Abstract
Malignant gliomas are invasive tumors with the potential to progress through current available therapies. These tumors are characterized by a number of abnormalities in molecular signaling that play roles in tumorigenesis, spread, and survival. These pathways are being actively investigated in both the pre-clinical and clinical settings as potential targets in the treatment of malignant gliomas. We will review many of the therapies that target the cancer cell, including the epidermal growth factor receptor, mammalian target of rapamycin, histone deacetylase, and farnesyl transferase. In addition, we will discuss strategies that target the extracellular matrix in which these cells reside as well as angiogenesis, a process emerging as central to tumor development and growth. Finally, we will briefly touch on the role of neural stem cells as both potential targets as well as delivery vectors for other therapies. Interdependence between these varied pathways, both in maintaining health and in causing disease, is clear. Thus, attempts to easily classify some targeted therapies are problematic.
Introduction
Like all cancers, malignant gliomas are characterized by a wide number of genetic and epigenetic abnormalities that account for their properties: unregulated growth, invasion, and resistance to apoptosis. These include but are not limited to gene mutation, amplification, rearrangement and loss as well as promoter methylation and other mechanisms controlling gene expression. Furthermore, as these tumors develop and spread within a complex extracellular matrix (ECM), therapies that target this milieu are also under investigation. Finally, angiogenesis, a critical component of the neoplastic process, is emerging as a powerful area for targeted therapy of malignant gliomas.
As more is learned of the mechanisms underlying both malignant transformation and tumor–host relationships, new anti-cancer treatments are emerging. Many of these newer therapies target components of specific pathways that contribute to the malignant phenotype. These include various signal transduction pathways, those involved in angiogenesis, and still others that regulate the ECM. Collectively, these agents have come to be known as “targeted therapies.” The distinction is useful in that it distinguishes them from other cancer therapies, for example, covalent DNA-binding agents, whose mechanisms of action may be viewed as less specific. However, it should be noted that new agents targeting diverse aspects important in tumorigenesis such as the nucleosome and transcriptome as well as hormones and their receptors continue to emerge and may also prove valuable in anti-cancer therapy.
Malignant gliomas are a heterogeneous group of tumors arising from either macroglial cells – astrocytes and oligodendroglia – or more likely their precursors. They infiltrate brain parenchyma thus existing within the cells and ECM of the normal central nervous system (CNS). The term ‘malignant glioma’ is not recognized under the World Health Organization (WHO) tumor classification scheme.Citation1 In clinical practice, however, the term is commonly used to distinguish between gliomas of relatively lower and higher histologic grades. In this review of our clinical experience with these tumors to date, some studies report findings based upon strict histologic criteria while others describe the experience with a variety of tumor types subsumed under the category ‘malignant glioma.’
As more is learned of the molecular phenotypes of these heterogeneous neoplasms, our methods for classifying them are likely to change. Indeed, the rationale for the development of targeted therapies depends upon our ability to define molecular characteristics that will predict responses to specific interventions. The tumors discussed in this review, their WHO classification, and select details regarding their molecular phenotypes are outlined in .
Table 1 WHO classification of tumors subsumed under the category “Malignant glioma”
Some non-CNS tumors have proven to be quite sensitive to specific targeted therapies when used as single agents. Malignant gliomas have proven largely resistant to this approach. The reasons underlying our experience to date are complex but may relate to the molecular heterogeneity of malignant gliomas as well as redundancy in aberrant signaling pathways active in these complex tumors. Finally, their origin and residence within the CNS, a tissue that tightly regulates its intra- and extracellular milieu, may explain, in part, the modest results observed with targeted therapies to date. New compounds are under development and existing ones are now being used in combinations, both with one another and with other therapeutic modalities including conventional chemotherapy and radiotherapy.
Epidermal growth factor receptor
Our discussion begins with epidermal growth factor receptor (EGFR), one of the most studied potential targets in gliomas. EGFR is a human EGF receptor (HER)-family tyrosine kinase. It consists of an extracellular ligand-binding domain, a single transmembrane domain, and an intracellular tyrosine kinase domain. EGFR dysregulation can occur at many levels in malignant gliomas. Often more than one alteration is present in the same tumor. This is especially true with glioblastoma multiforme (GBM), the most common and aggressive of these tumors. As shown in , GBM are classified as either primary, arising de novo; or secondary, arising as a consequence of progression from a tumor of lower grade.Citation1 Structural and/or functional abnormalities in the EGFR gene and protein expression differ substantially between these two subsets. For example, EGFR gene amplification is seen in 40%–60% of primary GBM with increased protein expression in 50%–60%.Citation2 This phenomenon is seldom encountered in secondary GBM. Similarly, EGFR gene mutations are common in primary GBM, encountered in 40%–50%, but rare in secondary cases. The majority of the mutations are truncations. Five are recognized and classified as variants I–V (vI–vV). Of these, the constitutively active, ligand-independent vIII mutation is most common.Citation3
The EGFRvIII mutation has been reported in 27% to 50% of GBM patients.Citation3–Citation5 It is found only in tumors with EGFR overexpression or amplification. The presence of EGFR overexpression and amplification is closely correlated.Citation6 Similar alterations in EGFR gene structure and protein expression are uncommon in both lower grade astrocytomas and oligodendroglioma.Citation7 EGFRvIII is formed by an in-frame 268 amino acid deletion of exons 2 to 7 in the extracellular domain of the receptor. This creates a unique epitope which can serve as a target for therapies discussed further on. EGFRvIII does not bind to the EGFR ligands. It remains surface-bound and does not internalize. There is some evidence demonstrating that EGFR amplification and EGFRvIII overexpression confer a poor prognosis for overall survival.Citation6 Total expression levels of EGFR do not appear to have prognostic significance.Citation8
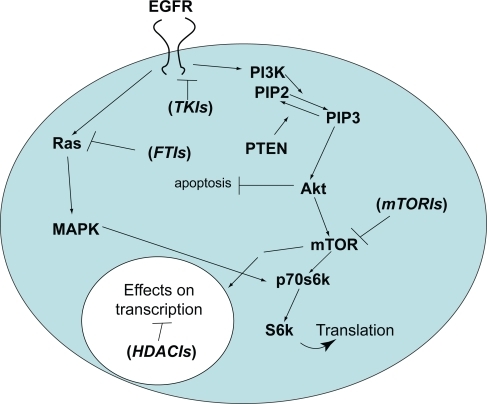
Upon binding one of several potential ligands (EGF, transforming growth factor-α [TGFα], and others), EGFR homo- or heterodimerizes with other HER-family receptors leading to autophosphorylation of the tyrosine residues on its intracellular domain.Citation9 This leads to downstream activation of several pathways important in the development of the malignant phenotype, outlined in . As more is learned about these downstream pathways, new areas for the development of targeted therapies are emerging. We will discuss agents that directly target EGFR first and then proceed to its varied potential downstream targets in the sections that follow.
Figure 1 Simplified diagram of the two major pathways affected by EGFR-mediated signaling in malignant glioma cells. The PI3 kinase pathway is shown on the right and the Ras/Map kinase pathway on the left. Following EGFR activation, a series of sequential phosphorylation events result in cell proliferation and survival through increased transcription and translation of key proteins while inhibiting pro-apoptotic pathways. Agents that target key points in the pathway that are discussed in the text are shown in parentheses at their respective sites of action.
EGFR small molecule inhibitors
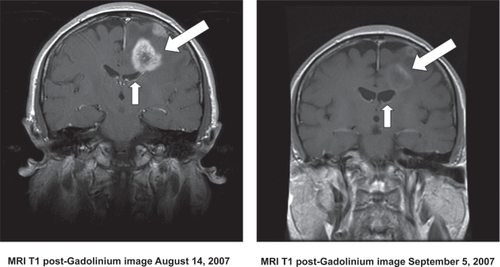
Several tyrosine kinase inhibitors (TKIs) that downregulate EGFR-mediated signaling by blocking its intracellular ATP domain have been developed and tested as therapeutic agents against a wide variety of cancers, including malignant glioma. Two of the most studied agents – erlotinib (OSI-774, Tarceva®; Genentech, San Francisco, CA) and gefitinib (ZD1839, Iressa®; AstraZeneca, Washington, DC) – have been employed in both phase I and II clinical trials involving malignant gliomas of varying grades and histologies. These are summarized in . Although neither agent met the predetermined efficacy endpoints in any study, striking and durable responses to treatments were seen in a subset of patients. An example of such a response is shown in . Because of these remarkable responses in tumors largely refractory to all therapy, attempts have been made to understand the molecular mechanisms that underlie the success or failure of these agents in order to improve their therapeutic impact. There are many potential causes for both the success and failure of single-agent TKIs as antiglioma therapy. In order to understand these influences we must move downstream of EFGR to some of the pathways influenced by EGFR activation.
