Abstract
Persistent polyclonal B-cell lymphocytosis (PPBL) is a rare and recently described entity. The review of the literature show PPBL is diagnosed predominantly but not exclusively in women, usually smokers. PPBL is recognized by a moderate, chronic and absolute lymphocytosis (>4 × 109/l) in the peripheral blood. In 10% of cases without lymphocytosis, the PPBL diagnosis has to be suggested by peripheral blood examination showing in all cases atypical binucleated lymphocytes. A polyclonal serum IgM is also associated and HLA-DR7 expression is present in most cases. Contrary to B-cell chronic lymphoproliferative disorders (B-CLPD), peripheral B cells are polyclonal with kappa and lambda light-chain expression and no clonal rearrangement of immunoglobulin heavy chain genes is usually demonstrated. The detection of an extra isochromosome for the long arm of chromosome 3 +i(3)(q10) has to be considered as a specific marker of PPBL. We performed conventional cytogenetic analysis (CCA) in 111 patients with typical PPBL we followed-up more than 4 years. +i(3q) was detected in 34% (33/98), PCC in 8% (8/98) and both abnormalities in 31% (30/98). CCA showed neither +i(3q) nor PCC in 28% (27/98). Fluorescence in situ hybridization (FISH) was also performed in 84 cases and +i(3q) was detected in 71% (60/84). When combining both procedures in 84 patients, +i(3q) was detected in 17 patients with negative CCA and was confirmed in 43 patients with positive CCA. CCA and FISH were both negative in 24 cases. Whether patients with PPBL are at increased risk of hematological malignancy remains unclear. After a median follow-up of 4.4 years, most PPBL patients presented a stable clinical and biological course. Six patients died from pulmonary cancer, myocardial infarction, cerebral aneurysm rupture or diffuse large B-cell lymphoma. Two patients had IgM monoclonal gammopathy of undetermined significance (MGUS) at the time of PPBL diagnosis and two other patients developed IgM MGUS respectively 12 and 22 years after PPBL diagnosis. A malignant non Hodgkin's lymphoma (NHL) appeared in 3 additional patients: two patients presented diffuse large B cell lymphoma and 1 patient a splenic marginal zone lymphoma. In conclusion, the possibility of PPBL to evolve toward a clonal proliferation, malignant lymphoma or secondary solid cancer lead us to consider PPBL not as a benign pathology. We recommend a careful and continued clinical and biological long-term follow-up in all PPBL patients.
Introduction
Benign or reactive lymphocytosis with an absolute lymphocyte count >4 × 109/l is common in children and young adults and is usually related to infectious mononucleosis or other viral infections. In contrast, benign lymphocytosis in adults is unusual: transient absolute lymphocytosis has been described in “stress”-related emergency trauma and medical conditions but it lasts for only a few hours (CitationThomassen et al 1986). Persistent lymphocytosis is usually related to chronic lymphoproliferative disorders (CLPD), most commonly of B-cell origin (B-CLPD). A lymphocytosis in this age group indicates B-chronic lymphocytic leukemia (B-CLL), B-chronic prolymphocytic leukemia (B-PLL), hairy cell leukemia (HCL), Waldenström’s macroglobulinemia, follicular lymphoma with leukemic phase, splenic lymphoma with villous lymphocytes (SLVL), or intermediate lymphoma. These disorders are characterised by a chronic course, typical morphologic appearance of lymphocytes on peripheral blood smears, exclusively either kappa or lambda light-chain expression on B-cells surface, caryotypic abnormalities, and expansion of a clone of neoplastic lymphocytes. Conversely, polyclonal lymphocytosis can be observed in hyposplenic states (CitationWilkinson et al 1983), in nodular lymphocyte predominant Hodgkin’s disease (CitationMariette et al 1993), in rheumatoid polyarthritis (CitationPlater-Zirberck et al 1985) and in other medical conditions such as Gaucher disease (CitationMarty et al 1988).
CitationGordon and colleagues (1982) described a syndrome associating chronic absolute lymphocytosis, polyclonal B-cell lymphocytosis with binucleated lymphocytes (PPBL) in the peripheral blood and a polyclonal increase of serum IgM in three women, all of whom expressed the HLA-DR7 antigen. We subsequently described cytogenetic abnormalities in seven patients with PPBL, including an additional isochromosome for the long arm of chromosome 3 +i(3q) in 6 cases and premature chromosome condensation (PCC) in all 7 cases (CitationTroussard et al 1994). That cytogenetic profile was subsequently confirmed in a large number of case reports.
