Abstract
The prominence of the PI3K-Akt signaling pathway in several tumors indicates a relationship with tumor grade and proliferation. Critical cellular processes are driven through this pathway. More detailed knowledge of the pathogenesis of tumors would enable us to design targeted drugs to block both membrane tyrosine kinase receptors and the intracellular kinases involved in the transmission of the signal. The newly approved molecular inhibitors sunitinib (an inhibitor of vascular endothelial growth factor receptor, platelet-derived growth factor receptor, and other tyrosine kinase receptors), sorafenib (a serine–threonine kinase inhibitor that acts against B-Raf) and temsirolimus (an mTOR inhibitor) shown clinical activity in advanced kidney cancer. Chronic myeloid leukemia has changed its natural history thanks to imatinib and dasatinib, both of which inhibit the intracellular bcr/abl protein derived from the alteration in the Philadelphia chromosome. Intracellular pathways are still important in cancer development and their blockade directly affects outcome. Cross-talk has been observed but is not well understood. Vertical and horizontal pathway blockade are promising anticancer strategies. Indeed, preclinical and early clinical data suggest that combining superficial and intracellular blocking agents can synergize and leverage single-agent activity. The implication of the Akt signaling pathway in cancer is well established and has led to the development of new anticancer agents that block its activation.
Introduction
The phosphatidylinositol 3-kinase (PI3K)-Akt pathway is situated in downstream tyrosine kinase receptors (TKRs) and regulates essential cellular functions such as proliferation, growth, and survival.Citation1 Akt is a serine/threonine kinase that belongs to the AGC (protein kinase A/protein kinase G/protein kinase C-like) family of protein kinases. Because it shows high homology with protein kinases A and C, Akt is also referred to as protein kinase B (PKB).
PI3K-Akt signaling is frequently altered in human cancers.Citation1 We describe the main downstream effectors of PI3K-Akt pathway involved in its proliferative and survival responses. We also examine PI3K-Akt signaling pathway alterations associated with human cancers and their implications in the development of target-based anticancer drugs.
Structure and activation of Akt kinase
In mammals, three Akt genes encode for the isoforms Akt1 (PKBα), Akt2 (PKBβ), and Akt3 (PKBγ), and they all have a similar structure and size.Citation1 Akt isoforms contain a pleckstrin homology (PH) domain in the N-terminus, a central catalytic domain with kinase activity, and a C-terminal regulatory domain (). The PH domain binds phosphatidylinositol-3,4,5-trisphosphate (PIP3). Akt also contains two main phosphorylation sites: Threonine308 (Thr308) in the kinase domain and Serine473 (Ser473) in the regulatory domain.Citation1
Figure 1 Akt structure and regulation. A) Akt structure. There are three Akt isoforms (Akt1/2/3), all of which share a similar structure and size. These isoforms contain a PH (pleckstrin homology) domain in the N-terminus, a central catalytic domain with kinase activity, and a regulatory domain in the C-terminus. The PH domain binds inositol triphosphate (PIP3). Akt also contains two main phosphorylation sites: one threonine in the kinase domain (Thr308) and one serine the regulatory domain (Ser473). B) Dual regulatory mechanism of Akt activation: translocation to the plasma membrane followed by phosphorylation. After their activation by specific growth factors (GF), tyrosine kinase receptors (TKRs) can promote the activation of the PI3K complex (p85 plus p110 subunits) directly or by means of the small GTPase monomeric protein Ras. Active PI3K converts inositol biphosphate (PIP2) into PIP3, which triggers the translocation of Akt and PDK1 to the plasma membrane, where they interact with PIP3 through their PH domain. Subsequently, Akt is phosphorylated by PDK1 in its Thr308 residue and by other kinases such as mTOR in its Ser473 residue. C) Negative regulation of Akt activation. The phosphatases PTEN and SHIP (in grey) promote the blockade of Akt translocation to the plasma membrane by dephosphorylation of PIP3 (in red) in PIP2 (in green). Additionally, CTMP (in blue) negatively regulates Akt activation preventing its phosphorylation in Ser473 and Thr308.
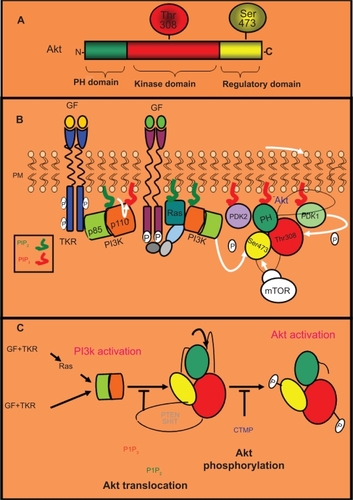
After binding their growth factors, TKRs can directly or indirectly, via an adaptor molecule such as IRS1, activate PI3K, a molecular complex composed of a p85 regulatory subunit and a p110 catalytic subunit.Citation1 Phosphotyrosine residues on the cytoplasmic tail of the activated TKRs or on their associated adaptor proteins recruit PI3K complex to the membrane by binding its p85 regulatory subunit and removing the inhibitory effect of p85 (). The GTPase RAS can also directly activate PI3K through the binding to its p110 catalytic subunit. Once on the cell membrane, PI3K phosphorylates phosphatidylinositol and converts inositol 4,5 biphosphate (PIP2) into PIP3. Subsequently, Akt and the serine/threonine kinase PDK1 translocate to the membrane where they interact with PIP3 through their PH domain. This interaction promotes conformational changes in Akt, resulting in exposure of Thr308 and Ser473. Thr308 is phosphorylated by PDK1, leading to the stabilization of Akt.Citation1 The second phosphorylation event associated with Akt activation occurs at Ser473, and is required for full activation (). Recently, a second protein complex with kinase activity, the mammalian target of rapamycin (mTOR)-rictor complex has been shown necessary for this Ser473 phosphorylation of Akt.Citation2 Therefore, Akt activation implies a dual mechanism: translocation to the plasma membrane after PI3K activation followed by two phosphorylation events mediated by PDK1 and mTOR-rictor complex. This pathway is also negatively regulated at the level of PIP3 by phospholipid phosphatases, such as the phosphatase and tensin homologue PTEN.Citation3
Biological functions of Akt signaling
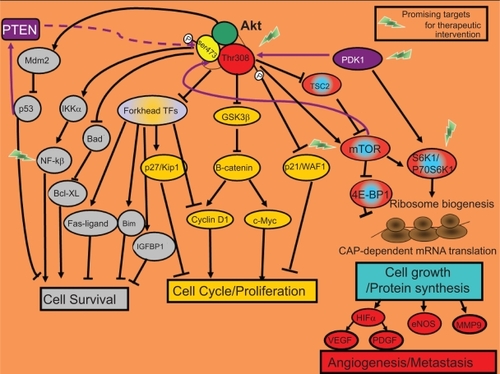
Although, differences in the signaling capabilities of Akt isoforms have been established, as we review below, the biological consequences of Akt activation relevant for cancer progression are survival, proliferation, growth, angiogenesis, and metastasis ().Citation3–Citation5
Figure 2 Biological functions of Akt signaling. Most of the cellular responses controlled by Akt are related to cancer. Once active, Akt regulates the activity of other intracellular signaling proteins that will trigger changes in the expression of specific genes that encode for proteins involved in cell survival (in grey) or proliferation/cell cycle (in yellow). Additionally, by activation of the mTOR protein, Akt will enhance the translation of specific mRNAs. TOP-dependent mRNAs that encode for proteins involved in ribosome biogenesis and also the translation of CAP-dependent mRNAs that encode for proteins with key cellular functions such as angiogenesis and metastasis (in red-blue).
Akt activation and cell survival
Promotion of cell survival is the most studied function of the Akt pathway. Akt develops its anti-apoptotic role through phosphorylation of downstream substrates that control the apoptotic machinery.
Akt indirectly downregulates activation of the pro-apoptotic protein p53 by activating Mdm2 (murine double minute-2), which promotes p53 degradation.Citation5 In response to genetic damage, p53 stimulates the expression of pro-apoptotic proteins to ensure that damaged genetic information is not passed to descending cells. Thus, tumor cells develop the ability to survive after not repairing DNA damage through activation of the PI3K-Akt pathway ().
Additionally, Akt signaling leads to transcription of NF-κB (nuclear factor κB) anti-apoptotic target genes. Akt activates IKK (IκB kinase), which induces degradation of IκB (the NF-κB inhibitor). The unmasked NF-κB can then enter the nucleus and activate the expression of pro-survival target genes ().Citation3,Citation5
Akt also restores the anti-apoptotic function of BCL-XL through inactivation of its inhibitor, BAD ().Citation3,Citation5
The inactivation of the Forkhead family of transcription factors by Akt inhibits the transcription of their death target genes (Fas-ligand, Bim, and immunoglobulin-binding protein-1 [IGBP-1]; ).Citation3,Citation5
Akt also phosphorylates p21/WAF, which increases p21 stability. Elevated p21 protein levels have been observed in various aggressive tumors linked to chemoresistance. Glioblastoma cell lines with active Akt showed higher p21 stability and were more resistant to paclitaxel.Citation6
Akt activation and proliferation
Akt signaling can also affect proliferation by regulating proteins of the cell-cycle machinery.