Figure 2 MRI scans showing a typical prolonged response to single-agent erlotinib in a patient with progressive GBM. The image in the left panel is consistent with a partially necrotic enhancing mass and surrounding peri-tumoral edema. The image on the right, obtained 16 months after the initiation of erlotinib, reveals a decrease in the mass and associated edema. The patient was on high-dose dexamethasone treatment (to control cerebral edema) at the time of erlotinib treatment initiation and was off all dexamethasone at the time of the follow-up scan.
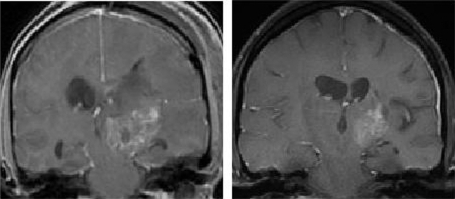
Table 2 Epidermal growth factor receptor tyrosine kinase inhibitor trials in adult malignant gliomas
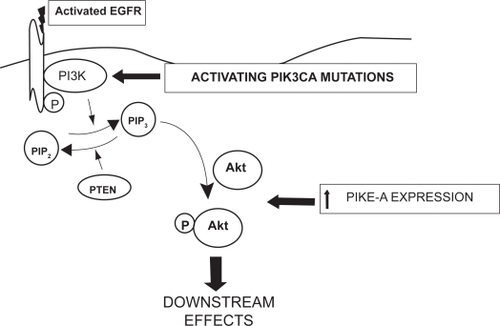
The first pathway which we will discuss will be the phosphatidylinositol-3-kinase (PI3K)/Akt pathway. Following EGFR activation, PI3K catalyzes the conversion of phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3), as shown on the right side in . PI3K has been shown in other cancers to be activated by the Ras pathway, which will be discussed later.Citation10 Subsequent phosphorylation of the serine/threonine kinase, Akt, promotes cell growth, and proliferation through the mammalian target of rapamycin (mTOR) while simultaneously inhibiting apoptosis.Citation11 Additional perturbations at several points in this pathway could render the single-agent TKIs discussed above inactive. For example, as shown in and , PIP3 is dephosphorylated back to PIP2 by the tumor suppressor gene, phosphatase and tensin homologue deleted on chromosome 10 (PTEN). PTEN function is affected by gene deletion and/or mutation in the majority of GBM.Citation12 Again, this is more commonly seen in primary than either secondary GBM or malignant gliomas of lower grade.Citation12
Figure 3 Schematic diagram of the potential sites in the PI3 kinase pathway where resistance to single-agent TKIs directed against the EGFR might occur. 1) The phosphatase activity of the PTEN gene product is lost in the majority of GBM, favoring accumulation of the triphosphate form of phosphatylinositol, PIP3; 2) Activating mutations in the PIK3CA gene, controlling the catalytic subunit of PI3K are well-documented, also favoring accumulation of PIP3;3) In addition, increased expression of the phospho-AKT enhancer, PIKE-A, is found. These alterations may occur in isolation or in any combination in GBM.
Two retrospective studies using selected samples from two of the clinical trials discussed above where tumor responses were observed, attempted to determine the predictive value of EGFR status (wild-type or EGFRvIII) in combination with PTEN activity to TKI response. Each study used different methods to investigate this hypothesis, reaching partially contradictive conclusions. In one report, only those tumors expressing EGFRvIII in the presence of intact PTEN were responsive to inhibition by these TKIs.Citation13 In the other, only tumors expressing wild-type EGFR (not tumor-expressing EGFRvIII) responded to therapy.Citation14 In the latter study, PTEN activity was implied by studying activation of Akt, its downstream target. In this case, no tumor thought to lack PTEN activity responded to therapy with these agents. Based upon these observations, it would appear that functional PTEN is necessary (but perhaps not sufficient) for TKI activity in malignant glioma. Particulars surrounding EGFR status remain unclear, however. There are no reported mutations in the EGFR TKI-binding domain in gliomas that would render EGFR TKIs ineffective as have been demonstrated in other cancers.Citation15
Additional, untested hypotheses that might explain these results also exist. For example, potentially oncogenic mutations in a PIK3CA gene encoding the p110α catalytic subunit of PI3K are well documented in GBMCitation16 (see ). Activation by Ras binding to PIK3CA has been noted in other solid tumors.Citation10 Furthermore, increased expression of the Akt modulator, PI3K-enhancer (PIKE-A), is also seen in gliomas.Citation17,Citation18 Activity at one or both of these sites could render TKIs unsuccessful, independent of EGFR and/or PTEN status. On the other hand, it does not appear that direct dysregulation or mutation of Akt plays a significant role in mediating this resistance. Of note, there has been only one report of mutation of the Akt1 isoform in a gliosarcoma.Citation19 Finally, alterations in the Ras/mitogen-activated protein kinase (MAPK) pathway (shown on the left in and discussed in detail below) might also influence susceptibility of malignant gliomas to inhibition by TKIs. In summary, given the complexity of the pathways involved and the many points at which important alterations can occur, it is not surprising that a straightforward predictive test for response to single-agent TKIs eludes us.
Anti-EGFR antibodies
Some of the same proteins integral to molecular signaling pathways may also serve as either tumor-enriched or – specific targets of antibodies. Monoclonal antibodies (mAb) are used with increasing frequency in cancer therapy. They have the advantage of high specificity for their targets, yet may exert their effects through several mechanisms. mAb receptor binding can result in either receptor activation or inhibition. Alternatively, mAb may cause a cell’s death via antibody-dependent cellular toxicity (ADCC). Furthermore, they may be used as carriers of toxins to enhance their specificity and reduce toxicity. Targeted antibody therapy has been used successfully in breast cancer with the anti-HER2 antibody trastuzumab (Herceptin®; Genentech) and in colorectal cancer with the anti-wtEGFR and anti-EGFRvIII antibody cetuximab (IMC-C225, Erbitux®; Bristol-Myers Squibb, New York, NY).
Evidence for mAb uptake in malignant gliomas following intravenous (IV) administration comes from several sources. Two studies using the EGFR-specific murine monoclonal antibody, MAb 425 (EMD55900; Merck and Co., Inc., Whitehouse Station, NJ), demonstrated binding in human brain tissue. These studies provided proof of concept but were not powered to evaluate efficacy.Citation20,Citation21 A phase I/II trial using the radiolabelled EGFR-specific humanized monoclonal antibody nimotuzumab (hR3, OSAG 101, Therolac®; Oncoscience AG, Wedel, Germany) also demonstrated some tumor uptake.Citation22 EGFR antibodies have also been combined with radiotherapy with the antibody serving as a directed carrier. A small randomized study using MAb 425 bound to iodine125 administered IV in patients with grade III and IV gliomas noted no benefit.Citation23 Other trials using antibodies to target EGFR in gliomas are ongoing and include: IV cetuximab, in combination with radiation therapy (RT) and temozolomide,Citation24 nimotuzumab either in combination with RT in diffuse pontine gliomas or alone at recurrence, as well as a phase III trial in GBM adding it to temozolomide after RT/temozolomide, and a large phase II trial of MAb 425 bound to I125.
The relatively large size of mAbs has raised concerns about their potential for use in the treatment of malignant glioma. The blood–brain barrier (BBB) is usually cited as the main concern as it may limit access of mAb, administered systemically, to their targets. It should be noted, however, that the blood–brain tumor barrier (BBTB) differs significantly from the BBB. Furthermore, as discussed above, there is evidence for at least some delivery of mAb, administered systemically, to brain tumors. Thus, the failure of an antibody-directed therapy may or may not be due to issues of intra-tumoral mAb concentration. Nevertheless, this concern has led to a variety of techniques aimed at “circumventing” the BBB. Preclinical data include the use of cetuximab, either alone or in combination with boronated EGFRvIII-specific antibodies (L8A4) given intratumorally by convectionenhanced delivery (CED) in rat glioma models.Citation25 Thus far, one phase I clinical trial using intracavitary administration of radiolabelled nimotuzumab in the treatment of progressive GBM and AA has been published. Of the 11 patients enrolled, one with an AA demonstrated a sustained complete radiographic response (CR). The complications noted in the study include those associated with intracranial catheters as well as radiation necrosis.Citation26
EGFR vaccines
Vaccines capable of producing both humoral (antibody mediated) and cell-mediated anti-brain tumor responses are an area of basic, translational, and clinical research. True for most cancers, the identification of brain tumor-specific antigens has proven challenging. In the case of malignant glioma, the EGFRvIII mutant has proven a highly specific target in those cases where it is expressed. An antigenic epitope on this mutant receptor stimulates highly specific humoral and cellular immune responses. This has been studied in both pre-clinical models and phase I and II studies in patients with recurrent malignant glioma.Citation27,Citation28 An immune attack in the CNS focused on a tumor-specific epitope raises concern for collateral damage. The relative immune privilege of the CNS also makes one question the ability to raise an adequate immune response to the tumor target. To the surprise of many, this approach has proven to be safe and also appears effective for some patients.Citation27 Based on these findings, a phase II clinical trial is ongoing in patients with newly diagnosed GBM.