The real incidence of PPBL has never been calculated because PPBL is probably underdiagnosed, as most patients are asymptomatic. Nevertheless PPBL appears to be a rare haematological disorder and underreported (CitationDelannoy et al 1993; CitationAgrawal et al 1994; CitationMossafa et al 1999). In this review, we focus our interest on the clinical, morphologic, immunologic and karyotypic findings in patients with PPBL. We had the opportunity to collect the clinical data and the specimens of a very large cohort of 111 patients with typical PPBL in France. We will present the specific clinical and biological data in that very large cohort and also discuss the aetiology of PPBL.
The identification of PPBL is important to distinguish PPBL from others B-CLPD. Whether this syndrome represents a premalignant (CitationCasassus et al 1987) or benign disease remains unclear. Although long-term follow-up suggests that whenever malignant progression occurs, it is after a long latency. The benign clinical course and lack of biological evolution in the majority of PPBL cases suggest that the recognition is so important that aggressive therapy could be avoided.
Clinical data
PPBL affects mainly adults but it was reported in a 3-month-old baby (CitationGomez et al 2000). The median age is approximately 40–50 years. Most patients are asymptomatic: the presence of splenomegaly is observed in 10% of cases and only a very few patients had hepatomegaly or adenopathy based on clinical examination and/or thoracic and abdominal computerised tomography. Antiphospholipid/cofactor antibodies of immunoglobulin (IgM) isotype or lupus anticoagulant was also reported in a few cases (CitationGranel et al 2002).
The majority of PPBL patients present with an absolute and moderate lymphocytosis >4 × 109/L. In 10% of patients without absolute lymphocytosis, PPBL diagnosis has to be suggested by cytology and confirmed by detection of typical binucleated lymphoid cells and +i(3q). Neutrophil and platelet counts are usually normal. Analysis of serum immunoglobulin reveal persistent, stable and polyclonal increase of IgM levels, with low to normal levels of IgA and IgG. No increase incidence of infection or any clinical evidence of defective humoral immunity was notified. Ninety percent of PPBL patients are HLA-DR7-positive (CitationTroussard et al 1996) but no patient complained adult coeliac disease.
Cytology: presence of binucleated lymphocytes in the peripheral blood
Binucleated lymphocytes are detected in the peripheral blood of all PPBL patients. The majority of lymphocytes are large with abundant faintly, basophilic cytoplasm and in all patients a characteristic nuclei demonstrating a rounded or more commonly irregular form with variable numbers of binucleated cells (). Nuclei range from those demonstrating a slight indentation to bilobed forms with the inconstant presence of an internuclear linkage in a few cells. Nuclear chromatin is dense and nucleoli usually visible. Binucleated lymphocytes are not specific of PPBL and have been described after irradiation (CitationRoy-Taranger et al 1965) and more rarely in B-CLPD, as HCL Japanese variant (CitationTroussard et al 1997), B-CLL or marginal zone B-cell lymphoma (CitationSamson et al 2002).
Figure 1 Morphologic features showing typical binucleated cells in a PPBL patient.
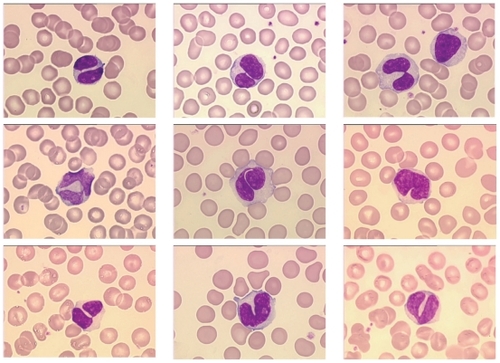
The investigation of bone marrow biopsies is limited in PPBL. The bone marrow is infiltrated by a slight or moderate intravascular and at a lesser extent interstitial lymphocytic pattern (CitationFeugier et al 2004).