Akt blocks transcription of the cell cycle inhibitor p27/KIP1 by inactivating Forkhead transcription factors.Citation3 It also inhibits the anti-proliferative effects of p21 and p27, thus impairing their entry into the nucleus.Citation3
Furthermore, Akt inhibits glycogen synthase kinase-3 (GSK3), thus Akt directly prevents β-catenin degradation. Once inside the nucleus, stabilized β-catenin activates expression of pro-proliferative target genes such as cyclin D1 and c-Myc ().Citation3,Citation5
Akt-mTOR activation and cell growth, angiogenesis, and metastasis
The target of rapamycin (TOR) is an evolutionary conserved Ser/Thr kinase that represents the catalytic subunit of two distinct signaling complex: the mTOR-ractor complex (mTOR complex 1) and mTOR-rictor and SIN1 complex (mTOR complex 2).Citation7 In the presence of growth-promoting signals such as nutrients and growth factors, mTOR complex 1 promotes growth by upregulation of the protein synthesisCitation8 and it also induces the biogenesis of the machinery for the protein synthesis, the ribosome.Citation9 The function of mTOR complex 2 is less well defined, it is known that is required for phosphorilation of AktCitation2 () and it is also involved in actin cytoskeleton reorganization and cell survival.Citation10 mTOR complex 1 is inhibited by rapamycin and its derivates everolimus and tenserolimus.Citation7 Therefore, rapamycin analogs are not able to block mTOR complex 2 effects. In fact, in response to these drugs, an increase in Akt phosphorylation is detected in tumor byopsies and tumor samples from animal models as a result of a feedback activation loop of Akt signaling through an IGF-1R-dependent mechanism.Citation11–Citation13
Figure 3 PI3K-Akt-mTOR pathway and cross-talk with other signaling cascades: (Ras/Raf/MAPK and BCR-ABL). PI3K-Akt and Ras/Raf/MAPK pathways are common routes that control key cellular responses. The large amount of cross-talk between these pathways is often responsible for treatment resistance.
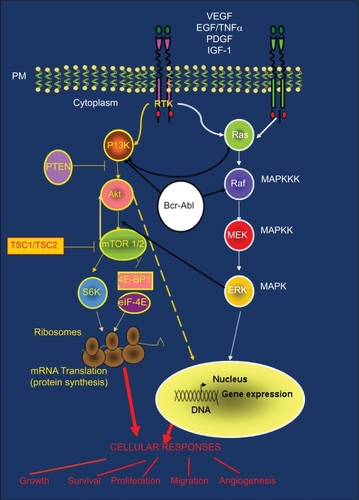
The TSC1/TSC2 (tuberous sclerosis complex) protein complex is involved in the negative regulation of the mTOR kinase ( and ). mTOR is activated by the GTPase Rheb which in turn is controlled by the TSC1/TSC2 complex. As a result of growth-stimulating signals, Akt phosphorylates TSC2 and causes the dissociation of the TSC1/TSC2 complex. This dissociation reduces the inhibitory function of the TSC1/TSC2 complex on GTPase Rheb thus enabling the activation of the mTOR complex 1. mTOR complex 1 controls cell growth in part by phosphorylating of the kinase 70 S6K1 (S61) and the protein 4EBP-1 (4E-binding protein 1), both of them known regulators of protein synthesis ( and ). p70 S6K1 is activated by two phosphorylation events: phosphorylation on Ser473 by mTOR complex 2 and on Thr308 by PDK1.Citation7,Citation14 Subsequently, phosphorylated p70 S6K1 activates the ribosomal protein S6 that stimulates the translation of 5′-TOP messenger ribonucleic acids (mRNAs). These mRNAs encode for proteins of the translation machinery, resulting in a high protein translation rate ( and ).
Besides activating p70 S6K1, mTOR controls the association of the translation initiation factor eIF-4E with its inhibitor 4EBP-1. mTOR phosphorylates the 4EBP-1 inhibitor. Thus, eIF-4E can be released from 4EBP-1 and stimulate the translation of the CAP-dependent mRNAs () that encode for proteins with key cellular functions such as hypoxia-inducible factor-α (HIF-α), a transcription factor that controls the expression of approximately 30 hypoxia-regulated genes.Citation7 These target genes include pro-angiogenic genes, such as VEGF (vascular endothelial growth factor), PDGF (platelet-derived growth factor), and genes that encode proteases associated with local invasion such as matrix metalloproteinase 9 (MMP9). In fact, active p70 S6K1 promotes invasion in ovarian cancer cell lines by stimulating metalloproteinase MMP9 expression.Citation15
VEGF is considered the most potent stimulator of angiogenesis within tumors. HIF-α protein levels are controlled by the von Hippel–Lindau (pVHL) protein complex.Citation16 Absence and/or inactivation of pVHL has been documented in many tumors, thus leading to HIF-α accumulation. Subsequent high VEGF expression promotes angiogenesis.Citation15 PDGF is considered at least as important as VEGF in the stabilization and maturation of newly formed vessels. In fact, PDGF over-function may also cause tumors.
Akt isoforms and specific biological functions
It has been suggested that different Akt isoforms show different signaling capabilities.Citation17 Different roles for Akt isoforms in the physiological response to insulin (the storage of nutrients in muscle and adipose tissues) has been already suggested. In fact, Akt2 levels increase during the differentiation of cells into insulin-responsive adipocytes, whereas the expression of Akt1 decreases in these cells.Citation18,Citation19 In addition, Akt2 is placed in the plasmatic membrane near the insulin-responsive GLUT4 glucose transporter whereas Akt1 is placed mainly in the cytosolic compartment. Phosphorylation of GLUT4 transporter by Akt2 has been also reported.Citation20,Citation21 These data suggest that Akt2 might be involved in insulin-induced GLUT4 translocation and glucose uptake.
Additional data supporting a different role for Akt isoforms came from transgenic mice. An impairment in insulin ability to maintain normal glucose homeostasis is known as insulin resistance and predisposes to the development of type 2 diabetes, hypertension, and cardiovascular disease.Citation22 Accordingly, mice lacking Akt2 displayed insulin resistance and a diabetes-like syndrome.Citation23 Mice lacking Akt1 showed normal glucose homeostasis but were small throughout life,Citation24,Citation25 which suggests that Akt1 may be mainly involved in control of growth and proliferation.
The knowledge of the biological functions mediated by each Akt isoform may be useful to predict the toxicities that may be associated with those inhibitors that target a specific Akt isoform or with pan-Akt inhibitors. As we review below, high grade hyperglycemia has been already observed in animal models and clinical trials with novel Akt inhibitors.
Akt activation in cancer
The PI3K-Akt pathway is often aberrantly activated in cancer (see ) due to genetic and epigenetic alterations. It is also associated with poor prognosis in a variety of tumor types (pancreas, gastric, breast, tongue, glioma, non-small cell lung cancer [NSCLC], and others) and resistance to chemotherapy, radiotherapy, and/or target-based anticancer agents.Citation26,Citation27 Alterations in signaling proteins placed upstream PI3K and genetic alterations in genes that encode for proteins belonging to the pathway such as PTEN deletions and “hot-spot” mutations in PI3KCitation28 lead to an aberrant activation of this pathway and are associated with the development and progression of human cancers.
Table I Akt signaling deregulation in tumors. Association with resistance to treatment
Alterations upstream of PI3K-Akt
The most common alterations upstream of PI3K-Akt include amplification/activating mutations, an increase in the expression of genes that encode for TKRs, and higher expression of genes that encode for growth factors. Activating mutations in intracellular signaling proteins placed upstream of Akt (eg, Ras) also promote the aberrant activation of Akt signaling in cancer cells, thus leading to inappropriate survival and proliferation ().
The PI3K/Akt/mTOR and the Ras/Raf/MAPK pathways are commonly activated by TKRs, and there is a large amount of cross-talk between them ().Citation29 Indeed, defects in the signaling molecules belonging to one pathway may also alter the other pathway through these interconnections. Due to this cross-talk, gain of function mutations in Ras that occur in many tumors (90% of pancreas, 50% of colon, 50% of thyroid, 30% of lung cancers) could also be associated with an increase in Akt signalling.Citation29 The Bcr-Abl fusion protein, generated after chromosomal translocation associated with chronic myeloid leukaemia, is also able to constitutively activate PI3K-Akt and Ras/Raf/MAPK pathways ().Citation30
Alteration in PI3K or Akt
Several isoforms of the PI3K family are implicated in disease. In particular, members of the class 1A PI3Ks (heterodimers with a p85 regulatory and a p110 catalytic subunit). Many tumors show activating mutations in the genes that encode for the catalytic and regulatory subunits of PI3K.Citation3,Citation27
Akt amplification or overexpression occur at the mRNA level in various tumor cell lines and in a number of human cancers (see ). Amplification in the Akt gene has been associated with poor prognosis and resistance to radiotherapy and/or chemotherapy.Citation3 Nevertheless, activation of Akt isoforms by phosphorylation appears to be more clinically significant than Akt amplification or overexpression.Citation31
Alteration in phosphatases belonging to the pathway
Two protein phosphatases, PTEN and SHIP (SH2 containing inositol phosphatase), negatively regulate Akt activation by blocking Akt translocation to the plasma membrane ().