mTOR inhibitors
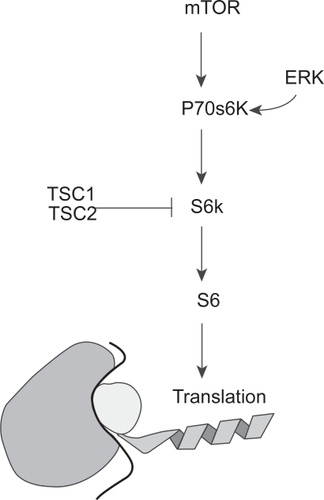
As shown in , the serine/threonine kinase, mTOR, lies downstream of Akt in the PI3K pathway. mTOR is involved in the regulation of cell growth, proliferation, motility, and survival. mTOR, in turn, activates another serine-threonine kinase, p70s6 kinase (p70s6K). p70s6K serves as a point of convergence for the Ras/MAPK/extracellular signal regulated kinase (ERK) pathway (see ). p70s6K in turn activates S6K which phosphorylates the ribosomal protein S6. Activated S6 upregulates translation. Of potential importance in glioma biology and therapy, the tuberous sclerosis complex 1 and 2 gene products (TSC1 and TSC2, respectively) inhibit S6K activation ().Citation30 TSC1 and TSC2 mutations are involved in the development of subependymal giant cell astrocytomas, a low grade tumor found in patients with tuberous sclerosis. Although mutations of TSC 1 and 2 are not found in high-grade gliomas, loss of function of these proteins may prove to play a role in the development of a small percentage of high-grade gliomas as well, making them potential targets for directed therapies. Loss of heterozygosity (LOH) for either the TSC1 or TSC2 locus was found in less than 10% of grade III and IV astrocytomas. No mutations of the intact allele were found. It is possible that LOH at these loci is merely an expression of the myriad genetic aberrancies in malignant gliomas. It is also possible that there may be transcriptional silencing of the gene. It has been recently demonstrated that TSC1 and TSC2 mRNA levels in GBM are decreased in comparison to normal brain or grade II astrocytomas.Citation31 While there are a number of points in this pathway where modulation may be of benefit for the treatment of gliomas, we will limit our focus to mTOR inhibitors before proceeding to the RAS/MAPK pathway.
Figure 4 A schematic showing the convergence of the PI3K pathway (through mTOR) and the Ras/MAPK pathway (through ER K) on p70s6K and its effect on translation. Note the inhibitory effect of TSC1 and 2, an area not fully explored in gliomagenesis.
In malignant gliomas a number of mTOR inhibitors have been studied in the treatment of malignant glioma. These include: sirolimus (Rapamune®; Wyeth Pharmaceuticals, Madison, NJ) US Food and Drug Administration (FDA)-approved for the prophylaxis of solid organ transplant rejection and temsirolimus (CCI-779, Torisel®; Wyeth Pharmaceuticals) FDA-approved for use in renal cancer. Both everolimus (RAD001; Novartis Oncology, East Hanover, NJ) and deferolimus (AP23573; ARIAD/Merck), still experimental, have all been evaluated in clinical trials alone or in combination with other therapies in gliomas.
Sirolimus has been evaluated in combination with the EGFR TKIs erlotinib or gefitinib in a retrospective case series of patients with recurrent malignant gliomas. The six-month progression-free survival (PFS-6) was encouraging and the majority of patients had either stable disease or partial response on radiographic imaging.Citation29,Citation32 A phase I trial using it in combination with gefitinib demonstrated tolerability and with over 40% of the patients demonstrating stable disease (SD) or partial response (PR), the results are promising.Citation30 There is currently one active phase I/II trial in gliomas using sirolimus as a single agent and two trials evaluating sirolimus in combination with erlotinib. The use of mTOR inhibitors in combination with EGFR-specific TKIs is rational given the evidence, discussed above, for resistance to single agent EGFR-specific TKIs in the majority of those with GBM.
Temsirolimus, an ester analogue of sirolimus, was evaluated as a single agent in a phase II trial of recurrent GBM. No patients had radiographic response based on the criteria used.Citation33 However, approximately a third of patients had a qualitative decrease in enhancement or mass effect while on a stable or decreased dose of steroids. Unfortunately PFS-6 in these heavily pretreated patients was low. Correlative studies demonstrated potential to predict response prior to treatment by detecting elevated tumor levels of the downstream phosphorylated p70s kinase. In addition, early onset hyperlipidemia may have been predictive of a response.Citation33 Another phase II trial evaluating temsirolimus in recurrent primary GBM found radiographic stabilization with SD or PR but no prolonged survival.Citation34 There are currently a number of ongoing clinical trials further evaluating temsirolimus alone or in combination with various agents including targeted therapies such as erlotinib, sorafenib (a multiple kinase inhibitor) (Nexavar®; Bayer, Pittsburgh, PA), the farnesyl transferase inhibitor (FTI) tipifarnib (R115777, Zarnestra®; Johnson and Johnson, Langhorne, PA), and the antiangiogenic agent bevacizumab (Avastin®; Genentech) as well as the alkylating agent temozolomide for malignant gliomas.
Everolimus may, in addition to its mTOR activity, enhance the effects of radiotherapy.Citation35 Thus, its further evaluation in gliomas is warranted. Furthermore, when everolimus was used in combination with the EGFR/vascular endothelial growth factor receptor 2 (VEGFR2) TKI, AEE788 (Novartis) it demonstrated improved growth inhibition and median survival greater than with single agents in human glioma xenografts in mice.Citation36 Studies evaluating its use as a single agent or in combination with temozolomide or with other targeted agents including the multiple kinase inhibitor imatinib (Gleevec®; Novartis), gefitinib, AEE788, and bevacizumab are ongoing. There are no published clinical trial results to date using everolimus in any brain tumor setting.
RAS
The Ras/MAPK signal transduction pathway represents another key pathway mediated by EGFR (see , left side). Ras, consists of a family of guanosine triphosphatases (GTPases): H-ras, K-ras, N-ras, activated when they move from the cytoplasm to a membrane bound state. This process is modulated in part by prenylation, the adding of farnesyl or geranylgeranyl to the carboxy-terminal cysteine residue of the proteins, and known respectively as farnesylation or geranylation Citation38 Unlike many other solid tumors, there are only rare activating mutations of Ras described in human GBMs.Citation37,Citation39 However, amplification of Ras is commonly seen in a variety of human gliomas, making it an apt therapeutic target.Citation40 Furthermore, over-activation of EGFR in both its wild-type and mutant forms as well as over-activation of platelet-derived growth factor (PDGF) may lead to over-activation of Ras.Citation41,Citation42 Upstream TKI activity is therefore likely adequate for upregulation of Ras.Citation58 Ras, in turn activates the MAPK pathway as well as the PI3K/Akt pathway discussed earlier.
Farnesyl transferase inhibitors
Farnesyl transferase inhibitors (FTIs), although typically classified as targeted therapies, have potential effects on a broad range of proteins (~300) which are controlled by farnesylation. This leads to the conjecture that FTIs may be of benefit in multiple subtypes of malignant gliomas. Their broad effects may be of benefit not only in the tumors with Ras pathway upregulation, the context in which they were developed, based on data from in vitro models.Citation40,Citation57
Tipifarnib is a FTI that has been used during and following RT in phase I trials with malignant gliomas of varying grades, including GBMs and pediatric diffuse brain stem gliomas.Citation43–Citation45 Results of two phase II studies for progressive brain tumors have been published to date: one in adults with recurrent malignant gliomas, the other in children tumors of several types, including malignant gliomas.Citation46,Citation47 There was suggestion of some benefit in adults who were thought to have received adequate doses. Another recently published trial of tipifarnib did not demonstrate benefit when used prior to radiation.Citation49 Lovastatin® (Merck), used primarily in the treatment of dyslipidemia, affects prenylation and has been evaluated as a single agent in a phase I/II trial of recurrent malignant glioma.Citation48 The novel route of intranasal administration of perillyl alcohol for its FTI properties has been evaluated in a phase I/II clinical trial.Citation51 It is likely that FTIs such as tipifarnib, and agents with FTI effects such as lovastatin, will be investigated further in gliomas in combination with other agents, particularly those that target the PI3K pathway as these converge on p70s6K (see ).