Immunophenotyping: a polyclonal expansion of memory B-cells
The lymphocytosis is clearly of the B-cell type. Lymphocytes react with the CD19, CD20 and CD22 antigens. Both kappa and lambda light-chain are expressed, indicating a polyclonal expansion of the B-lymphocyte pool (CitationTroussard et al 1994; CitationMossafa et al 1996). Double labeling performed on cytocentrifuge slides with kappa and lambda monoclonal antibodies by immunoenzymatic methods using ABC (avidin biotin peroxydase complex) and APAAP (alkaline phosphatase antialkaline phosphatase) procedures showed both kappa and lambda light-chain in binucleated lymphocytes and nonbinucleated lymphoid cells (CitationTroussard et al 1994). When using cytofluorometry, the cellular DNA content determination and proliferation index analysis is not different from that of the controls and diploidy can be demonstrated in binucleated lymphocytes by image analysis and manual cell sorting.
Peripheral B cells co-express IgM and IgD, suggesting that they derive from a naive B-cell subset (CitationHimmelmann et al 2001; CitationLoembé et al 2002). CD27 is a type I glycoprotein expressed on some B cells and the majority of T cells, and is a member of the tumor necrosis factor (TNF) receptor family, with unique cysteine-rich motifs. CD27+ B cells exhibit several features of primed (memory) B cells. In five PPBL patients, a large increase in the number of IgD+, CD27+ double positive B cells, (60%–88% versus 8%–15% in the controls) was detected in the peripheral blood. The number of CD27+ which also coexpress receptor-type protein tyrosine phosphatase CD148 (CD27+ CD148+ double-positive B cells) was also increased (CitationHimmelmann et al 2001). PPBL B cells express CD25 and CD23 with usually no or low CD5 antigens (CitationSalcedo et al 2002). An unusual CD5 positive phenotype can be observed (CitationReeder et al 1999). The cells also express CD24, CD79, FMC7, and CD21 at levels higher than normal B lymphocytes. Taken together, all those data suggest B-cell population is a memory B-cell population distinguished by the expression of CD27, IgMhigh, CD21high, CD5low, and CD23low, a profile associated with a normal marginal B-cell compartment (CitationSalcedo et al 2002).
An associated abnormality of T cells has not been found: not only was the absolute number of T-cells normal but the CD4 and CD8 cells subsets were quantitatively normal as well.
Chromosome analysis in PPBL patients: an unique cytogenetic profile associating i(3q) and premature chromosome condensation
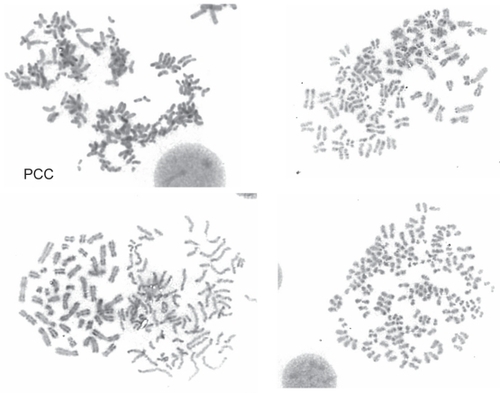
Following stimulation of peripheral blood lymphocytes with pokeweed, the presence of an extra chromosome 3 long arm i(3q) was identified for the first time in 1989. +i(3)(q10) was present in 3/26 metaphases in only 1/6 patients with PPBL (CitationPerreault et al 1989). However, when using the combinations of the tetrad-canoyl-o-phorbol 13 acetate (TPA) and lipopolysaccharide (LPS) from Escherichia coli used in some samples, which encourages B lymphoid proliferation, +i(3q) () was detected more frequently and was observed in 6/7 PPBL patients (CitationMossafa et al 1996). +i(3q) is distributed and restricted in nonbinucleated B cells whatever the light chain expression (CitationTroussard et al 1999). In B-CLPD, distinct, recurring and highly consistent chromosomal changes are observed: however +i(3q)(q10) is rarely identified and to our knowledge was detected in only one case of Waldenstrom macroglobulinemia (CitationWong et al 2002) and in a few cases of chronic lymphocytic leukemia (CitationSpecchia et al 2002; CitationWong et al 2002). G1 premature chromosome condensation (PCC) () is also a characteristic of PPBL patients. PCC, originally described in virally infected cells, is also detected in normal cells treated with mould extract, cytochalasin B. The common feature is the presence of asynchronous mitosis in multinucleated cells. One nucleus must be in metaphase and its presence results in the condensation of the chromatin of an adjacent interphase nucleus. The morphology depends on the interphase cells. In PPBL, the morphology of the PCC indicate condensation of G1 and G2 cells, exhibiting single and double chromatids, respectively. PCC of S-phase cells is not observed in any case. Interestingly PCC can be associated with +i(3q). As both abnormalities, either +i(3q) or PCC, are rarely present in other benign or malignant proliferations, a strong correlation between PPBL and i(3q) and/or PCC is probable.