PTEN prevents elevated levels of PIP3 by dephosphorylating its 3′ position. Thus, PTEN leads to the blockade of Akt translocation to the plasma membrane ().Citation3 PTEN is one of the most frequently mutated or deleted genes in human cancer (see ),Citation32 and its absence in tumors correlates strongly with activation of Akt.Citation3 Pre-clinical models showed that mice with conditional tissue disruption of PTEN develop tumors in the affected tissue.Citation3 Furthermore, PTEN loss also predicts for resistance to anticancer drugs such as the anti-HER2 (human epidermal growth factor receptor-2) agent trastuzumab.Citation33
SHIP is another negative regulator of Akt that dephosphorylates PIP3 at the 5′ position (). Loss of SHIP can also occur in cancer cells. Peripheral T cells isolated from acute and chronic T cell leukemia/lymphoma patients showed lower levels of PTEN and SHIP, which correlates with upregulation of Akt signaling.Citation34
In addition, Akt phosphorylation is negatively controlled by carboxyl-terminal modulating protein (CTMP), which acts downstream of PTEN and SHIP, at the plasma membrane level, and binds specifically to the carboxyl-terminal regulatory domain of Akt. CTMP reduces the activity of Akt by inhibiting its phosphorylation at Ser473 and Thr308 (). Loss of CTMP by hypermethylation of its promoter has also been detected in glioblastoma and glioma cell lines.Citation35
Akt pathway deregulation in selected cancers
As reviewed above, isolated or combined defects in PI3K/Akt pathway components and/or molecules in interconnected signaling cascades are associated with many cancers and lead to an increase in Akt signaling. Aberrant Akt activation mediates the signals for survival, proliferation, angiogenesis, and/or metastasis in tumor cells. Furthermore, resistance to cancer therapies is often associated with abnormal activation of this pathway.
Here, we focus on the PI3K-Akt alterations associated with specific tumors ().
Renal cell carcinoma
The most frequent histopathologic variant of renal cell carcinoma (RCC) is the clear-cell type (75% of cases).Citation36
In familial and in most sporadic (40%–60%) clear-cell RCC tumors, pVHL is disrupted.Citation36 and the PI3K-Akt-mTOR pathway plays a critical role in survival, proliferation, and angiogenesis.Citation16
The transcription factor, HIF, which controls the expression of hypoxia-inducible genes, is a heterodimer of two subunits (HIF-α and HIF-β). In each cell, HIF-β is constitutively expressed, whereas the intracellular amount of HIF-α is highly controlled at two levels: PI3k/Akt signaling through mTOR at the translational level and pVHL at the post-translational level.Citation36,Citation37
pVHL is a component of a ubiquitin ligase complex that promotes the destruction of specific cellular proteins through proteosome degradation. In normal tissue, under normoxic conditions, pVHL recognizes and binds hydroxylated HIF-α, which, once bound by pVHL, will be ubiquitinated to be degraded by the proteosome. However, under hypoxic conditions, HIF-α, is not hydroxylated, pVHL is not able to recognize HIF-α and, consequently, is not degraded. Stabilized HIF-α is translocated to the nucleus, binds with HIF-β, and forms a heterodimer that promotes expression of hypoxia-response target genes.Citation37
pVHL is lost or inactivated in most clear-cell RCC, and HIF-α, although hydroxylated, is not degraded. Accumulated HIF-α, leads to increased expression of hypoxia-response genes.Citation37,Citation38 HIF-inducible genes are associated with tumorogenesis because they are involved mainly in angiogenesis (VEGF, PDGF, others), proliferation and survival (transforming growth factor alpha [TGF-α], insulin-like growth factor 2 [IGF2], and others), metabolism, and pH regulation.Citation37,Citation38
The characterization of PI3K-Akt-mTOR in metastatic RCC patients using the UCLA RCC tissue array showed that loss of PTEN increased in all RCC patients with a greater loss in clear-cell tumors and sarcomatoid tumors. The presence of active Akt (pAkt) was greatest in collecting duct carcinoma followed by clear-cell carcinoma and loss of PTEN correlated with pAkt (p = 0.028) and HIF-α expression (p < 0.0001). Active p70 S6K (pS6K) was also highly expressed by sarcomatoid and clear-cell tumors (higher expression in high-grade and in high-stage).Citation36 The mTOR inhibitor temsirolimus has recently shown efficacy in non-clear–cell RCC histologic subtypes.Citation39 One proposed resistance mechanism for mTOR inhibition is the compensatory upregulation of Akt through the activation of the IGF receptor (IGF-1R) pathway as we will explain in the last section of this review. Accordingly, preclinical data showed by Holland and colleagues established a rationale for the combination of therapies that target Akt and mTOR in RCC patients.Citation40
Additionally, immunohistochemical analyses have also revealed that no patient who had low expression of pS6K or pAkt was a responder to this mTOR inhibitor.Citation41 Pre-clinical assays with RCC cell lines have also shown that a loss of pVHL sensitized cells to temsirolimus, both in vitro and in mouse xenograft models, suggesting that loss of pVHL could be a predictive marker of the response to mTOR blockade.Citation42 Furthermore, PTEN-deficient tumor cell lines with high levels of pAkt were also more sensitive to the effects of temsirolimus in vitro.Citation43 In this molecular scenario, the Akt pathway is a promising target in the treatment of metastatic RCC. In fact, the mTOR inhibitor temsirolimus has been already approved for the first line treatment of poor risk advanced RCC patientsCitation44 and clinical trials with the mTOR inhibitor everolimus are currently underway to show its efficacy in RCC patients.Citation45
Breast cancer
Hormone receptor- and HER2-positive breast cancer currently account for 75%–80% and 15%–20% of breast cancer cases, respectively, with about half of HER2-positive cases co-expressing hormone receptors. The remaining 10%–15% of breast cancer cases do not express HER2 and hormone receptors and are called triple-negative breast cancers.Citation46 Evidence shows PI3K pathway aberrations in breast tumors and suggests that this pathway may play a distinct role in the pathogenesis of different breast cancer molecular subtypes.
The analysis of 64 hormone receptor-positive-Her2-negative breast cancer samples from patients with stage I to III tumors managed with hormonotherapy ± chemotherapy showed that the activation of the PI3K/Akt pathway was higher in those tumors with less hormone receptor levels and was also associated with adverse outcome.Citation47
Recently, a genomic analysis of PIK3CA, PTEN, and Akt mutations in a collection of 547 breast cancer samples showed that PIK3CA mutations were more common in hormone receptor-positive (34.5%) and HER2-positive (22.7%) than in triple-negative tumors (8.3%). Akt1 (1.4%) and PTEN (2.3%) mutations were restricted to hormone receptor-positive cancers. However, PIK3CA mutations did not have a significant effect on outcome after adjuvant tamoxifen therapy in a population of 157 hormone receptor-positive breast cancer patients.Citation48
HER2 overexpression in breast cancer is associated with a high recurrence rate, poor survival, and resistance to chemotherapy and endocrine therapy.Citation49,Citation50 PI3K/Akt and Ras/MAPK pathways are the major mediators of HER2 signaling.Citation51 Accordingly, tumor cells in which HER2 is overexpressed exhibit constitutive Akt activity.Citation52 Furthermore, immunohistochemical analysis of breast cancer samples indicated a possible role of PI3K/Akt/mTOR signaling in HER2-mediated breast cancer progression and has suggested its association with poor disease-free survival. Additionally, it has been suggested that the anti-HER2 agent trastuzumab depends on the intact PTEN for its action in HER2 overexpressing breast cancer cell lines and that the loss of PTEN predicts for trastuzumab resistance.Citation33 Therefore, Akt blockade could represent a promising strategy to treat breast cancer patients who are trastuzumab resistant.Citation53,Citation54 Moreover, an oral dual tyrosine kinase inhibitor (TKi) of epidermal growth factor receptor (EGFR) and HER2, as a single agent, promotes, apoptosis in trastuzumab-resistant breast cancer cell lines. In these cells, its cytotoxic effect is correlated with the blockade of the activation of Akt, p70 S6K, and MAPK.Citation53 Furthermore, lapatinib also sensitizes HER2-positive breast cancer cells to other therapies such as radiation and also restores tamoxifen sensitivity in tamoxifen-resistant breast cancer models suggesting that this ability of lapatinib may be due to the inhibition of the PI3K-Akt signaling pathway. Accordingly, pre-clinical models also show that failure to inhibit Akt activation leads to resistance to therapies with TKIs against HER-family.Citation26,Citation55–Citation58
Akt activity also promotes resistance to chemotherapy in breast cancer cell lines,Citation54 and recent findings correlate pAkt levels and HER2 status with resistance to endocrine therapy in metastatic breast cancer.Citation59 A retrospective study with tumor samples from 36 metastatic breast cancer patients treated with endocrine therapy showed 12 cases (33.4%) with pAkt. In the pAkt-positive patients, endocrine therapy proved less efficacious than in pAkt-negative patients (p < 0.01)Citation54 and pAkt was associated with a poor objective response (p < 0.05). Clinical benefit was also lower in HER2-positive patients (P<0.05) and was the lowest in the HER2 and pAkt-positive patients (p < 0.01). Therefore, evidence suggests that pAkt may be a useful predictor of resistance to chemotherapy and endocrine therapy for breast cancer. These data suggest that inhibition of Akt activation in breast cancer patients may increase the efficacy of endocrine therapy and chemotherapy.