Histone deacetylase inhibitors
Gene transcription is controlled, in part, by histone acetylation. Two enzymes, histone acetyl transferase and histone deacetlyase (HDAC), mediate this state and are in dynamic equilibrium in the cell. HDAC inhibitors (HDACI) are thought to mediate their anti-tumor effect by causing an accumulation of acetylated histones, leading to a relaxation of chromatin. This leads in turn to increased transcription.Citation52 HDACIs may facilitate preferential expression of tumor suppressor genes, in part, by releasing epigenetically controlled gene silencing.Citation52 HDACI may also affect the cytoskeleton in turn interfering with the mitotic process. Thus, the potential exists for HDACIs to influence the cancer cell at many levels.Citation53
SAHA (suberoylanilide hydroxamic acid, vorinostat, Zolinza®; Merck), FDA-approved for cutaneous T-cell lymphoma, is being evaluated in clinical trials of multiple cancers, including malignant gliomas. Upregulation of pro-aptotic genes such as TNFα and downregulation of pro-growth and anti-apoptotic genes has been demonstrated in glioma cell lines treated with SAHA.Citation54 It has also been demonstrated that SAHA can cross the BBB in mouse glioma xenografts.Citation54 There are yet no published glioma trials using SAHA. However, there are a number of ongoing phase I and II clinical trials with this agent. The majority of these are using it in combination with nontargeted therapies such as temozolomide, carboplatin, as well as bevacizumab, isotretinoin (cis-retinoic acid, Accutane®; Roche, Nutley, NJ) the proteosome inhibitor bortezomib (Velcade®; Millennium Pharmaceuticals, Cambridge, MA).
The anticonvulsant valproic acid (Depakote®) has demonstrated HDACI activity preclinically in models of brain tumors as well as many other types of cancer.Citation56 A synergistic effect between valproic acid and RT has been shown as well.Citation55 There is an ongoing trial adding valproic acid to the standard of care regimen of RT and temozolomide in newly diagnosed GBM. Thus far, no clinical data have been published specifically regarding valproic acid’s HDAC inhibition. However, a limited number of patients in other clinical trials for brain tumors have had their seizures treated with valproic acid. There have been no retrospective evaluations of prior brain tumor trials to look for additive or synergistic effects in the patients taking valproic acid as well as the study agent.
Angiogenesis
The next therapeutic target we will discuss is angionesis, the process of new blood vessel growth in both normal as well as pathologic states. These investigations began with the hypothesis that both tumor growth and spread is angiogenesis-dependant. The first successful use of an angiogenesis inhibitor involved the cytokine, interferon-alpha (IFNα).Citation60,Citation61 Since then, the anti-angiogenic properties of many agents have been explored. Gliomas differ from many other solid tumors in that they seldom metastasize to other sites. However, some malignant gliomas are defined, in part, by endothelial proliferation. GBM demonstrates a greater degree of angiogenesis than most other tumors.Citation63 It stands to reason that the inhibition of angiogenesis would become a focus of anti-glioma therapy, perhaps controlling the growth of the highest grade tumors and preventing the progression of tumors from lower to higher grades. Finally, anti-angiogenic strategies may also alter the vascular niche, which in turn may directly affect the neural stem cells which may be the progenitors of tumor cells.Citation64
IFNα and β
The first anti-angiogenic agent studied in humans was IFNα.Citation61 IFNα has been tested in a large phase III clinical trial in grade III or IV gliomas in combination with the nitrosurea, carmustine (BCNU) vs carmustine alone. There was no improvement in overall survival (OS) or time to progression (TTP).Citation66 IFNα has also been evaluated in combination with a number of other agents including isotretinoin and tamoxifen without notable benefit.Citation67,Citation68 Interferon-β (IFNβ) has also been used, both alone and in combination with other agents in the treatment of newly diagnosed and recurrent gliomas. Results have been modest.Citation65
Thalidomide and related compounds
Thalidomide is a therapeutic agent with a long history. Initially used as a sedative, it was found to be teratogenic, causing the maldevelopment of limbs in children born to mothers taking it while pregnant. It is thought that the anti-angiogenic activity of thalidomide, only discovered many years later, was at least partially responsible for the birth defects.Citation69 Thalidomide is thought to work through a number of mechanisms. One of the primary anti-angiogenic effects is deemed to be secondary to its inhibition of basic fibroblast growth factor (bFGF). A number of additional mechanisms may have both antiangiogenic as well as more broad anti-tumor activity including: modulation of the expression of adhesion molecules, downregulation of TNF, as well as stimulation of CD8+ T-cell responses.Citation69,Citation70
A number of studies using thalidomide, either alone or in combination with cytotoxic chemotherapies have been completed in malignant gliomas both at recurrence as well as in the newly diagnosed setting. When compared to historical controls, thalidomide used in combination with the alkylating agent temozolomide in the newly diagnosed setting demonstrated benefit when compared to nitrosoureas.Citation71 When compared to temozolomide alone, the benefit is less apparent.Citation71 When used for GBM at first diagnosis in combination with temozolomide and another agent with potential antiangiogenic properties, celecoxib there was no significant improvement in progression free survival.Citation72
Lenalidomide (CC-5013, Revlimid®; Celgene), a thalidomide analogue used to treat multiple myeloma and myelodysplastic syndrome, has been used in the treatment of malignant gliomas. In preclinical studies, the anti-angiogenic effects of lenalinomide are more pronounced than those of thalidomide.Citation77 Decreased VEGF secretion and decreased phosphorylated Akt have been found in cellular models of cancers treated with the agent.Citation78 A phase I trial of single-agent lenalidomide in recurrent primary brain tumors mostly GBMs, has been completed.Citation79 It was found to be tolerable. It is currently being investigated either alone or in combination with other therapies in primary brain tumors in both phase I and phase I/II trials,
Anti-VEGF antibodies
VEGF is a potent angiogenic cytokine. Several VEGF isoforms are recognized. They play variable roles in embryogenesis, neo-angiogenesis, and lymphatic channel formation.Citation73 The interaction between isoform VEGF-A and its primary transmembrane receptor, VEGFR-2 is the predominant driving force for angiogenesis. The process, however, is more complicated. Interactions between the various VEGF subtypes and their respective receptors as well as other stimulatory molecules (ie, platelet derived growth factor) and their receptors, also influence this process. These pathways may be modulated by a number of other signals, including angiopoietin-1 (Ang1) and angiopoietin-2 (Ang2), molecules that are integral to the stabilization/destabilization of blood vessels.Citation74
VEGF is produced by most (60%) human tumors. Its expression is upregulated in only some AAs, but in most GBMs.Citation75 This is likely a necessary, but not alone sufficient, stimulus in driving the progression of secondary GBM from tumors of lower grade. For this reason, anti-VEGF therapies are under active investigation. Bevacizumab, a recombinant humanized monoclonal antibody directed against VEGF-A, is being evaluated both alone and in combination with other agents in the treatment of both newly diagnosed and recurrent malignant gliomas. Considerable interest has been generated in the remarkable radiographic responses, (reduced enhancement and tumor-associated edema), seen following its use (see ). While encouraging, and often correlated with an improvement in symptoms, this effect has clouded attempts to determine the response rate in these tumors when compared to historical controls and is leading to new paradigms for determining both tumor response and its duration.Citation76 Those agents that either have been or are now being used in combination with bevacizumab include both temozolomide and irinotecan (CPT-11, Camptosar®, Pfizer), a topoisomerase-I inhibitor, at recurrence or at initial diagnosis of high grade gliomas.Citation76,Citation80–Citation84 The results of these trials are promising in this group of relatively refractory patients. Both 6PFS and OS are significantly increased when compared to matched historical controls. Median OS has been reported between 9.7 to 11.5 months in GBM. 6PFS for GBM has been reported between 30%–63%. Median OS and 6PFS in the patients with grade III gliomas has been longer than those with grade IV, as would be expected. This is a stark improvement when compared to 6PFS of 21% and OS of 7.5 months (30 weeks).Citation80–Citation83
Figure 5 MRI scans showing a typical response of a recurrent GBM to bevacizumab. The image on the right shows the tumor-associated enhancement (large arrow) and accompanying compression of the ventricle by mass effect (small arrow). The image on the right shows the same region following a one month course of bevacizumab. Note the marked reduction in contrast enhancement and mass effect.
