, as has been well described,Citation7,Citation28 and are also consistent with those previously shown in these cells with regard to UV induced damage.Citation27
UV radiation induces defining changes in survivin protein expression
Exposure of DNA-PK+/+ M059K fibroblast cells to low-dose (50 J/m2) UV radiation resulted in a sustained expression of survivin that was associated with enhanced p53 and p21 protein expression similar to what was recorded in MCF-7 cells ().
Figure 4 Survivin, p53 and p21, after UV radiation, are differentially expressed in PI3K related kinase competent versus incompetent cells. A) M059K (DNA-PK+/+), M059J (DNA-PK−/−), B) GM02184E (ATM+/+), GM03189D (ATM−/−), C) GM847 (ATR+/+) and GM847Kd (ATR kinase dead) were exposed to 50 or 100 J/m2 UV and analyzed by Western blotting after 24, 48, and 72 hours.
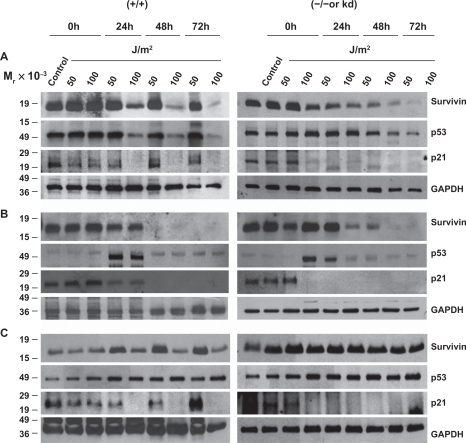
In contrast, high-dose (100 J/m2) UV radiation treatment resulted in a reduced p53 and p21 protein expression and a concomitant decline in survivin expression (). DNA-PK−/− M059J cells irradiated with either low- or high-dose UV responded with a time- and dose-dependent reduction in survivin, p53 and p21 () with levels of cell death in these cells increasing dramatically compared with their DNA-PK+/+ M059K counterparts ().
Low- and high-dose UV radiation on ATM+/+ GM02184E and ATM−/− GM03189D fibroblast cells was investigated with results mirroring more the Saos2 cells with and without wortmannin than the DNA-PK cell lines. High and low dose radiation treatments resulted in time dependent cellular reduction in survivin, p53 and p21 levels with modest levels of apoptotic protection measured but increasing from 24 h to 48 h after UV irradiation ( and ). Interestingly, GM847/ATRkd-(ATR+/+) cells were UV irradiated, survivin, p53, and p21 levels responded most similar to how MCF-7 cells without wortmannin and the DNA-PK+/+ M059K fibroblast cells had (). Survivin protein levels increased after 50 J/m2 UV as did p21. P53 levels remains mostly unchanged. GM837/ATRkd+(ATR−/−) cells showed an increase in survivin and p53 levels and a complete loss of p21 expression after UV treatments (). Cells expressing the mutant form of ATR(ATRkd+) were not more susceptible to UV DNA damage induced apoptosis even after 48 hours ().
Cellular redistribution of survivin, such as was recorded after wortmannin treatment, occurred in cells lacking any one of the three DNA damage-sensing protein kinases: DNA-PK, ATM or ATR (). However, failed survivin redistribution from the mitochondria in response to 50 J/m2 UV occurred only in the cells lacking ATM (). ATM preferentially phosphorylates Chk2,Citation8,Citation9 and mitochondria release of survivin to counteract DNA damage induced cell death has been shown to be Chk2-dependent,Citation10 further implying that ATM may be the primary kinase involved in this process.
Figure 5 UV radiation repartitions survivin from the cytosol to the mitochondria and nucleus. M059K (DNA-PK+/+), M059J (DNA-PK−/−), GM02184E (ATM+/+), GM03189D (ATM−/−), GM847 (ATR+/+) and GM847Kd (ATR kinase dead), exposed to 50 or 100 J/m2 UV for 24 hours were subcellularly fractionated into cytosol (A), mitochondria (B) and nuclear (C) fractions and analyzed by Western blotting.