Based on the above evidence, clinical trials that explore the efficacy of the mTOR inhibitors temsirolimus and everolimus alone or in combination with aromatase inhibitors in hormone receptor positive breast cancer patients are underway.Citation60–Citation62
Regarding triple-negative breast cancer, one of its molecular features is the overexpression of genes that encode for the TKRs EGFR and c-KIT that lead to abundant MAPK and Akt signaling activation downstream. Therefore, Akt could be also a promising target for the treatment of triple-negative breast cancer.Citation46
In conclusion, these findings suggest that Akt activation should be used as a molecular marker in breast cancer patients to predict treatment response. The blockade of Akt activation in combination with current therapies may be also considered a novel therapeutic approach in most breast cancer molecular subtypes.
Non-small cell lung cancer
Pre-clinical data have shown that pAkt is also active in most NSCLC cell lines and that the modulation of Akt activity (by pharmacological or genetic approaches) sensitizes cells to chemotherapy and radiation.Citation58 A correlation between pAkt and poor prognosis has been also established for all stages of NSCLC.Citation63,Citation64 Tang and colleagues reported that loss of PTEN and the presence of pAkt is correlated with poor differentiation, lymph node involvement, distant metastasis, and late stages in NSCLC patients.Citation65 Therefore, the combinations of drugs that modulate Akt activation with the therapeutic modalities typically used in NSCLC are thought to improve treatment response.
Additionally, while some authors have reported the possible role of pAkt as a predictive marker of response to gefitinib, an oral small molecule that blocks EGFR, in NSCLC patients with specific EGFR mutations, others have failed to find any association. Sordella and colleagues showed that these EGFR mutants selectively activate the Akt survival pathway.Citation66 Cappuzo and colleagues reported that gefitinib was more effective in patients with pAkt-positive tumors. These patients showed better response rates, disease control rates, and times to progression than patients with pAkt-negative tumors.Citation67 Other authors have indicated that although no significant correlation between EGFR mutations and expression of pAkt was detected in gefitinib-treated patients, pAkt overexpression was associated with prolonged time to progression.Citation68 Consistent with these findings, pre-clinical studies have shown that those EGFR mutations that sensitize to gefitinib selectively activated the Akt pathway. High sensitivity to EGFR TKIs in NSCLC seemed to be closely correlated with dependence on Akt activation in response to EGFR signaling.Citation65,Citation69 Therefore, further studies are necessary to confirm whether the assessment of pAkt status could be a useful complementary test for the identification of EGFR-positive NSCLC patients with the highest possibility of profiting from TKIs.
Exposure to cigarette smoke (active or passive) is the main cause of lung cancer.Citation70 The presence of nicotinic acetylcholine receptors in NSCLC cell lines and primary tissues has been documented.Citation71,Citation72 After exposure to nicotine, Akt is phosphorylated in NSCLC cells. The Akt activation induced by nicotine leads to an increase in its downstream effectors and confers resistance to NSCLC cells treated with chemotherapeutic agents.Citation69,Citation71 Furthermore, it has been shown that nicotine in NSCLC cells promotes resistance to TKIs such as gefitinib.Citation69
Pancreatic cancer
High levels of pAkt have been detected in 40%–70% of pancreatic cancers and 60% of tumors show EGFR amplification. Therefore, these proteins and their downstream effectors could be new therapeutic targets for treatment, as well as useful molecules for diagnosis and prognosis.Citation73 In fact, erlotinib, an oral small-molecule anti-EGFR, has been approved by the US Food and Drug Administration (FDA) for the treatment of locally advanced or metastatic pancreatic cancer. EGF receptor-related protein (ERRP), a novel EGFR-negative modulator has also shown anti-tumor activity in pre-clinical studies through its ability to downregulate molecular markers such as Akt, NF-κB, and MAPK.Citation74,Citation75 Buck and colleagues also showed that the combination of the mTOR inhibitor rapamycin with erlotinib has anti-tumor activity in human pancreatic cancer cell lines that do not respond to erlotinib in monotherapy. These authors suggest that although erlotinib could not downregulate baseline Akt activity in pancreatic cancer-resistant cell lines, it could synergize with rapamycin by inhibiting the stimulated Akt activity induced by rapamycin.Citation76 Indeed, strategies aimed at blocking Akt activation could be promising for the treatment of pancreatic cancer.
Hormone-refractory prostate cancer
Metastatic hormone-refractory prostate cancer (HRPC) patients show enhanced Akt activity. Some also show loss of PTEN, which leads to the activation of downstream Akt anti-apoptotic effectors such as Bcl-xl, Bcl-2 (B-cell lymphoma 2), and NF-κB, and other Akt effectors such as mTOR/p70 S6K.Citation77,Citation78
The current reference treatment for HRPC consists mainly of chemotherapy with docetaxel. However, interference with cell survival, through pro-apoptotic drugs, combined with docetaxel is a new approach.Citation78 Clinical trials with agents that reduce the expression of anti-apoptotic Bcl-2 proteins are ongoing (a phase II study with oblimersen sodium plus docetaxel), studies with anti-NF-κB therapies have been reported (phase I/II studies with thalidomide, arsenic trioxide, and bortezomib), and also the mTOR inhibitors (rapamycin, temsirolimus, and everolimus) have also produced good results in pre-clinical models. Nevertheless, a phase II clinical study with the Akt inhibitor perifosine showed poor results.Citation76,Citation78
Gastric cancer
In the last American Society of Clinical Oncology (ASCO) annual meeting, Xu and colleagues showed that 148 (78.7%) were positive for p-Akt among 188 gastric adenocarcinoma samples. High expressions of p-Akt were significantly correlated with pTNM stage (p = 0.031), depth of invasion (p < 0.001), lymph node metastasis (p < 0.001) and differentiated levels (p < 0.012).Citation79
Additionally, Oki and colleagues have shown a direct correlation between Akt activation and chemoresistance in gastric cancer. Analysis of primary gastric carcinoma tissue and corresponding normal mucosa from gastric cancer patients who underwent surgery showed that Akt activation and loss of PTEN were associated with increased resistance to several chemotherapeutic agents.Citation80,Citation81 A pre-clinical study also showed that those chemotherapeutic agents that induced apoptosis in human gastric cancer cell lines downregulated the PI3K-Akt pathway.Citation80
Additionally, pAkt levels were associated with the efficacy of some adjuvant chemotherapy regimens and indicated the association between pAkt levels with poor prognosis for T3/T4 gastric cancer patients.Citation82
Although further studies are necessary to establish the predictive value of Akt signaling in sensitivity/resistance to gastric cancer therapies, these data suggest that drug combinations including Akt inhibitors may improve response in patients with gastric cancer.
Hepatocellular carcinoma
Alterations in the TKR c-Met have been involved in HCC (hepatocellular carcinoma) progression.Citation83 In human HCC cell lines, hepatocyte growth factor (HGF) uses its association with c-Met to promote the activation of the PI3K-Akt pro-survival pathway, which led to suppression of the proapoptotic signals mediated by Fas.Citation84 Fas is a membrane receptor that induces apoptosis after binding its ligand FasL onto the surface of T-cells. Fas/FasL signaling seems to mediate chemosensitivity in pre-clinical models of HCC.Citation85 Those HCC cell lines that respond to chemotherapy show increased Fas/FasL-mediated apoptosis. c-Met, through its ability to activate Akt, may be involved in resistance to those chemotherapeutic agents that mediate its cytotoxic effect by stimulating the Fas-FasL apoptotic pathway.
EGFR, IGF2, and its receptor IGF-1R are also detected in HCC cell lines and cross-talks between EGFR and IGF-1R have been reported in different tumor types. IGF2-IGF-1R activates proliferation through Ras/MAPK pathway and survival by means of PI3K/Akt.Citation86,Citation87 In fact, gefitinib alone was not able to completely prevent the development of HCC in pre-clinical modelsCitation86 and IGF2-IGF-1R may contribute to gefitinib resistance in HCC cells through the activation of PI3K/Akt pathway. Treatment of HCC cell lines with gefitinib blocked Akt activation induced by EGFR itself, although gefitinib had no effects on the Akt activation induced by IGF2-IGF1R; therefore, it could not cause the death of the tumor cells by itself.
Additionally, a selective IGF-1R inhibitor was able to induce apoptosis in these HCC cells and its cytotoxic effect was boosted by gefitinib.Citation88 Therefore, the IGF2/IGF-1R survival signal promoted by the activation of Akt may contribute to gefitinib resistance in HCC. In this setting, the pharmacological combination of gefitinib with anti-IGF-1R antibody or inhibition downstream in the pI3K-Akt pathway could enhance the anti-tumor effects reached by gefinitib as a single agent in HCC.