Retrospective review of tissue from selected study participants (AA and GBM) from the time of diagnosis, prior to treatment with bevacizumab and irinotecan, in one of the phase II trials noted a correlation between specific biomarkers and patients responses. VEGF levels in the tumor at time of diagnosis correlated with radiographic response, but not over-all survival. The pretreatment levels of the hypoxia marker, hypoxia induced carbonic anhydrase 9 (CA9) corresponded to a poor one year survival. The median survival for patients with tumors demonstrating high CA9 expression on IHC was 37 weeks. Those with low or no CA9 expression had a median survival (74 weeks) which was twice as long.Citation85 A retrospective evaluation of the clinical outcomes of patients with recurrent high grade gliomas treated with bevacizumab and irinotecan at a single institution demonstrated promising results. Six month progression-free survival (PFS6) for patients with GBM was almost 64% with a median overall survival (OS) in these patients of 11.5 months. Over half of the patients demonstrated a diffuse pattern of disease spread at recurrence.Citation83 Refer to for the published trials of bevacizumab in glioma. There are numerous ongoing phase II clinical trials evaluating the role of bevacizumab in newly diagnosed GBM in combination with either temozolomide, or irinotecan, during or after RT, The results from these studies are still pending. There are also ongoing clinical trials using bevacizumab in combination with targeted therapies including other anti-angiogenic agents, such as enzastaurin (LY317615, Lilly), a serine threonine kinase affecting protein kinase C β, in the recurrent setting.
Table 3 Clinical trials of bevacizumab in gliomas
VEGF decoy receptors
Aflibercept (VEGF Trap®, Regeneron) is a soluble decoy receptor for VEGF-A and placental growth factor (PGF). It is composed of the domains of VEGF receptor (VEGFR) 1 and 2 bound to the constant domain of human immunoglobulin G1. Recently published studies in mice with glioma xenografts reveal an improved overall survival. They also demonstrate what appears to represent the development of a more invasive diffusely infiltrative phenotype.Citation86 This resembles the diffuse pattern of disease recurrence which is being seen with bevacizumab and may be found with all anti-VEGF therapies. Aflibercept is being actively investigated in malignant gliomas both at initial diagnosis and recurrence.
VEGFR inhibitors
Cediranib (AZD2171, Recentin™, AstraZeneca) is a small molecule tyrosine kinase inhibitor with activity against multiple VEGFRs, PDGFa, PDGFb, and c-kit. This “targeted” therapy has effects on multiple arms of the angiogenic pathway. Correlative studies from a phase II clinical trial demonstrate potential biomarkers, which may prove important in guiding anti-angiogenic therapy in the future. When patients progressed through cediranib there was an elevation of serum angiogenic factors bFGF and SDF1a. However, when there was evidence of radiographic changes to the vasculature during a drug holiday the serum levels of those factors did not increase. These findings suggest that the timing of dosing of anti-VEGF or VEGFR agents may be of significant importance. In this same study of patients with recurrent GBM, there was a modest increase in OS (211 days vs 175 days) when compared to historical controls. A benefit that was also noted was a decrease in the vascular permeability which is associated with cerebral edema.Citation87 This in turn may help patients decrease their steroid requirements and improve their quality of life.
Endothelin
Interactions between endothelin-1 (ET-1), ET-2, and ET-3 with their receptors ETa and ETb are important in normal cell growth and proliferation as well as in malignancy. ETa and ETb are present in human GBM.Citation88 ET-1 facilitates increased VEGF. It also increase HIF-1a expression in normoxic and hypoxic states.Citation89
A phase I study through the NABTT of atrasentan (Xinlay®, Abbott), an ETa receptor antagonist, used in malignant gliomas (almost all recurrent), demonstrated its tolerability. It blocks ET-1 induced increases in VEGF. It has also been shown to block ET-1 induced activation of HIF-1a. In contrast to bevacizumab, where there is a marked decrease in the vascular permeability, many of the side effects with atrasentan such as edema, rhinorrhea can potentially be attributed to increased vascular permeability. In this study there were some partial responses as well as stable disease.Citation90 Because of these results, a phase II trial with RT/TMZ and atrasentan is planned.
THC
Δ9-tetrahydro cannabinoid (THC) has been shown to have effects on angiogenesis and tumor growth in mammalian xenograft models of gliomas. Its activity is thought to be at least in part secondary to its effect on the endogenous cannabinoid receptor-1 (CR-1) and/or CR-2. A phase I clinical trial in recurrent GBM patients treated with administration of intracavitary THC solution administration proved to be relatively safe. However, in this study, the investigators were not able to demonstrate a significant anti-angiogenic effect due to the therapy. As with most clinical trials in gliomas, there were a few long term survivors.Citation91
COX-2 pathway
The cyclooxegenase (COX) pathway plays a role in angiogenesis. Arachidonic acids are converted to prostaglandins and thromboxanes via the activity of COX-2. Prostaglandin E2 (PGE2) has been shown to induce angiogenesis in animal models of cancer. Selective inhibition of COX-2, using celecoxib (Celebrex®, Pfizer) has been looked at extensively in a number of cancers including gliomas. In preclinical models of glioma the effect of celecoxib has been only modest. In turn it has been used primarily in combination with other agents in the clinical setting without any marked improvement in clinical or radiographic parameters.Citation72,Citation92
Integrins
The final targeted anti-angiogeneic therapies we will discuss are those which target the integrins involved in angiogenesis. Integrins are cell surface receptors composed of alpha and beta subunits and are involved in binding and signaling between cells. There are 24 unique integrin heterodimers. Of particular interest as therapeutic targets in many types of cancer are αvβ3 and αvβ5 found on endothelial cells as well as tumor cells and know to play a role in angiogenesis. The integrins, unlike some of the targets mentioned previously in this review are not kinases. They are however involved in activating signaling pathways originating from the focal adhesion complexes of which they are a component. αvβ3 binds to a number of ligands including fibrinogen and fibronectin and is found to be present in both tumor angiogenesis and chronic inflammation. Its expression is stimulated by basic fibroblast growth factor (bFGF). Antagonism of αvβ3 induces apoptosis of endothelial cells. TGFα and VEGF activate a parallel angiogenic pathway involving αvβ5.Citation93
The two above mentioned integrins serve as very reasonable targets in gliomas. Cilengitide (EMD121974; Merck) a αvβ3/αvβ5 antagonist has been evaluated in a number of solid tumors including gliomas. Two phase I studies have been published evaluating its use in gliomas in the adult and pediatric populations and was found to be extremely tolerable.Citation94,Citation95 Some of the preclinical data in pancreatic cancer demonstrated benefit only with continuous administration. This may be of importance in the phase II setting. A recently closed phase I trial which included glioma patients used continuous infusion cilengitide; however, the results are still pending. A phase II study using bolus scheduling of cilengitide was recently completed and is awaiting publication.
Table 4 A selection of ongoing clinical trials in malignant gliomas
Neural stem cells
There is growing evidence supporting the role of neural stem cells (NSCs) as well as the more differentiated glial progenitor cells in gliomagenesis.Citation96 The exact cell(s) of origin has(have) not been discovered. NSC have been isolated from the subventricular zone and the hippocampus in human brains.Citation97,Citation98 NSC and tumor progenitor cells bear markers including the glycoprotein CD133, the carbohydrate antigen CD15, the transcription factor OLIG2, and the intermediate filament protein nestin, distinguishing them from the remaining tumor cell population. The resilience of these stem/progenitor cells may underlie the refractoriness of tumors to our current therapies such as RT.Citation99,Citation100 It is likely that our treatment regimens will need to incorporate therapies with activity against both this extremely specialized cell population as well as to the non-pluripotent cells that compromise the tumors. Some of the molecular pathways discussed earlier may remain important when targeting glial tumor progenitor cells. For example, there is preclinical evidence that EGFR-specific TKIs discussed earlier cause cell death in some glioma populations exhibiting stem cell markers. It appears intact PTEN is essential for this.Citation101 Akt inhibition using small molecule kinase inhibitors (SH-6) of cancer stem cells leads to decreased cell motility and death. It also leads to improved survival in a mouse glioma xenograft model. There is also some evidence for decreased cell viability with the use of the mTOR inhibitor rapamycin and the PI3K inhibitor LY290042.Citation102 Another promising target in glioma stem cells, not discussed earlier in this review, is the neural cell adhesion molecule L1CAM, which is important for cell growth, migration, and survival. CD133+ glioma cells are enriched for L1CAM. Use of lentivirus shRNA targeting L1CAM has demonstrated growth inhibition in cellular models and improved survival in mouse glioma xenograft models.Citation103 There is very active preclinical work investigating the targeting of glioma stem cells. The bulk of clinical trial activity using stem cells however is not as a therapeutic target, but in their use as a rescue after high-dose chemotherapy.