Failed survivin redistribution from the nucleus in response to 50 J/m2 UV occurred in the cells lacking DNA-PK and those expressing the kinase dead ATR (), implying that DNA-PK and ATR may be the primary kinases involved in this process as well as possible overlapping functions between these two kinases.
Interestingly, cells lacking DNA-PK had a significant increase in cytosolic survivin protein () that was not associated with reduced cellular apoptosis (). It is also probable that this survivin redistribution is p53-dependent as similar experiments performed in the p53-deficient Saos-2 cell line, failed to redistribute as was recorded in the MCF-7 cells and those cells lacking DNA-PK, ATM or ATR.
Based upon our results, it is likely that loss of any one of these kinases may lead to survivin dysregulation and an increased risk of cancer. It will be of significant interest to investigate further the role these kinases play in cancer and its onset given cancer’s genomic instability. The work we show here was mostly accomplished in fibroblast cell lines that inherently have a more stable genome than do cancer cells.
Conclusion
We have shown the important discovery that survivin’s cellular transport is modified depending upon the presence or absence of specific DNA damage-sensing proteins like DNA-PK, ATM and ATR.
Our data show that survivin is differentially expressed, and localized as a result of UV light initiated DNA damage and that its localization changes from one subcellular locale to another as a result of the presence or absence of PIK kinases.
In addition, our data suggests that there might be a common underlying mechanism by which ATM, ATR and DNA-PK detect and transduce UV damage signals and in order to unravel a possible role for survivin in this pathway, the ability to eliminate more than one of the DNA damage-sensing proteins will be necessary. The overall underlying mechanism may also involve interactions with other DNA repair or maintenance proteins or function through a common effector such as p53. Furthermore, we have shown that these three DNA damage sensors can interact with each other directly or indirectly, and regulate the activities of each other.
These findings are further confirmed by the differences observed between the genetically modified cell lines and the cells treated with the non-specific pan-PIK kinase inhibitor wortmannin.
Consistent with survivin’s association with unfavorable clinicopathological parameters, trafficking survivin throughout the tumor cell could potentially be responsible for driving the aggressive status of the tumor, prohibiting or minimizing therapeutic results. Thus, treatment of a tumor, using modalities that can inhibit one or more of these sensors may prove useful in the treatment of cancer.
Many questions remain unanswered regarding the cellular response to DNA damage. How a cell decides to choose between the repair of damaged DNA and the initiation of apoptosis is just one of those questions.
It is our belief that how the multifunctional protein survivin is activated and then trafficked within the cell may play an important role in these decisions. The continued elucidation of survivin’s role within these pathways will continue to challenge our future research.
Acknowledgements
The author’s would like to thank especially, Dr. William A. Cliby (Mayo Clinic, Rochester, MN USA) for the kind gift of the GM 847kd fibroblast cells. Grant Support: NCMHD Project EXPORT Program 5P20MD001632/Project 3 (N.R. Wall). Funding was also obtained as part of a start-up package from Loma Linda University’s Center for Molecular Biology and Gene Therapy, now the Center for Health Disparities Research and Molecular Medicine (NRW) and a National Merit Test Bed (NMTB) award sponsored by the Department of the Army under Cooperative Agreement Number DAMD17–97–2–7016 (NRW).
Supplement
Supplemental Figure 1 (A) Low dose genotoxic stressors such as Adriamycin (Adria., 100 nM), Cisplatin (C.P., 3 mM) and UV (50 J/m2) arrest MCF-7 cells in the G2/M phase of the cell cycle while high-dose UV (doses ≥ 100 J/m2) or the tubulin poison Taxol (2 mM) induce cellular death. (B) Breast carcinoma MCF-7 cells were irradiated with 50–400 J/m2 UV under minimal 1X PBS. PBS was immediately removed and complete culture medium was added. After 24 hours, cells were harvested and analyzed for DNA content by propidium iodide staining and flow cytometry. Percentages of apoptotic cells with hypodiploid (sub-G1) DNA content are indicated per each condition tested. Data are representative of one of two independent experiments with comparable results.