Figure 2 Partial karyotype showing +i(3q) -R-banding from patient with persistent polyclonal B-cell lymphocytosis.
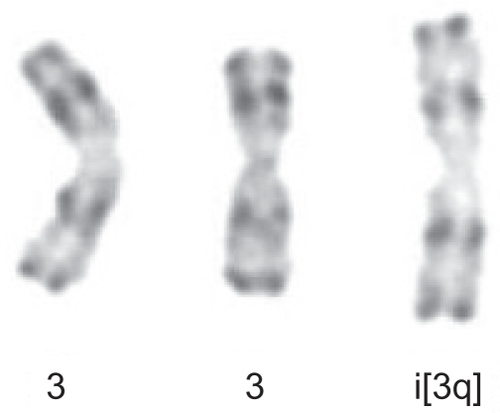
Figure 3 Premature chromosome condensation (PCC) R-banding of an early G1-PCC with condensed single chromatide.
In a large series of 111 patients with PPBL we follow-up, conventional cytogenetic analysis (CCA) allowed us to identify extra isochromosome +i(3)(q10) in 34% (33/98), PCC in 8% (8/98) and both abnormalities in 31% (30/98). CCA showed neither +i(3)(q10) nor PCC in 28% (27/98). Isochromosome +i(3q) was also studied in interphase with alpha-satellite chromosome 3 specific (SO CEP3) and LSI 3’BCL6 (3q27) break apart probes obtained from Abott Molecular (Rungis, France) and was detected in 71% (60/84). We also combined CCA and FISH analysis in 84 patients: after FISH, +i(3q) was detected in 17 patients with negative CCA and was confirmed in 43 patients with positive CCA. CCA and FISH were both negative in 24 cases.
Trisomy 3 can be detected in addition to extra +i(3)(q10), suggesting that PPBL is associated with an increase frequency of chromosome 3 instability (CitationCallet-Bauchu et al 1999). When excluding +i(3)(q10) and PCC, chromosomal instability is also a common occurrence in PPBL and is observed in more than two thirds of PPBL patients. It is characterised by the presence of others independent clonal abnormalities, del(6q), +der(8) or +8, poliploid karyotype, structural changes, aneuploidy and/or non clonal chromosomal aberrations with either loss or more frequently gain of chromosomes (CitationMossafa et al 2004).
The long-term follow-up of PPBL remains to be determined and the role of i(3q) in the pathogenesis of PPBL is debatable. The gain of chromosome 3q was demonstrated as a pivotal genetic aberration transition from severe dysplasia/carcinoma in situ to invasive cervical carcinoma of the uterine cervix (CitationHeselmeyer et al 1996). It could have a role in the possibility of emergence of a clonal and/or malignant subpopulation. On the other hand, isochromosome, i(3)(q10) inhibits muscle differentiation, when transferred into myoblasts. The i(3q) is in that model able to induce an abnormal phenotype of the cells including abnormal centrosome amplification, aneuploidy and loss of G1 arrest following γ-irradiation (CitationSmith et al 1998).
Molecular studies
To determine if the lymphocytosis observed in PPBL patients represented a clonal, biclonal, polyclonal or random expansion of cells, immunoglobulin gene rearrangements were examined. In almost all cases, molecular studies show a polyclonal pattern. Southern hybridization of DNA with immunoglobulin probes was performed in 14 patients and failed to show any clonal population in 12 patients; however two patients curiously showed an immunoglobulin gene rearrangement (CitationChan et al 1990; CitationDelage et al 1992). Polymerase chain reaction (PCR) procedures using either consensus primers for the FR3 portion of the variable region and/or VH family specific primers for the FR1 region (12,16) show also a polyclonal expansion of B cells.