Brain tumors
The PI3K-Akt pathway is relevant in glioma initiation and progression.Citation89 PTEN is frequently lost or mutated in high-grade gliomas and most of these tumors have elevated Akt activity. Gliomas also seem to be good targets for therapies based on inhibitory compounds of the PI3K-Akt-p70 S6K pathway.
Recently, activation of the Akt pathway in 101 neuroblastoma samples was also tested: 60% of the tumor samples expressed PI3K, 80% Akt, and 73% pAkt. In addition, p70 S6K was expressed in 95% of tumors. Akt and pAkt also showed higher expression in metastases than in primary tumors.Citation90
Akt inhibitors
As we described above, PI3K-Akt-mTOR pathway plays a critical role in proliferation and survival in tumor cells and is also linked with resistance to radiotherapy, chemotherapy, endocrine therapy and novel anticancer therapies.Citation91–Citation94 In this setting, this pathway seems to bring together all the characteristics of a good target for the treatment of cancer. Currently, clinical trials with inhibitors of the PI3K-Akt pathway in monotherapy or in combination with other anti cancer drugs are underway in cancer patients. In addition, these novel inhibitors are being also tested in chemo- and hormonal therapy refractory patients.Citation95,Citation96
Although in this review we will focus on inhibitors of Akt, currently PI3K and mTOR inhibitors are more advanced in their development than Akt inhibitors. In fact, a new generation of PI3K inhibitors is emerging. Some of them, such as NVP-BEZ235, a synthetic low molecular weight compound belonging to the class of imidazoquinolins, which potently and reversively inhibits class I PI3K catalytic activity by competing with the ATP is in early phase clinical trials.Citation97 And as we mentioned above, the rapamycin-derivated temsirolimus and everolimus, as mTOR inhibitors, have been already approved for the treatment of same cancer patients and clinical trials to explore their efficacy in new tumor types are currently underway.Citation16,Citation41,Citation98 Furthermore, rapamycin analogs lead to an increase in PI3K and Akt activation, through a feedback mechanism mediated by IGF-1R. Therefore, this Akt activation may counteract the inhibition of mTOR. Combined treatment with mTOR inhibitors and anti-IGF-1R monoclonal antibodies (MAbs) are currently being tested in caner patients.Citation99 In addition, due to the cross-talk between ER and PI3K/Akt/mTOR pathways, clinical trials with mTOR inhibitors and endocrine therapy are also underway.Citation60
In this context, the use of anti-target agents that block selectively each Akt isoforms or the use of dual inhibitors that block this pathway in two levels (pg, dual Akt-S6K small inhibitors, dual mTOR complex 1 and 2 inhibitors, etc) may improve the results reached with the current mTOR inhibitors. During recent years, intense efforts made in the search for Akt inhibitors have yielded several promising candidates, such as lipid-based inhibitors that compete with PIP3 to bind the PH domain of Akt, ATP-competitive inhibitors, small-molecule inhibitors, and peptide-based inhibitors reviewed elsewhere.Citation100,Citation101
The lipid-based inhibitors include perifosine (KRX-0401), an orally active membrane-permeable ether lipid with a single long alkyl chain that inhibits the translocation of all Akt isoforms to the membrane. In vitro, perifosine has anti-proliferative effects in many tumor cell lines and sensitizes tumor cells to radiation and chemotherapy. Perifosine has also shown efficacy and tolerability in phase I clinical trials.Citation102 However, the lack of objective responses as a single agent has been observed in several recent phase II trials in patients with malignant melanoma, prostate cancer, head and neck cancer, and pancreatic adenocarcinoma.Citation101 Phase II trials with perifosine combined with radiotherapy, chemotherapy or other anticancer agents, such as imatinib mesylate and trastuzumab, are ongoing.Citation100 In the last ASCO annual meeting, preliminary results from a phase I study from a multicenter trial of perifosine plus in patients with advanced solid tumors were reported. Accrual is in the last cohort. To date no unexpected toxicities and clinical activity has been noted within the first three cohorts with four of six (67%) evaluable patients with advanced cancer achieving at least stable disease for more than six months. One partial response has been also seen.Citation103
Triciribine phosphate monohydrate (TCN-P; VQD-002, VioQuest Pharmaceuticals, Basking Ridge, NJ, USA) is an old anticancer agent discovered more than 35 years ago that was discontinued due to severe side effects at high doses. The discovery of triciribine as a potent and selective inhibitor of Akt has led to renewed interest in its further development. VQD-002 works as a pan-Akt inhibitor that targets the PH domain of Akt. It inhibits the growth of human tumors that overexpress Akt in mice at low doses without visible side effects. VQD-002 has demonstrated compelling preclinical activity in combination with other targeted therapies (trastuzumab and TKIs) in many tumor types, such as glioblastoma, lymphomas, refractory leukemia, breast, colon, NSCLC, and prostate.Citation104 VQD-002 is in Phase I/IIa clinical trials in solid and hematological tumors.Citation101
GSK690693 is an Akt ATP-pocket binder developed by GSK with ICCitation50 values of 2 nM, 13 nM, and 9 nM against Akt1, Akt2, and Akt3, respectively.Citation101 In vitro, GSK690693 showed a strong additive effect in a variety of tumor cells when combined with other anti-target drugs such as lapatinib. It also showed significant efficacy in xenograft models from ovarian (SK-OV-3), prostate (LNCaP), and breast (BR474, HCC-1954) carcinoma cell lines. Phase I clinical trials in patients with solid tumors and lymphomas treated with intravenous GSK690693 are underway.Citation105,Citation106
XL418 is an orally active and low-nanomolar dual Akt and p70 S6K inhibitor developed by Exelisis Inc. (San Francisco, CA, USA) that also acts as an ATP-pocket binder.Citation101 It has already demonstrated good in vivo efficacy in several xenograft models, including lung (A549) and breast adenocarcinoma (MCF7) tumor cell lines as a single agent or in combination with chemotherapeutic drugs such as paclitaxel. It has also been shown to enhance apoptosis in combination with TKIs that target EGFR, HER2, and VEGFR. A phase I clinical trial with this drug is ongoing.
Therefore, we can see that perifosine and VQD-002 block Akt activation through their interaction with the PH domain of Akt,Citation101 whereas GSK690693 and XL418 inhibit the kinase activity of the active Akt through binding to its ATP pocket.
Treatment with pan-Akt inhibitors has shown unexpected toxicity. One of these side effects includes hyperglycemia. GSK690693 leads to increased blood glucose and insulin levels, although these return to baseline levels as the circulating drug concentration decreases. This side effect was also observed in clinical trials with perifosine and triciribine phosphate.Citation101 The hyperglycemia induced by these inhibitors is consistent with the phenotype of the Akt2 knockout mice.Citation107 Another pan-Akt inhibitor developed by Abbott, a series of 3,5-disubstituted pyridines (A-443654 and A-6745639), was reported to cause severe hypotension in rats and dogs that could be partly due to inhibition of other kinases.Citation100
Regarding the feedback mechanism mediated by IGF1-R, the inhibition of Akt or mTOR complex 1 leads to subsequent suppression of phosphorylation of p70 S6K, which inhibits the phosphorylation and subsequent inactivation of insulin receptor substrate 1 (IRS-1) by p70 S6K. Activation of IRS-1 promotes the insulin-stimulated activation of Akt via PI3K.Citation12,Citation101,Citation108–Citation110 Therefore, this Akt activation might attenuate the therapeutic effects reached with the mTOR complex 1 inhibitors, although the combination rapamycin analogs with therapies that simultaneously prevent subsequent Akt activation (pg, MAbs against IGF1-R, and others) might be more effective. In this sense, significant synergy between mTOR inhibitors and anti-TKR drugs has also been observed.Citation101 In fact, dual inhibition with the mTOR inhibitor everolimus combined with a monoclonal antibody against IGF-1R results in a supra-additive growth inhibitory effect both in vitro and in vivo in breast cancer models.Citation13 Clinical studies with this combination in breast cancer patients are planned.