There is also active ongoing work in the use of NSCs as delivery mechanisms for other targeted therapies. It has been demonstrated that NSCs and bone marrow derived mesenchymal stem cells (MSC) home in on glial tumor cells in animal models.Citation104 This may prove to be an efficacious vehicle for administering and adequately distributing a targeted therapy to tumor cells. NSCs and MSCs have been used to deliver via viral vectors a number of genes including interleukins, INFβ, as well as genes effecting the extracellular matrix, apoptosis, and pro-drug activating enzymes.Citation105 NSCs appear to have a tropism towards the necrotic center as well as invading edge of glioma xenografts.Citation106 There is concern that many therapies do not reach adequate concentrations in regions of necrosis within the tumor, and using stem cells may be a means to circumvent this. Although the xenograft model does not accurately represent the invasive nature of gliomas in humans, the tumor cells which are most actively invading the surrounding parenchyma are an important target. It has recently been shown that NSCs and MSCs can deliver oncolytic adenoviruses to tumor cells with good distribution throughout the tumor, and not relying on passive diffusion of the adenoviruses.Citation107,Citation108
Gene therapy
The final means of addressing specific targets in gliomas which we will touch on is gene therapy. Gene therapy is a means for either replacing defective or silent tumor suppressor genes in tumors or of adding in new genes which may serve an oncolytic purpose. While we will only address this subject briefly, further reading can be found in a recent review.Citation109 Components of the molecular pathways such as p53 or PTEN, could serve as a potential way to reverse the malignant phenotype and potentially make the tumor cells more susceptible to other specific therapies. Transduction of p53 has been explored in clinical trials, however, many of the tumor cells further from the administration site of the vector did not take up the virus and express p53.Citation110 Efforts are also ongoing using the insertion of methylguanine methyl transferase (MGMT) into hematopoetic cells in conjunction with traditional chemotherapies in combination with O-6-benzylguanine (O6BG). The majority of more recent scientific effort, however, has been to introduce genes into the tumor that would lead to cell death. Viral thymidine kinase enzyme, which is targeted by the antivirals acyclovir (Zovirax®; GlaxoSmithKline, Philadelphia, PA), has been incorporated into the tumor cells DNA. Treatment with the antiviral agents leads to DNA chain termination, potentially leading to cell death.Citation111,Citation112 Various cytokines including IFNβ, interleukin 2 (IL-2), IL-4, IL-12, as well as others are being evaluated.Citation113–Citation116
Viral vectors have been used as the primary means of introducing genes into tumor cells. Nonreplicating or conditionally replicating adenoviruses and herpes viruses have been primarily used. One of the limitations of delivery of genes via viral vectors has been an inability to infect all tumor cells. As discussed above, NSCs are being used to circumvent this problem. There are currently clinical trials, both ongoing as well as in development, using gene therapy to treat malignant gliomas.
Gene therapy continues to hold significant promise in the treatment of cancers, including malignant gliomas. Tremendous efforts have been made to overcome the various obstacles which impede the efficacy of this treatment modality.
Conclusion
We have reviewed the experience to date with targeted therapies in the treatment of malignant gliomas. The data presented reflect years of work by basic scientists, pharmacologists, and clinical investigators. Many human subjects have participated in studies. In reviewing the clinical outcomes, readers may reach the conclusion that little impact has been made on malignant gliomas to date. This may hold for these tumors as a whole. However, the impressive responses seen in subsets of patients following treatment with either single-agent tyrosine kinase inhibitors or EGFRvIII vaccines speak to the potential of this approach. Given these findings, it is tempting to ponder the relevance of increasingly personalized medicine as the molecular heterogeneity of these tumors is well documented. Alternatively, the promising results emerging in the majority of patients treated with anti-VEGF antibodies upon tumor progression argues in favor of the continued search for an “Achilles’ heel.” Ultimately, these approaches are not mutually exclusive and offer new hope for people suffering the consequences of malignant glioma. Future directions include combinations of various targeted agents, either with one another or with more conventional chemotherapies and/or radiotherapy. These studies are underway. In addition, new agents, as yet untested, are in active development. Thus, after decades of limited progress in advancing the treatment of malignant gliomas, breakthroughs are occurring co-incident with the development of rationally targeted agents derived from an improved understanding of cancer biology.
Disclosure
The authors report no conflicts of interest in this work.
References
- LouisDNOhgakiHWiestlerODCaveneeWKWHO Classification of Tumors of the Central Nervous System4th editionLyon, FranceInternational Agency For Research on Cancer2007
- WongAJBignerSHBignerDDKinzlerKWHamiltonSRVogelsteinBIncreased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplificationProc Natl Acad Sci U S A19878419689969033477813
- WickstrandCJMcClendonREFriedmanAHBignerDDCell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIIICancer Res19975718413041409307304
- BiernatWHuangHYokooHKleihuesPOhgakiHPredominant expression of mutant EGFR (EGFRvIII) is rare in primary glioblastomasBrain Pathol200414213113615193025
- PelloskiCEBallmanKVFurthAFEpidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastomaJ Clin Oncol200725162288229417538175
- ShinojimaNTadaKSiraishiSPrognostic value of epidermal growth factor receptor in patients with glioblastoma multiformeCancer Res200363206962697014583498
- ReifenbergerJReifenbergerGIchimuraKSchmidtEEWechlerWCollinsVPEpidermal growth factor expression in oligodendroglial tumorsAm J Pathol1996149129358686753
- ChakravartiASeiferheldWTuXImmunohistochemically determined total epidermal growth factor receptor levels not of prognostic value in newly diagnosed glioblastoma multiforme: report from the Radiation Therapy Oncology GroupInt J Radiat Oncol Biol Phys200562231832715890570
- YardenYShilohBZSnapshot: EGFR signaling pathwayCell20071315101818045542
- YuanTLCantleyLCPI3K pathway alterations in cancer: variations on a themeOncogene200827415497551018794884
- VivancoISawyersCLThe phosphatidylinositol 3-kinase Akt pathway in human cancerNat Rev Cancer20022748950112094235
- EndersbyRBakerSJPTEN signaling in brain: neuropathology and tumorigenesisOncogene2008275416543018794877
- MellinghoffIKWangMYVivancoIMolecular determinants of the response of glioblastomas to EGFR kinase inhibitorsN Engl J Med2005353192012202416282176
- Haas-KoganDAPradosMDTihanTEpidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinibJ Natl Cancer Inst2005971288088715956649
- KobayashiSBoggonTJDayaramTEGFR mutation and resistance of non-small-cell lung cancer to gefitinibN Engl J Med2005352878679215728811
- GalliaGLRandVSiuI-MPIK3CA gene mutations in pediatric and adult glioblastoma multiformeMol Cancer Res200641070971417050665
- BroderickDKDiCParrettTJMutations of PI3KCA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomasCancer Res200464155048505015289301
- LiuRTianBGearingMHunterSYeKMaoZCdk5-mediated regulation of the PIKE-A-Akt pathway and glioblastoma cell invasionProc Natl Acad Sci U S A2008105217570757518487454
- SchullerURuiterMHermsJKretzschmarHAGrabson-FrodlEAbsence in the mutations to AKT1 oncogene in glioblastomas and medulloblastomasActa Neuropathol2008115336736818172656
- FaillotTMagdelenatHMadyEA phase one study of an anti-epidermal growth factor receptor monoclonal antibody for the treatment of malignant gliomasNeurosurgery19963934784838875477
- StragliottoGVegaFStasieskiPGroppPPoissonMDelattreJYMultiple infusions of anti-epidermal growth factor receptor (EGFR) monoclonal antibody (EMD 55,900) in patients with recurrent malignant gliomasEur J Cancer199632A46366408695267
- RamosTCFigueredoJCatalaMTreatment of high-grade glioma patients with the humanized anti-epidermal growth factor receptor (EGFR) antibody h-R3: report from a phase I/II trialCancer Biol Ther20065437537916575203
- WigodaZKulaDBierzynska-MacyszynzGUse of monoclonal anti-EGFR antibody in the radioimmunotherapy of malignant gliomas in the context of EGFR expression in grade III and IV tumorsHybridoma (Larchmt)200625312513216796458