The study of Ig-V gene somatic mutations was rarely performed in the B cells isolated from PPBL patients (CitationLoembé et al 2002; CitationSalcedo et al 2002). However, the relevant marker of memory B cells thus far identified is the presence of somatically mutated high-affinity antigen receptors. When combining both studies, 90 clones were analysed from 6 PPBL patients and 85.5% clones (77/90) were mutated, a proportion of sequences higher than that reported for normal blood lymphocytes. Mutations in PPBL cells are present in all three framework regions (FR) as well as CDR1 and CDR2 but the number of observed R mutations is usually lower than that expected for FR but higher than expected for CDR, suggesting no evidence of positive antigenic selection following somatic hypermutation.
Using nested PCR assay, BCL-2/IgH rearrangements are identified in PPBL patients (CitationDelage et al 1997, Citation1998, Citation2001; CitationGranados et al 1998). We also studied the presence of BCL-2/ IgH rearrangements in a series of eight PPBL patients and demonstrated the constant presence of BCL-2/IgH rearrangements in 8/8 DNA samples, multiple rearrangements in 3/8 cases and also normal BCL-2 protein expression, as compared to BCL-2 level in B lymphocytes from healthy population (CitationLancry et al 2001). When pooling all the published results, BCL-2/IgH rearrangements are detected both in the major breakpoint region (MBR) and the minor cluster region (mcr). When using a quantitative real-time PCR, CitationHimmelmann and colleagues (2001) demonstrated the presence in the peripheral blood of a high number of 1055–3487 t(14; 18)-positive cells per 106 B cells, as compared to only nine positive cells in 106 B cells obtained from a buffy coat from a healthy donor. BCL-2/IgH rearrangements were also identified in nearly 50% of normal individuals: the frequency of translocations increases with age and heavy smoking. In those cases, the frequency of the BCL-2/IgH rearrangements is estimated to be equal or less than one in 105 to 106 B cells. Despite the presence of BCL-2/ IgH rearrangements, the accumulation of B lymphocytes in PPBL is probably not related to an overexpression of BCL-2 protein (CitationHimmelmann et al 2001; CitationLancry et al 2001).
Functional defect in PPBL: conflicting signals from the CD40 survival and Fas death pathways
The accumulation of polyclonal binucleated mature B lymphocytes in the peripheral blood could be explained by defects in apoptosis and/or uncontrolled cell growth. Overexpression of the antiapoptotic protein Bcl-2 in CLL is associated with an aggressive clinical course, chemotherapy resistance and shortened survival. In PPBL, Bcl-2 is equivalent to that detected in lymphocytes of healthy individuals (CitationLancry et al 2001). The polyclonal B-cells are resistant to Fas stimulation despite a high expression of Fas receptor and apoptosis is induced after etoposide treatment (CitationRoussel et al 2003). In contrast to normal B cells, defects of the CD40 survival pathway has been described in PPBL patients (CitationLoembé et al 2001). B-cell accumulation observed in PPBL could result from conflicting signals from the CD40 survival and Fas death pathways.
PPBL requires a careful a long-term follow-up
Whether patients with PPBL are at increased risk of hematologic malignancy remains unclear (CitationTroussard et al 1999). One patient developed a malignant pulmonary blastoma 20 years after PPBL was diagnosed (CitationLawlor et al 1991). One additional PPBL patient developed B large cell lymphoma 19 years after PPBL diagnosis (CitationRoy et al 1998). After a median follow-up of 4.4 years (0.5–29 years), PPBL patients presented a stable clinical and biological course in 99/111 cases (89%). In two patients, we noticed a decrease of lymphocytosis and the number of binucleated lymphocytes but isochromosome +i(3q) persisted 2 years after stopping tobacco. Six patients died: one heavy smoker patient from pulmonary cancer nine years after PPBL diagnosis, 1 from myocardial infarction, 1 from cerebral aneurysm rupture, 2 deaths from unknown causes and 1 from diffuse large B-cell lymphoma described later. Two patients had IgM monoclonal gammopathy of undetermined significance (MGUS) at the time of PPBL diagnosis. Two other patients developed IgM MGUS (serum IgM, 6.9 g/L and 12.88 g/L) 12 and 22 years after PPBL diagnosis, respectively. In those 4 cases, no bone marrow biopsy was performed. IgM gammopathy remained asymptomatic and stable after a median follow-up of 102 months (range 52–348). Two patients presented pulmonary cancer in the clinical course of PPBL and one patient developed cervical cancer 12 years after diagnosis of PPBL. A malignant non-Hodgkin’s lymphoma (NHL) was also observed in 3 additional patients: two patients presented diffuse large B cell lymphoma (DLBCL) and 1 patient a splenic marginal zone lymphoma (SMZL). It therefore appears that, while the majority of patients demonstrate a polyclonal B lymphocytosis based on immunological and genotypic analysis, the emergence of a clonal sub-population is possible (CitationCornet et al 2008).