The search for small molecule inhibitors that interact with the allosteric site of Akt represents a new frontier in drug discovery. In this regard, Merck and Co, Inc. (Whitehouse Station, NJ, USA) has synthesized allosteric isoform-selective Akt inhibitors that are able to inhibit Akt kinase activity (like the ATP pocked binders) and in addition the activation of Akt by phosphorylation.Citation101 In this setting, the activation of Akt via the feedback mechanism would be abolished. Therefore, these allosteric Akt inhibitors should be more efficient than the ATP-pocket binders in attenuating Akt activity. However, preliminary studies have revealed that the effect of single isoform inhibition is not superior to the simultaneous inhibition of Akt1 and Akt2 with pan-Akt inhibitors.Citation101
Another trend in this area is the search for novel Akt inhibitors that bind to other sites of Akt, based on the fact that activity and function of Akt are also regulated by many interacting proteins.Citation101 Akt-in is a peptide composed of 15 amino acids designed to mimic the interaction between Akt and a physiological coactivator, T-cell leukemia/lymphoma 1 (TCL1). The Akt-in sequence encompasses a portion of the TCL1 protein that binds Akt in its PH domain and inhibits Akt kinase activity and membrane translocation in cell lines. It also inhibits tumor cell proliferation in preclinical models but has poor oral bioavailability and cellular penetration. Other approaches use the consensus protein sequence preferred by Akt to develop pseudopeptide substrates that have been shown to inhibit Akt and the growth of cancer cells in vitro.Citation111,Citation112
Akt antisense inhibitors that block Akt translation are also being developed. RX-0201 is a 20-mer oligonucleotide with a sequence complementary to that of Akt-1 mRNA that inhibits the expression of Akt-1 in tumor cell lines and bears significant in vitro and in vivo anticancer activity with good safety. A phase I trial with RX-0201 that aimed to determine the maximum tolerated dose and pharmacokinetic and safety profile of RX-0201 was reported at the last ASCO meeting and phase II clinical trials are being planned.Citation113
Summary
A more in-depth knowledge of the molecular pathogenesis of cancer has led to the discovery of new tumor targets. These are aimed at targets located on the cell membrane and inside the cell. The newly approved target-based anticancer agents (erlotinib, gefitinib, lapatinib, trastuzumab, cetuximab, sunitinib, sorafenib, imatinib, dasatinib, and temsirolimus) have not only shown efficacy in terms of response rate and progression-free survival, but also a good safety profile.
The PI3K/Akt pathway emerges as one of the most promising targets in the immediate future. Pharmaceutical companies are developing new agents against this pathway. Hopefully, these new agents will become part of the anti-tumor armamentarium.
Abbreviations
| 4EBP-1 | = | 4E-binding protein 1 |
| AGC | = | protein kinase A/protein kinase G/protein kinase C-like |
| Bcl-2 | = | B-cell lymphoma 2 |
| CAP | = | catabolite activator protein |
| CTMP | = | carboxyl-terminal modulating protein |
| DNA-PK | = | DNA-dependent protein kinase |
| EGFR | = | epidermal growth factor receptor |
| ERRP | = | EGF receptor-related protein |
| GSK3 | = | glycogen synthase kinase-3 |
| HCC | = | Hepatocellular carcinoma |
| HER2 | = | human epidermal growth factor receptor-2 |
| HGF | = | hepatocyte growth factor |
| HIF-a | = | hypoxia-inducible factor a |
| HRPC | = | hormone-refractory prostate cancer |
| IGBP-1 | = | immunoglobulin binding protein-1 |
| IGF-1R | = | insulin-like growth factor receptor 1 |
| IGF2 | = | insulin growth factor-2 |
| IKK | = | I kappa B kinase complex |
| IRS-1 | = | insulin receptor substrate 1 |
| Mdm2 | = | murine double minute-2 |
| MMP9 | = | matrix metalloproteinase 9 |
| mTOR | = | mammalian target of rapamycin |
| NF-kB | = | nuclear factor κB |
| NSCLC | = | non-small cell lung cancer |
| p70 S6K1 | = | S6 kinase-1 |
| PDGF | = | platelet-derived growth factor |
| PH | = | pleckstrin homology |
| PI3K | = | phosphatidylinositol 3-kinase |
| PIP2 | = | inositol 4, 5 biphosphate |
| PIP3 | = | phosphatidylinositol-3, 4, 5-trisphosphate |
| PKB | = | protein kinase B |
| PTEN | = | phosphatase and tensin homologue |
| pVHL | = | von Hippel–Lindau protein complex |
| RCC | = | renal cell carcinoma |
| SHIP | = | SH2 containing inositol phosphatise |
| TCL1 | = | T-cell leukemia/lymphoma 1 |
| TGF-a | = | transforming growth factor alpha |
| TKI | = | tyrosine kinase inhibitor |
| TKRs | = | tyrosine kinase receptor |
| TSC1/TSC2 | = | tuberous sclerosis complex |
| VEGF | = | vascular endothelial growth factor. |
Acknowledgements
We thank Fátima Cruz, Luisa Martín and Nieves Ruíz-Ayllón for editorial assistance. The authors report no conflicts of interest in this work.
References
- VivancoISawyersCLThe phosphatidylinositol 3-kinase Akt pathway in human cancerNat Rev Cancer2002248950112094235
- SarbassovDDGuertinDAAliSMSabatiniDMPhosphorylation and regulation of Akt/PKB by the rictor-mTOR complexScience20053071098110115718470
- OsakiMOshimuraMItoHPI3K-Akt pathway: its functions and alterations in human cancerApoptosis2004966767615505410
- ManningBDCantleyLCAkt/PKB signaling: navigating downstreamCell20071291261127417604717
- LuoJManningBDCantleyLCTargeting the PI3K-Akt pathway in human cancer: rationale and promiseCancer Cell2003425726214585353
- LiYDowbenkoDLaskyLAAkt/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survivalJ Biol Chem2002277113521136111756412
- WullschlegerSLoewithRHallMNTOR signaling in growth and metabolismCell200612447148416469695
- MamaneYPetroulakisELeBacquerOSonenbergNmTOR, translation initiation and cancerOncogene200625486416642217041626
- MartinDEHallMNThe expanding TOR signaling networkCurr Opin Cell Biol200517215816615780592
- JacintoELoewithRSchmidtAMammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitiveNat Cell Biol20046111122112815467718
- WanXHarkavyBShenNGroharPHelmanLJRapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanismOncogene2007261932194017001314
- O’ReillyKERojoFSheQBmTOR inhibition induces upstream receptor tyrosine kinase signaling and activates AktCancer Res2006661500150816452206
- Di CosimoSScaltritiMValDPI3-K/Akt/mTOR pathway as a target for breast cancer therapy2007 ASCO Annual Meeting. Abstract No. 3511
- BjornstiMAHoughtonPJThe TOR pathway: a target for cancer therapyNat Rev Cancer2004433534815122205
- SeeligerHGubaMKleespiesAJauchKWBrunsCJRole of mTOR in solid tumor systems: a therapeutical target against primary tumor growth, metastases, and angiogenesisCancer Metastasis Rev20072661162117713840
- PantuckAJSeligsonDBKlatteTPrognostic relevance of the mTOR pathway in renal cell carcinoma: implications for molecular patient selection for targeted therapyCancer2007109112257226717440983
- ChanTORittenhouseSETsichlisPNAkt/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylationAnnu Rev Biochem199968965101410872470
- HillMMClarkSFTuckerDFBirnbaumMJJamesDEMacaulaySLA role for protein kinase Bbeta/Akt2 in insulin-stimulated GLUT4 translocation in adipocytesMol Cell Biol1999197771778110523666
- SummersSAWhitemanELChoHLipfertLBirnbaumMJDifferentiation-dependent suppression of platelet-derived growth factor signaling in cultured adipocytesJ Biol Chem1999274238582386710446150
- CaleraMRMartinezCLiuHJackAKBirnbaumMJPilchPFInsulin increases the association of Akt-2 with Glut4-containing vesiclesJ Biol Chem1998273720172049516411
- KupriyanovaTAKandrorKVAkt-2 binds to Glut4-containing vesicles and phosphorylates their component proteins in response to insulinJ Biol Chem1999274145814649880520
- HunterSJGarveyWTInsulin action and insulin resistance: diseases involving defects in insulin receptors, signal transduction, and the glucose transport effector systemAm J Med19981053313459809695
- ChoHMuJKimJKInsulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta)Science20012921728173111387480
- ChenWSXuPZGottlobKGrowth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 geneGenes Dev20012203220811544177
- ChoHThorvaldsenJLChuQFengFBirnbaumMJAkt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in miceJ Biol Chem2001276383493835211533044
- SerginaNVRauschMWangDEscape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3Nature200744543744117206155
- BellacosaAKumarCCDi CristofanoATestaJRActivation of Akt kinases in cancer: implications for therapeutic targetingAdv Cancer Res200594298616095999
- SamuelsYDiazLAJrSchmidt-KittlerOMutant PIK3CA promotes cell growth and invasion of human cancer cellsCancer Cell20057656157315950905
- Rodriguez-VicianaPWarnePHDhandRPhosphatidylinositol-3-OH kinase as a direct target of RasNature19943705275328052307
- MaruYMolecular biology of chronic myeloid leukemiaInt J Hematol20017330832211345196
- CicenasJThe potential role of Akt phosphorylation in human cancersInt J Biol Markers20082311918409144
- EngelmanJALuoJCantleyLCThe evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolismNat Rev Genet2006760661916847462
- NagataYLanKHZhouXPTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patientsCancer Cell2004611712715324695
- FukudaRHayashiAUtsunomiyaAAlteration of phosphati-dylinositol 3-kinase cascade in the multilobulated nuclear formation of adult T cell leukemia/lymphoma (ATLL)Proc Natl Acad Sci U S A2005102152131521816217039
- KnobbeCBReifenbergerJBlaschkeBReifenbergerGHypermethylation and transcriptional downregulation of the carboxyl-terminal modulator protein gene in glioblastomasJ Natl Cancer Inst20049648348615026474
- CohenHTMcGovernFJRenal-cell carcinomaN Engl J Med20053532477249016339096
- HaaseVHThe VHL/HIF oxygen-sensing pathway and its relevance to kidney diseaseKidney Int2006691302130716531988
- SemenzaGLTargeting HIF-1 for cancer therapyNat Rev Cancer2003372173213130303
- HudesGCarducciMTomczakPA phase III, randomized, 3-arm study of Temsirolimus (TEMSR) or interferon-α (IFN) or the combination of TEMSR + IFN in the treatment of first-line, poor-risk patients with advanced renal cell carcinoma. ASCO Annual Meeting ProceedingsJ Clin Oncol200624Abstract LBA4.