- CombsSEHeegerSHaselmannREdlerLDebusJSchulz-ErtnerDtreatment of primary glioblastoma multiforme with cetuximab, radiotherapy and temozolomide (GERT) – phase I/II trial: study protocolBMC Cancer2006613316709245
- YangWWuGBarthRFMolecular targeting and treatment of composite EGFR and EGFRvIII-positive gliomas using boronated monoclonal antibodiesClin Cancer Res200814388389118245552
- CasacoALopezGGarciaIPhase I single-dose study of intracavitary-administered Nimotuzumab labeled with 188 Re in adult recurrent high-grade gliomaCancer Biol Ther20087333333918094616
- SampsonJHArcherGEMitchellDAHeimbergerABBignerDDTumor-specific immunotherapy targeting the EGFRvIII mutation in patients with malignant gliomaSemin Immunol200820526727518539480
- HeimbergerABCrottyLEArcherGEHessKRWickstrandCJFriedmanAHEpidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumorsClin Cancer Res20039114247425414519652
- DohertyLGigasDCKesariSPilot study of the combination of EGFR and mTOR inhibitors in recurrent malignant gliomasNeurology200667115615816832099
- ErmoianRPKaprealianTLambornKRSignal transduction molecules in gliomas of all gradesJ Neurooncol2009911192618759130
- ParryLMaynardJHPatelAMolecular analysis of the TSC1 and TSC2 tumor suppressor genes in sporadic glial and glioneuronal tumorsHum Genet2000107435035611129334
- SorensonAGBatchelorTTWenPYZhangWTJainRKResponse criteria for gliomaNat Clin Pract Oncol200851163464418711427
- GalanisEBucknerJCMaurerMJPhase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group studyJ Clin Oncol200523235294530415998902
- ChangSMWenPCloughesyTPhase II study of CCI-779 in patients with recurrent glioblastoma multiformeInvest New Drugs200523435736116012795
- ShinoharaETCaoCNiermannKEnhanced radiation damage of tumor vasculature by mTOR inhibitorsOncogene200524355414542215940265
- GoudarRKShiQHjelmelandMDCombination therapy of inhibitors of epidermal growth factor receptor/vascular endothelial growth factor receptor 2 (AEE788) and the mammalian target of rapamycin (RAD001) offers improved glioblastoma tumor growth inhibitionMol Cancer Ther20054110111215657358
- KnobbeCBReifenbergerJReifenbergerGMutation analysis of the Ras pathway genes NRAS, HRAS, KRAS, and BRAF in glioblastomasActa Neuropathol2004108646747015517309
- SebtiSMProtein farnesylation: implications for normal physiology, malignant transformation, and cancer therapyCancer Cell20057429730015837619
- JeukenJvan den BroeckeCGijsenSBoots-SprengerSWesselingPRas/Raf pathway activation in gliomas: the result of copy number gains rather than activating mutationsActa Neuropathol2007114212113317588166
- FeldkampMMLauNRoncariLGuhaAIsotype-specific Ras.GTPlevels predict the efficacy of farnesyl transferase inhibitors against human astrocytomas regardless of Ras mutational statusCancer Res200161114425443111389071
- FeldkampMMLalaPLauNRoncariLGuhaAExpression of activated epidermal growth factor receptors Ras-guanosine triphosphate and mitogen-activated protein kinase in human glioblastoma multiforme specimensNeurosurgery19994561442145310598712
- ChakravartiAChakladarADelaneyMALathamDELoefflerLSThe epidermal growth factor receptor pathway mediates resistance to sequential administration of radiation and chemotherapy in primary human glioblastoma cells in a Ras-dependant mannerCancer Res200262154307431512154034
- Haas-KoganDABanerjeeAKocakMPhase I trial of tipifarnib in children with newly diagnosed intrinsic diffuse brainstem gliomaNeuro Oncol200810334134718417739
- MoyalECLaprieADellanesMPhase I trial of tipifarnib (R115777) concurrent with radiotherapy in patients with glioblastoma multiformeInt J Radiat Oncol Biol Phys20076851396140117570606
- CloughesyTFKuhnJRobbinsHIPhase I trial of tipifarnib in patients with recurrent malignant glioma taking enzyme-inducing anti-epileptic drugs: a North American Brain Tumor Consortium StudyJ Clin Oncol200624223651365616877733
- CloughesyTFWenPYRobbinsHIPhase II trial of tipifarnib in patients with recurrent malignant glioma either receiving or not receiving enzyme-inducing antiepileptic drugs: a North American Brain Tumor Consortium StudyJ Clin Oncol200624223651365616877733
- FouladiMNicholsonHSZhouTA phase II study of the farnesyl transferase inhibitor, tipifarnib, in children with recurrent or progressive high-grade glioma, medulloblastoma/primitive neuroectodermal tumor, or brainstem glioma: a Children’s Oncology Group studyCancer2007110112535254117932894
- LarnerJJaneJLawsEPackerRMyersCShaffreyMA phase I–II trial of lovastatin for anaplastic astrocytoma and glioblastoma multiformeAm J Clin Oncol19982165795839856659
- LustigRMikkelsonTLesserGPhase II pre-radiation R115777 (tipifarnib) in newly diagnosed GBM with residual enhancing diseaseNeuro Oncol20081061004100918725460
- KazantsevAGThompsenLMTherapeutic application of histone deacetylase inhibitors for central nervous system disordersNat Rev Drug Discov200871085486818827828
- da FonescaCOLindenRFuturoDGattassCRQuirico-SantosTRas pathway activation in gliomas: a strategic target for intranasal administration of perillyl alcoholArch Immunol Ther Exp2008564267276
- SmithKTWorkmanJLHistone deacetylase inhibitors: anticancer compoundsInt J Biochem Cell Biol2008doi:10.1016j.biocel.2008.09.008
- SudakinVYenTJTargeting mitosis for anti-cancer therapyBiodrugs200721422523317628120
- YinDOngGMHuJSuberoylanilide hydroxamic acid, a histone deacetylase inhibitor: effects on gene expression and growth of glioma cells in vitro and in vivoClin Cancer Res20071331045105217289901
- CamphausenKCernaDScottTEnhancement of in vitro and in vivo tumor cell radiosensitivity by valproic acidInt J Cancer2005114338038615578701
- PhielCJZhangFHuangEYGuentherMGLazarMAKleinPSHistone deacetlyase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogenJ Biol Chem200127639367343674111473107
- AppelsNMBeijnenJHSchellensJHDevelopment of farnesyl transferase inhibitors: a reviewOncologist200510856557816177281
- ZujewskiJHorakIDBolCJPhase I and pharmacokinetic study of farnesyl protein transferase inhibitor R115777 in advanced cancerJ Clin Oncol200018492794110673536
- JonesHAHahnSMBernhardEMcKennaWGRas inhibitors and radiation therapySemin Radiat Oncol200111432833711677657
- FolkmanJTumor angiogenesis: therapeutic implicationsN Engl J Med197128521118211864938153
- WhiteCWSondheimerHMCrouchECWilsonHFanLLTreatment of pulmonary hemangiomatosis with recombinant interferon alpha-2aN Engl J Med198932018119712002710192
- FrederickLWangXYEleyGJamesCDDiversity and frequency of epidermal growth factor receptor mutations in human glioblastomasCancer Res20006051383138710728703
- BremSCotranRFolkmanJTumor angiogenesis: a quantitative method for histologic gradingJ Natl Cancer Inst19724823473564347034
- DasSSrikanthMKesslerJACancer stem cells and gliomaNat Clin Pract Neurol20084842743518628751
- WakabayashiTKayamaTNishikawaRA multicenter phase I trial of interferon-beta and temozolomide combination therapy for high-grade gliomas (INTEGRA Study)Jpn J Clin Oncol2008381071571818845525
- BucknerJCSchombergPJMcGinnisWLA phase III study of radiation therapy plus carmustine with or without recombinant interferon-alpha in the treatment of patients with newly diagnosed high-grade gliomaCancer200192242043311466698
- DillmanROSheaWMTaiDFInterferon-alpha2a and 13-cis-retinois acid with radiation treatment for high grade gliomaNeuro Oncol200131354111305415
- ChangSMBarkerFG2ndHuhnSLHigh dose oral tamoxifen and subcutaneous interferon alpha-2a for recurrent gliomaJ Neurooncol19983721691769524096
- D’AmatoRJLoughnanMSFlynnEFolkmanJThalidomide is an inhibitor of angiogenesisProc Natl Acad Sci U S A1994919408240857513432
- SampaioEPSarnoENGalillyRCohnZAKaplanGThalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytesJ Exp Med199117336997031997652
- ChangSMLambornKRMalecMPhase II study of temozolomide and thalidomide with radiation therapy for newly diagnosed glioblastoma multiformeInt J Radiat Oncol Biol Phys200460235335715380566
- KesariSSchiffDHensonJWPhase II study of temozolomide, thalidomide, and celecoxib for newly diagnosed glioblastoma in adultsNeuro Oncol200810330030818403492
- KowanetzMFerraraNVascular endothelial growth factor signaling pathways: therapeutic perspectiveClin Cancer Res200612175018502216951216
- ShibuyaMVascular endothelial growth factor-dependant and – independent regulation of angiogenesisBMB Rep200841427828618452647
- KaurBTanCBratDJPostDEVan MeirEGGenetic and hypoxic regulation of angiogenesis in gliomasJ Neurooncol200470222924315674480
- ChenWDelaloyeSSilvermanDHPredicting treatment response of malignant gliomas to bevacizumab and irinotecan by imaging proliferation with [18F] fluorothymidine positron emission tomography: a pilot studyJ Clin Oncol200725304714472117947718