Etiology of PPBL
The aetiology of polyclonal B-cell lymphocytosis remains elusive. An association with cigarette smoking was suggested by CitationCarstairs and colleagues (1985). These authors described four patients with PPBL, all cigarette smokers, in one of whom the lymphocyte count and morphology were abnormal when she smoked and not when she abstained. However, the role and influence of environmental factors and particularly tobacco is debatable, as we observed one female patient who had never smoke and we were unable to detect binucleated lymphocytes on cytocentrifuge slides in 30 asymptomatic female heavy smokers. Lymphocyte binuclearity is not observed in normal individuals and has been described only after low-dose irradiation (CitationRoy-Taranger et al 1965).
The binucleated morphology of lymphocytes could suggest an association with persistent viral infection such as Epstein-Barr virus (EBV) (CitationMittereer et al 1995). EBV is responsible for a polyclonal lymphoproliferative process in patients with immune deficiency. CitationChow and colleagues (1992) demonstrated the presence of EBV in the peripheral blood lymphocytes (but not in binucleated lymphocytes) from one patient by in situ hybridization and PCR. These studies are insufficient to exclude and/or confirm the role of EBV in PPBL.
The association with HLA-DR-7 phenotype suggests a possible genetic disorder. Taken together with the presence of chararacteristic, binucleated lymphocytes in asymptomatic family members, we suggest a genetic predisposition as a more likely possibility. The description of PPBL in identical twins (CitationCarr et al 1997) and also in a family including PPBL in a brother and sister (CitationHimmelmann et al 2001) strengthens that hypothesis as well as the detection of multiple BCL-2/ IgH rearrangements in 8/10 individuals among first-degree relatives (CitationDelage et al 2001).
Conclusion
PPBL is characterised by persistent and stable lymphocytosis (in but not exclusively female patients) with binucleated lymphocytes on peripheral blood smears and a polyclonal increase of serum IgM. Immunophenotyping revealed in all cases a polyclonal expansion of B-lymphocytes. Whether this syndrome represents a premalignant disease process or an actual polyclonal lymphocytosis remains unsettled. The occasional reports of clonal Ig rearrangements in this disorder suggest that in a minority of cases the polyclonal expansion may be followed by the emergence of one predominant clone. The demonstration of clinically, benign, clonal lymphoid population is well recognized in several T cell disorders such as large granular lymphocytes expansion. The benign clinical course and the lack of biological evolution in the majority of cases suggest that the importance in recognition of this disorder lies in the avoidance of aggressive therapy for these cases. A careful follow-up of these patients is mandatory: prospective immunological and genetic studies performed at different stages of the disease may clarify this issue.
Disclosure
The authors report no conflicts of interest in this work.