- HollandWSPreclinical rationale for combination targeted therapy in advanced clear cell renal cell carcinoma (RCC): Abrogation of rapamycin-mediated induction of Akt phosphorylation by perifosine2008 ASCO Annual Meeting. Abstract-No. 16083
- ChoDSignorettiSReganMMierJWAtkinsMBThe role of mamma lian target of rapamycin inhibitors in the treatment of advanced renal cancerClin Cancer Res200713(2 Pt 2):758s763s17255306
- ThomasGVTranCMellinghoffIKHypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancerNat Med20061212212716341243
- ShiYGeraJHuLEnhanced sensitivity of multiple myeloma cells containing PTEN mutations to CCI-779Cancer Res2002625027503412208757
- HudesGCarducciMTomczakPGlobal ARCC TrialTemsirolimus, interferon alfa, or both for advanced renal-cell carcinomaN Engl J Med2007356222271228117538086
- MotzerJRAD001 vs placebo in patients with metastatic renal cell carcinoma (RCC) after progression on VEGFr-TKI therapy: Results from a randomized, double-blind, multicenter Phase-III study R2008 ASCO Annual Meeting. Abstract No. LBA5026.
- CleatorSHellerWCoombesRCTriple-negative breast cancer: therapeutic optionsLancet Oncol2007823524417329194
- Gonzalez-AnguloAMHennessyBTMeric-BernstamFActivation of the PI3K/Akt signal transduction pathway is inversely associated with estrogen receptor levels and correlates with survival in hormone receptor-positive Her2/neu-negative breast cancerJ Clin Oncol20072518SAbstract No. 10588
- Stemke-HaleKGonzalez-AnguloAMLluchAAn integrative genomic and proteomic analysis of PIK3CA, PTEN, and Akt mutations in breast cancerCancer Res200868156084609118676830
- Alaoui-JamaliMAPatersonJAl MoustafaAEYenLThe role of ErbB-2 tyrosine kinase receptor in cellular intrinsic chemoresistance: mechanisms and implicationsBiochem Cell Biol1997753153259493954
- KurokawaHArteagaCLInhibition of erbB receptor (HER) tyrosine kinases as a strategy to abrogate antiestrogen resistance in human breast cancerClin Cancer Res20017Suppl4436s4442sdiscussion 4411s–4412s11916237
- OlayioyeMANeveRMLaneHAHynesNEThe ErbB signaling network: receptor heterodimerization in development and cancerEMBO J2000193159316710880430
- ZhouXTanMStone HawthorneVActivation of the Akt/mammalian target of rapamycin/4EBP-1 pathway by ErbB2 overexpression predicts tumor progression in breast cancersClin Cancer Res2004106779678815501954
- NahtaRYuanLXDuYEstevaFJLapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: effects on insulin-like growth factor I signalingMol Cancer Ther2007666767417308062
- ClarkASWestKStreicherSDennisPAConstitutive and inducible Akt activity promotes resistance to chemotherapy, trastuzumab, or tamoxifen in breast cancer cellsMol Cancer Ther2002170771712479367
- SheQBSolitDBassoAMoasserMMResistance to gefitinib in PTEN-null HER-overexpressing tumor cells can be overcome through restoration of PTEN function or pharmacologic modulation of constitutive phosphatidylinositol 3’-kinase/Akt pathway signalingClin Cancer Res200394340434614555504
- BiancoRShinIRitterCALoss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitorsOncogene2003222812282212743604
- MoasserMMBassoAAverbuchSDRosenNThe tyrosine kinase inhibitor ZD1839 (“Iressa”) inhibits HER2-driven signaling and suppresses the growth of HER2-overexpressing tumor cellsCancer Res2001617184718811585753
- BrognardJClarkASNiYDennisPAAkt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiationCancer Res2001613986399711358816
- TokunagaEKataokaAKimuraYThe association between Akt activation and resistance to hormone therapy in metastatic breast cancerEur J Cancer20064262963516464571
- CarpenterJTRocheHCamponeMRandomized 3-arm, phase 2 study of temsirolimus (CCI-779) in combination with letrozole in postmenopausal women with locally advanced or metastatic breast cancer [abstract]Proc Am Soc Clin Oncol200523564
- TaberneroJRojoFBurrisHA phase I study with tumor molecular pharmacodynamic (MPD) evaluation of dose and schedule of the oral mTOR-inhibitor everolimus (RAD001) in patients (pts) with advanced solid tumors [abstract]Proc Am Soc Clin Oncol2005233007
- BaselgaJSemiglazovVvan DamPPhase II double-blind randomized trial of daily oral RAD001 (everolimus) plus letrozole (LET) or placebo (P) plus LET as neoadjuvant therapy for ER+ breast cancer [abstract]San Antonio Breast Cancer Symposium20072066
- TsurutaniJFukuokaJTsurutaniHEvaluation of two phosphorylation sites improves the prognostic significance of Akt activation in non-small-cell lung cancer tumorsJ Clin Oncol20062430631416330671
- TsurutaniJSteinbergSMBallasMPrognostic significance of clinical factors and Akt activation in patients with bronchioloalveolar carcinomaLung Cancer20075511512117097759
- TangJMHeQYGuoRXChangXJPhosphorylated Akt 1 and loss of PTEN expression in non-small cell lung cancer confers poor prognosisLung Cancer20065118119116324768
- SordellaRBellDWHaberDASettlemanJGefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathwaysScience20043051163116715284455
- CappuzzoFHirschFRRossiEEpidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancerJ Natl Cancer Inst20059764365515870435
- HanSWHwangPGChungDHEpidermal growth factor receptor (EGFR) downstream molecules as response predictive markers for gefitinib (Iressa, ZD1839) in chemotherapy-resistant non-small cell lung cancerInt J Cancer200511310911515386420
- TracySMukoharaTHansenMMeyersonMJohnsonBEJännePAGefitinib induces apoptosis in the EGFRL858R non-small-cell lung cancer cell line H3255Cancer Res2004647241724415492241
- VideticGMStittLWDarARContinued cigarette smoking by patients receiving concurrent chemoradiotherapy for limited-stage small-cell lung cancer is associated with decreased survivalJ Clin Oncol2003211544154912697879
- CarlisleDLLiuXHopkinsTMSwickMCDhirRSiegfriedJMNicotine activates cell-signaling pathways through muscle-type and neuronal nicotinic acetylcholine receptors in non-small cell lung cancer cellsPulm Pharmacol Ther20072062964117015027
- TsurutaniJCastilloSSBrognardJTobacco components stimulate Akt-dependent proliferation and NFkappaB-dependent survival in lung cancer cellsCarcinogenesis2005261182119515790591
- JimenoAHidalgoMMolecular biomarkers: their increasing role in the diagnosis, characterization, and therapy guidance in pancreatic cancerMol Cancer Ther2006578779616648548
- ZhangYBanerjeeSWangZAntitumor activity of epidermal growth factor receptor-related protein is mediated by inactivation of ErbB receptors and nuclear factor-kappaB in pancreatic cancerCancer Res2006661025103216424038
- WangZSenguptaRBanerjeeSEpidermal growth factor receptor-related protein inhibits cell growth and invasion in pancreatic cancerCancer Res2006667653766016885366
- BuckEEyzaguirreABrownERapamycin synergizes with the epidermal growth factor receptor inhibitor erlotinib in non-small-cell lung, pancreatic, colon, and breast tumorsMol Cancer Ther200652676268417121914
- NelsonECEvansCPMackPCDevere-WhiteRWLaraPNInhibition of Akt pathways in the treatment of prostate cancerProstate Cancer Prostatic Dis20071033133917471291
- WangYKreisbergJIGhoshPMCross-talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancerCurr Cancer Drug Targets2007759160417896924
- XuRQiuMHanBThe prognostic impact of hypoxia-inducible factor-1α and p-Akt expressions in gastric adenocarcinoma [abstract]J Clin Oncol200826suppl4560
- OkiEBabaHTokunagaEAkt phosphorylation associates with LOH of PTEN and leads to chemoresistance for gastric cancerInt J Cancer200511737638015900596
- OkiEKakejiYTokunagaEAkt-mediated YB-1 phosphorylation induces resistance for chemotherapy of gastric cancer [abstract]. ASCO Annual Meeting ProceedingsJ Clin Oncol20074554
- MurakamiDTsujitaniSOsakiTExpression of phosphorylated Akt (pAkt) in gastric carcinoma predicts prognosis and efficacy of chemotherapyGastric Cancer200710455117334718
- D‘ErricoAFiorentinoMPonzettoALiver hepatocyte growth factor does not always correlate with hepatocellular proliferation in human liver lesions: its specific receptor c-met doesHepatology19962460648707284
- SuzukiAHayashidaMKawanoHSugimotoKNakanoTShirakiKHepatocyte growth factor promotes cell survival from Fas-mediated cell death in hepatocellular carcinoma cells via Akt activation and Fas-death–inducing signaling complex suppressionHepatology200032(4 Pt 1):79680211003625
- NakamuraMNaganoHSakonMRole of the Fas/FasL pathway in combination therapy with interferon-alpha and fluorouracil against hepatocellular carcinoma in vitroJ Hepatol200746778817045692
- SchifferEHoussetCCacheuxWGefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosisHepatology20054130731415660382
- LundPSchubertDNiketeghadFSchirmacherPAutocrine inhibition of chemotherapy response in human liver tumor cells by insulin-like growth factor-IICancer Lett2004206859615019164
- Desbois-MouthonCCacheuxWBlivet-Van EggelpoëlMJImpact of IGF-1R/EGFR cross-talks on hepatoma cell sensitivity to gefitinibInt J Cancer20061192557256616988945
- HjelmelandABLattimoreKPFeeBEThe combination of novel low molecular weight inhibitors of RAF (LBT613) and target of rapamycin (RAD001) decreases glioma proliferation and invasionMol Cancer Ther200762449245717766837
- SarteletHCastainMFabreMActivation of the PI3K/Akt pathway in neuroblastoma2007 ASCO Annual Meeting. Abstract No. 9523
- JiangBHLiuLZRole of mTOR in anticancer drug resistance: perspectives for improved drug treatmentDrug Resist Updat2008113637618440854
- JohnstonSRLearyAMartinLASmithIEDowsettMEnhancing endocrine response with novel targeted therapies: why have the clinical trials to date failed to deliver on the preclinical promise?Cancer20081123 Suppl71071718072235
- KolasaIKRembiszewskaAFelisiakAPIK3CA amplification associates with resistance to chemotherapy in ovarian cancer patientsCancer Biol Ther200911781[Epub ahead of print].