Disclosure
The authors declare that they have no competing interests.
References
- LiFAmbrosiniGChuEYControl of apoptosis and mitotic spindle checkpoint by survivinNature19983965805849859993
- AltieriDCSurvivin, cancer networks and pathway-directed drug discoveryNat Rev Cancer20088617018075512
- AltieriDCNew wirings in the survivin networksOncogene2008276276628418931693
- DohiTBeltramiEWallNRMitochondrial survivin inhibits apoptosis and promotes tumorigenesisJ Clin Invest20041141117112715489959
- FortugnoPWallNRGiodiniASurvivin exists in immunochemically distinct subcellular pools and is involved in spindle microtubule functionJ Cell Sci200211557558511861764
- LiFSurvivin study: what is the next wave?J Cell Physiol200319782912942537
- YangJYuYHamrickHEDuerksen-HughesPJATM, ATR and DNA-PK: initiators of the cellular genotoxic stress responsesCarcinogenesis2003241571158012919958
- StiffTWalkerSACerosalettiKATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stallingEMBO J2006255775578217124492
- JazayeriAFalckJLukasCATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaksNat Cell Biol20068374516327781
- GhoshJCDohiTRaskettCMActivated checkpoint kinase 2 provides a survival signal for tumor cellsCancer Res200666115761157917178848
- LiFAckermannEJBennettCFPleiotropic cell-division defects and apoptosis induced by interference with survivin functionNat Cell Biol1999146146610587640
- WallNRO’ConnorDSPlesciaJSuppression of survivin phosphorylation on Thr34 by flavopiridol enhances tumor cell apoptosisCancer Res20036323023512517802
- RouseJJacksonSPInterfaces between the detection, signaling, and repair of DNA damageScience200229754755112142523
- LowndesNFMurguiaJRSensing and responding to DNA damageCurr Opin Genet Dev200010172510679395
- AbrahamRTCell cycle checkpoint signaling through the ATM and ATR kinasesGenes Dev2001152177279611544175
- ZhaJHaradaHYangESerine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L)Cell1996876196288929531
- LingYHTornosCPerez-SolerRPhosphorylation of Bcl-2 is a marker of M phase events and not a determinant of apoptosisJ Biol Chem199827318984189919668078
- ScatenaCDStewartZAMaysDMitotic phosphorylation of Bcl-2 during normal cell cycle progression and Taxol-induced growth arrestJ Biol Chem199827330777307849804855
- YamamotoKIchijoHKorsmeyerSJBCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/MMol Cell Biol1999198469847810567572
- FurukawaYIwaseSKikuchiJPhosphorylation of Bcl-2 protein by CDC2 kinase during G2/M phases and its role in cell cycle regulationJ Biol Chem2000275216612166710766756
- O’ConnorDSGrossmanDPlesciaJRegulation of apoptosis at cell division by p34cdc2 phosphorylation of survivinProc Natl Acad Sci U S A200097131031310711069302
- O’ConnorDSWallNRPorterACAltieriDCA p34(cdc2) survival checkpoint in cancerCancer Cell20022435412150824
- HoffmanWHBiadeSZilfouJTTranscriptional repression of the anti-apoptotic survivin gene by wild type p53J Biol Chem20022773247325711714700
- SakamuroDSabbatiniPWhiteEPrendergastGCThe polyproline region of p53 is required to activate apoptosis but not growth arrestOncogene1997158878989285684
- WalkerKKLevineAJIdentification of a novel p53 functional domain that is necessary for efficient growth suppressionProc Natl Acad Sci U S A19969315335153408986812
- VenotCMaratratMDureuilCThe requirement for the p53 proline-rich functional domain for mediation of apoptosis is correlated with specific PIG3 gene transactivation and with transcriptional repressionEMBO J199817466846799707426
- ClibyWARobertsCJCimprichKAOverexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpointsEMBO J1998171591699427750
- Burdak-RothkammSRothkammKPriseKMATM acts downstream of ATR in the DNA damage response signaling of bystander cellsCancer Res2008687059706518757420