References
- AgrawalSMatutesEVokeJ1994Persistent polyclonal B-cell lymphocytosisLeuk Res1879157934138
- Callet-BauchuEGazzoSPoncetC1999Distinct chromosome 3 abnormalities in persistent polyclonal B-cell lymphocytosisGenes Chromosomes Cancer26221810502320
- CarrRFishlockKMatutesE1997Persistent polyclonal B-cell lymphocytosis in identical twinsBr J Haematol9627249029012
- CarstairsKCFrancombeWHScottJG1985Polyclonal lymphocytosis of B-lymphocytes induced by cigarette smoking?Lancet1110942860302
- CassasusPLortholaryPKomaroverH1987Cigarette smoking related to persistent polyclonal B-lymphocytis: a premalignant stateArch Pathol Lab Med11110813499130
- ChanMABenedictSHCarstairsKC1990Expansion of B-lymphocytes with an unusual immunoglobulin rearrangment associated with atypical lymphocytosis and cigarette smokingAm J Respir Cell Mol Biol2549522346660
- ChowKCNacillaJQWitzigTE1992Is persistent polyclonal B lymphocytosis caused by Epstein-Barr virus? A study with polymerase chain reaction and in situ hybridizationAm J Hematol4127051337664
- CornetELesesveJFMossafaHthe Groupe Français d’Hématologie Cellulaire (GFHC)2008Long-term follow-up of 111 patients with persistent polyclonal b-cell lymphocytosis (ppbl) with binucleated lymphocytesLeukemiaIn press
- DelageBDarveauAJacquesL1992Chronic B-cell lymphocytosis of the young woman: clinical, phenotypic, and molecular studiesBlood80Suppl 1447a1627802
- DelageRJacquesLMassinga-LoembeM2001Persitent polyclonal B-cell lymphocytosis: further evidence for a genetic disorder associated with B-cell abnormalitiesBr J Haematol1146667011552996
- DelageRRoyJJacquesl1997Multiple bcl2/Ig gene rearrangements in persistent polyclonal lymphocytosisBr J Haematol97589959207405
- DelageRRoyJJacquesL1998All patients with persistent polyclonal B cell lymphocytosis present Bcl-2/Ig gene rearrangementsLeuk Lymphoma31567749922047
- DelannmoyCDijanDWallefG1993Cigarette smoking and chronic polyclonal B-cell lymphocytosisNouv Rev Fr Hématol351414
- FeugierPDe MarchAKLesesveJF2004Intravascular bone marrow accumulation in persistent polyclonal lymphocytosis: a misleading feature for B-cell neoplasmMod Pathol1710879615143340
- GomezPMatutesESanchezJ2000An unusual form of persistent polyclonal B lymphocytosis in an infantBr J Haematol110430310971403
- GordonDSJonesBMBrowingSW1982Persistent polyclonal lymphocytosis of B-lymphocytesN Engl J Med30723266979709
- GranadosELlamasPPinillaI1998Persistent polyclonal B lymphocytosis with multiple bcl-2/IgH rearrangement: a benign disorderHaematologica83369759592988
- GranelBSerratriceJDisdierP2002Anti-phospholipid/cofactor antibodies in three cases of persistent polyclonal B lymphocytosisBr J Haematol119875612437675
- HeselmeyerKSchrockEdu ManoirS1996Gain of chromosome 3q defines the transition from severe dysplasia to invasive carcinoma of the uterine cervixProc Natl Acad Sci U S A93479848552665
- HimmelmannAGautschiONawrathM2001Persistent polyclonal B-cell lymphocytosis is an expansion of functional IgD+ CD27+ memory B-cellsBr J Haematol114400511529864
- HimmelmannARüeggRFehrJ2001Familial persistent polyclonal B cell lymphocytosisLeuk Lymphoma411576011342368
- LancryLRoullandSRouéG2001No bcl-2 protein over expression but bcl-2/IgH rearrangements in B cells of patients with persistent polyclonal B-cell lymphocytosisHematol J22283311920254
- LarcherCFendFMittererM1995Role of Epstein-Barr virus and soluble CD21 in persistent polyclonal B-cell lymphocytosisBr J Haematol90532407646990
- LawlorEMurrayMO’BrianDS1991Persistent polyclonal B lymphocytosis with Epstein-Barr virus antibodies and subsequent malignant pulmonary blastomaJ Clin Pathol4434121851502
- LoembeMMLamoureuxJDeslauriersN2001Lack of CD40-dependent B-cell proliferation in B lymphocytes from patients with persistent polyclonal B-cell lymphocytosisBr J Haematol11369970511380461