- BartlettJBDredgeKDalgleishAGThe evolution of thalidomide and its IMiD derivatives as anticancer agentsNat Rev Cancer20044431432215057291
- DredgeKHorsfallRRobinsonSPOrally administered lenalidomide (CC-5013) is anti-angiogenic in vivo and inhibits endothelial cell migration and Akt phosphorylation in vitroMicrovasc Res2005691–2566315797261
- FineHAKimLAlbertPSA phase I trial of lenalidomide in patients with recurrent primary central nervous system tumorsClin Cancer Res200713237101710618056189
- VredenburghJJDesjardinsAHerndonJE2ndPhase II trial of bevacizumab and irinotecan in recurrent malignant gliomaClin Cancer Res20071341253125917317837
- VredenburghJJDesjardinsAHerndonJE2ndBevacizumab plus irinotecan in recurrent glioblastoma multiformeJ Clin Oncol200725304722472917947719
- CloughesyTFA phase II, randomized, non-comparative clinical trial of the effect of bevacizumab (BV) alone or in combination with irinotecan (CPT) on 6-month progression free survival (PFS6) in recurrent treatment-refractory glioblastoma (GBM)ASCO Meeting Abstract2008No. 2010b
- ZunigaRMTorcuatorRJainREfficacy, safety, and patterns of response and recurrence in patients with high-grade gliomas treated with bevacizumab plus irinotecanJ Neurooncol200991332933618953493
- LaiAFilkaEMcGibbonBPhase II pilot study of bevacizumab in combination with temozolomide and regional radiation therapy for up-front treatment of patients with newly diagnosed glioblastoma multiforme: interim analysis of safety and tolerabilityInt J Radiat Oncol Biol Phys20087151372138018355978
- SathornsumeteeSCaoYMarcelloJETumor angiogenic and hypoxic profiles predict radiographic response and survival in malignant astrocytoma patients treated with bevacizumab and irinotecanJ Clin Oncol200826227127818182667
- Gomez-ManzanoCHolashJFueyoJVEGF Trap induces antiglioma effect at different stages of diseaseNeuro Oncol200810694094518708344
- BatchelorTTSorensonAGdi TomasoEAZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patientsCancer Cell2007111839517222792
- HarlandSPKucREPickardJDDavenportAPCharacterization of endothelin receptors in human brain cortex, gliomas, and meningiomasJ Cardiovasc Phamacol199526Suppl 3S408S411
- SpinellaFRosanoSDiCastorVEndothelin-1 and endothelin-3 promote invasive behavior via hypoxia-inducible factor-1alpha in human melanoma cellsCancer Res20076741725173417308114
- PhuphanichSCarsonKAGrossmanSAPhase I safety study of escalating doses of atrasentan in adults with recurrent malignant gliomaNeuro Oncol200810461762318477765
- GuzmanMDuarteMJBlazquezCA pilot clinical study of Delta9-tetrahydrocannabinol in patients with recurrent glioblastoma multiformeBr J Cancer200695219720316804518
- ReardonDAQuinnJAVredenburghJPhase II trial of irinotecan plus celecoxib in adults with recurrent malignant gliomaCancer2005103232933815558802
- AvraamidesCJGarmy-SusiniBVarnerJAIntegrins in angiogenesis ad lymphangiogenesisNat Rev Cancer20088860461718497750
- NaborsLBMikkelsonTRosenfeldSSPhase I and correlative biology study of cilengitide in patients with recurrent malignant gliomaJ Clin Oncol200725131651165717470857
- MacDonaldTJStewartCFKocakMPhase I clinical trial of celengitide in children with refractory brain tumors: Pediatric Brain Tumor Consortium Study PBTC-012J Clin Oncol200826691992418281665
- SanaiNAlvarez-BullyaABergerMSNeural stem cells and the origin of gliomasN Engl J Med2005353881182216120861
- SanaiNTramontinADQuinones-HinojosaAUnique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migrationNature2004427697674074414973487
- ErikssonPPerfilievaEBjork-ErikksonTNeurogenesis in the adult human hippocampusNat Med19841113131317
- BaoSWuQMcClendonREGlioma stem cells promote radioresistance by preferential activation of the DNA damage responseNature2006444712075676017051156
- RichJNCancer stem cells in radiation resistanceCancer Res200767198980898417908997
- GrifferoFDagaAMarrubiDDifferent response of human glioma tumor-initiating cells to EGFR kinase inhibitorsJ Biol Chem2009284117138714819147502
- EylerCEFooWCLaFiuraKMMcLendonREHjelmelandABRichJNBrain cancer stem cells display sensitivity to Akt inhibitionStem Cells200826123027302618802038
- BaoSWuQLiZTargeting cancer stem cells through L1CAM suppresses glioma growthCancer Res200868156043604818676824
- AboodyKSBrownARainovNGNeural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomasProc Natl Acad Sci U S A20009723128461285111070094
- AboodyKSNajbauerJDanksMKStem and progenitor cell-mediated tumor selective gene therapyGene Ther2008151073975218369324
- ZhaoDNajbauerJGarciaENeural cell tropism to glioma: critical role of tumor hypoxiaMol Cancer Res20086121819182919074827
- TylerMAUlasovIVSonabendAMNeural stem cells target intracranial glioma to deliver an oncolytic adenovirus in vivoGene Ther200916226227819078993
- SonabendAMUlasovIVTylerMARiveraAAMathisJMLesniakMSMesenchymal stem cells effectively deliver an oncolytic adenovirus to intracranial gliomaStem Cells200826383184118192232
- DentPYacoubAParkMSearching for a cure: gene therapy for glioblastomaGene Ther20087913351340
- LangFFBrunerJMFullerGNPhase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: biological and clinical resultsJ Clin Oncol200321132508251812839017
- KlatzmannDValeryCABensimonGA phase I/II study of herpes simplex virus type 1 thymidine kinase “suicide” gene therapy for recurrent glioblastoma: Study Group on Gene Therapy for GlioblastomaHum Gene Ther1998917259526049853526
- RainovNGA phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiformeHum Gene Ther200011172389240111096443
- EckSLAlaviJBJudyKTreatment of recurrent or progressive malignant glioma with a recombinant adenovirus expressing human interferon-beta (H5.010CMVhIFN-beta): a phase I trialHum Gene Ther20011219711311177547
- ColomboFBarzonLFranchinECombined HSV-TK/IL-2 gene therapy in patients with recurrent glioblastoma multiforme: biological and clinical resultsCancer Gene Ther2005121083584815891772
- RenHBoulikasTLundstromKImmunogene therapy of recurrent glioblastoma multiforme with a liposomally encapsulated replication-incompetent Semliki forest virus vector carrying the human interleukin-12 gene – a phase I/II clinical protocolJ Neurooncol2008641–214715412952295
- OkadaHPollackIFLiebermanFGene therapy of malignant gliomas: a pilot study of vaccination with autologous irradiated glioma and dendritic cells admixed with IL-4 transduced fibroblasts to elicit an immune responseHum Gene Ther200112557559511268289
- LassmanABRossiMRRaizerJJMolecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01–03 and 00–01Clin Cancer Res200511217841785016278407
- PradosMDLambornKRChangSPhase 1 study of erlotinib HCl alone and combined with temozolomide in patients with stable or recurrent malignant gliomaNeuro Oncol200681677816443950
- PreusserMGelpiERottenfusserAEpithelial growth factor receptor inhibitors for treatment of recurrent or progressive high grade glioma: an exploratory studyJ Neurooncol200889221121818458820
- FranceshiECavalloGLonardiSGefitinib in patients with progressive high-grade gliomas: a multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO)Br J Cancer20079671047105117353924
- de GrootJFGilbertMRAldapeKPhase II study of carboplatin and erlotinib (Tarceva, OSI-774) in patients with recurrent glioblastomaJ Neurooncol2008901899718581057
- BrownPDKrishnanSSarkariaJNPhase I/II trial of erlotinib and temozolomide with radiation therapy in the treatment of newly diagnosed glioblastoma multiforme: North Central Cancer Treatment Group study N1077J Clin Oncol200826345603560918955445
- KrishnanSBrownPDBallmanKVPhase I trial of erlotinib with radiation therapy in patients with glioblastoma multiforme: results of North Central Cancer Treatment Group protocol N0177Int J Radiat Oncol Biol Phys20066541192119916626884
- SchwerALDamekDMKavanaghBDA phase I dose-escalation study of fractionated stereotactic radiosurgery in combination with gefitinib in patients with recurrent malignant gliomasInt J Radiat Oncol Biol Phys2008704993100117967517
- RichJNReardonDAPeeryTPhase II trial of gefitinib in recurrent glioblastomaJ Clin Oncol200422113314214638850
- ReardonDAQuinnJAVredenburghJJPhase I trial of gefitinib plus sirolimus in adults with recurrent malignant gliomaClin Cancer Res200612386086816467100
- PradosMDYungWKAWenPYPhase-I trial of gefitinib and temozolomide in patients with malignant glioma: a North American Brain Tumor Consortium studyCancer Chemother Pharmacol20086161059106717694310
- PreusserMGelpiERottenfusserAEpithelial growth factor receptor inhibitors for treatment of recurrent or progressive high grade glioma: an exploratory studyJ Neurooncol200889221121818458820