- LoembeMMNéronSDelageR2002Analysis of expressed VH genes in persistent polyclonal B cell lymphocytosis reveals absence of selection in CD27 + IgM + IGD + memory B cellsEur J Immunol3236788812516560
- MarietteXTsapisAOksenhendlerE1993Nodular lymphocyte predominance Hodgkin’s disease featuring blood atypical polyclonal B-cell lymphocytosisBr J Haematol8581357918051
- MartyGERyanETPapadopoulosNM1988Polyclonal B-cell lymphocytosis and hypergammaglobulinemia in patients with Gaucher diseaseAm J Haematol2918994
- MittererMPescostaNFendF1995Chronic active Epstein-Barr virus disease in a case of persistent polyclonal B-cell lymphocytosisBr J Haematol90526317646989
- MossafaHMalaureHMaynadieMfor the Groupe Française d’Hématologie Cellulaire (GFHC)1999Persistent polyclonal B lymphocytosis with binucleated lymphocytes: a study of 25 casesBr J Haematol1044869310086784
- MossafaHTapiaSFlandrinGfor the Groupe Français d’hématologie Cellulaire (GFHC)2004Chromosomal instability and ATR amplification gene in patients with persistent polyclonal B-cell lymphocytosis (PPBL)Leuk Lymphoma451401615359640
- MossafaHTroussardXValensiF1996Isochromosome i(3q) and premature chromosome condensation are recurrent findings in chronic B-cell lymphocytosis with binucleated lymphocytesLeuk Lymphoma20267738624466
- PerraultCBoileauJGygerM1989Chronic B-cell lymphocytosisEur J Haematol23617
- Plater-ZyberkCMainiRNLamK1985A rheumatoid arthritis B cell subset expresses a phenotype similar to that in chronic lymphocytic leukemiaArthritis Rheum2897163899124
- ReederCBConleyCR1999CD5+ persistent polyclonal B-cell lymphocytosis in a maleLeuk Lymphoma33593610342588
- RousselMRouéGSolaB2003Dysfunction of the Fas apoptotic signaling pathway in persistent polyclonal B-cell lymphocytosisHaematologica882394012604424
- RoyJRyckmanCBernierV1998Large cell lymphoma complicating persistent polyclonal B cell lymphocytosisLeukemia121026309665185
- Roy-TarangerMMayaudGDavydoff-AlibertS1965Binuclear lymphocytes in the blood of individuals irradiated by a low doseRev Fr Etud Clin Biol10958655852017
- SalcedoICampos-CaroASampaloA2002Persistent polyclonal B lymphocytosis: an expansion of cells showing IgVH gene mutations and phenotypic features of normal lymphocytes from the CD27+ marginal zone B-cell compartmentBr J Haematol116662611849229
- SamsonTMossafaHLusinaD2002Dicentric chormosome 3 associated with binucleated lymphocytes in atypical B-cell chronic lymphoproliferative disorderLeuk Lymphoma4317495412685827
- SmithLLiuSJGoodrichL1998Duplication of ATR inhibits MyoD, induces aneuploidy and eliminates radiation-induced G1 arrestNat Genet1939469590286
- SpecchiaGAlbanoFAnelliL2002Concomitant tetrasomy 3q and trisomy 18 in CD5-, CD13+ chronic lymphocytic leukemiaCancer Genet Cytogenenet1331603
- ThomassenHVBoykoWJMontanerJS1986Absolute lymphocytosis associated with nonsurgical traumaAm J Clin Pathol8648033766460
- TroussardXFlandrinG1996Chronic B-cell lymphocytosis with binucleated lymphocytes (LWBL): a review of 38 casesLeuk Lymphoma2027598624467
- TroussardXMossafaHFlandrinG1997Identity between hairy B-cell lymphoproliferative disorder and persistent polyclonal B lymphocytosis?Blood90211019292552
- TroussardXMossafaHSalaünV1999Persistent polyclonal lymphocytosisLeukemia13497810086750
- TroussardXValensiFDebertC1994Polyclonal lymphocytosis with binucleated lymphocytes: a genetic predispositionBr J Haematol88275807803270
- WilkinsonLSTangAGjedstedaA1983Marked lymphocytosis suggesting chronic lymphocytic leukemia in three patients with hyposplenismAm J Med75105366650537
- WongKFSoCCChanJCW2002Gain of chromosome 3/3q in B-cell chronic lymphoproliferative disorder is associated with plasmacytoid differentiation with or without IgM overproductionCancer Genet Cytogenet13682512165458