- GuerreiroASFattetSFischerBTargeting the PI3K p110alpha isoform inhibits medulloblastoma proliferation, chemoresistance, and migrationClin Cancer Res200814216761676918980969
- WolpinBMHezelAFAbramsTOral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancerJ Clin Oncol200927219319819047305
- GhayadSEBiecheIVendrellJAmTOR inhibition reverses acquired endocrine therapy resistance of breast cancer cells at the cell proliferation and gene-expression levelsCancer Sci200899101992200319016759
- StaufferFGarcia-EcheverirriaCFuretPBiochemical, cellular, and in vivo profiling of a new PI3K inhibitor from the imidazoquinoline seriesLos Angeles, CAAmerican Association for Cancer Research Annual Meeting 2007Apr 14–18, 2007
- HutsonTEFiglinRAExperimental therapy for advanced renal cell carcinomaExpert Opin Investig Drugs2008171116931702
- YaoJPhanAChangDPhase II study of RAD001 (everolimus) and depot octreotide (Sandostatin LAR) in patients with advanced low grade neuroendocrine carcinoma (LGNET) [abstract]. ASCO 2006 Annual Meeting ProceedingsJ Clin Oncol20062418 Suppl404216921066
- LoPiccoloJGranvilleCAGillsJJDennisPATargeting Akt in cancer therapyAnticancer Drugs20071886187417667591
- QunLiRecent progress in the discovery of Akt inhibitors as anticancer agentsExpert Opin Ther Patents20071710771130
- MollinedoFAntitumour ether lipids: proapoptotic agents with multiple therapeutic indicationsExpert Opin Ther Patents200717385405
- AllertonJPEbrahimiBSchreederMTPhase I report from a multicenter trial of perifosine (PERI) + sunitinib (SUT) in patients with advanced cancers including renal cell carcinoma (RCC)2008 ASCO Annual Meeting Abstract No. 14565.
- MohapatraSChuBZhaoXCombination inhibition of Cdk9 and Akt induces apoptosis of metastatic prostate cancer cells [abstract]Los Angeles, CAAnnual Meeting of American Association for Cancer Research20075399
- KumarRRhodesNKnickVBGSK690693, a pan-Akt kinase inhibitor has potent anti-tumor activity and shows additive effect with lapatinib [abstract]Los Angeles, CAAnnual Meeting of American Association for Cancer Research2007279
- RhodesNKnickVBMcConnellRGSK690693, a pan-Akt kinase inhibitor with potent pharmacodynamic and antitumor activity in vivo [abstract]Los Angeles, CAAnnual Meeting of American Association for Cancer Research2007277
- DummlerBHemmingsBAPhysiological roles of PKB/Akt isoforms in development and diseaseBiochem Soc Trans20073523123517371246
- DonaldAMcHardyTRowlandsMGRapid evolution of 6-phenylpurine inhibitors of protein kinase B through structure-based designJ Med Chem2007502289229217451235
- SunSYRosenbergLMWangXActivation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibitionCancer Res2005657052705816103051
- ShiYYanHFrostPMammalian target of rapamycin inhibitors activate the Akt kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascadeMol Cancer Ther200541533154016227402
- LuoYSmithRAGuanRPseudosubstrate peptides inhibit Akt and induce cell growth inhibitionBiochemistry2004431254126314756561
- ZhuGDGandhiVBGongJSyntheses of potent, selective, and orally bioavailable indazole-pyridine series of protein kinase B/Akt inhibitors with reduced hypotensionJ Med Chem2007502990300317523610
- MarshallJPoseyJHwangSA phase I trial of RX-0201 (Akt anti-sense) in patients with an advanced cancer2007 ASCO Annual Meeting. Abstract No. 3564.
- DavidOJettJLeBeauHDyGHughesJFriedmanMBrodyARPhospho-Akt overexpression in non-small cell lung cancer confers significant stage-independent survival disadvantageClin Cancer Res20041510(20)6865687115501963
- SoriaJCLeeHYLeeJIWangLIssaJPKempBLLack of PTEN expression in non-small cell lung cancer could be related to promoter methylationClin Cancer Res20025851178118412006535
- SteelmanLSStadelmanKMChappellWHHornSBäseckeJCervelloMAkt as a therapeutic target in cancerExpert Opin Ther Targets200812911391165Review.18694380
- HoriguchiAOyaMUchidaAMarumoKMuraiMElevated Akt activation and its impact on clinicopathological features of renal cell carcinomaJ Urol200316971071312544348
- Shin LeeJSeok KimHBok KimYCheol LeeMSoo ParkCExpression of PTEN in renal cell carcinoma and its relation to tumor behavior and growthJ Surg Oncol20038416617214598361
- BjornssonJShortMPKwiatkowskiDJHenskeEPTuberous sclerosis-associated renal cell carcinoma. Clinical, pathological, and genetic featuresAm J Pathol1996149120112088863669
- ZhouXActivation of the Akt/Mammalian Target of Rapamycin/4E-BP1 Pathway by ErbB2 Overexpression Predicts Tumor Progression in Breast CancersClin Cancer Res2004106779678815501954
- BacusSSAltomareDALyassLChinDMFarrellMPGurovaKAkt2 is frequently upregulated in HER-2/neu-positive breast cancers and may contribute to tumor aggressiveness by enhancing cell survivalOncogene200251621223532354012032855
- LiJYenCLiawDPodsypaninaKBoseSWangSIPucJPTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancerScience199728,275(5308)194319479072974
- SaalLHHolmKMaurerMMemeoLSuTWangXPIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinomaCancer Res2005652554255915805248
- FrancalanciPDiomedi-CamasseiFPurificatoCSantorelliFMGiannottiADominiciCMalignant pancreatic endocrine tumor in a child with tuberous sclerosisAm J Surg Pathol20031027101386138914508401
- SteelmanLSStadelmanKMChappellWHHornSBäseckeJCervelloMAkt as a therapeutic target in cancerExpert Opin Ther Targets200812911391165Review.18694380
- McCallPWittonCJGrimsleySNielsenKVEdwardsJIs PTEN loss associated with clinical outcome measures in human prostate cancer?Br J Cancer20089981296130118854827
- ItohNSembaSItoMTakedaHKawataSYamakawaMPhosphorylation of Akt/PKB is required for suppression of cancer cell apoptosis and tumor progression in human colorectal carcinomaCancer200261594123127313412115344
- FrattiniMSignoroniSPilottiSBertarioLBenvenutiSZanonCPhosphatase protein homologue to tensin expression and phosphatidylinositol-3 phosphate kinase mutations in colorectal cancerCancer Res200565231122716322273
- SamuelsYVelculescuVEOncogenic mutations of PIK3CA in human cancersCell Cycle2004103101221122415467468
- TaberneroJRojoFBurrisHA phase I study with tumor molecular pharmacodynamic (MPD) evaluation of dose and schedule of the oral mTOR-inhibitor everolimus (RAD001) in patients (pts) with advanced solid tumors –lsqb;Abstr]Proc Am Soc Clin Oncol200523Abstr 3007.
- CappuzzoFHirschFRRossiEEpidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancerJ Natl Cancer Inst20059764365515870435
- HanSWHwangPGChungDHEpidermal growth factor receptor (EGFR) downstream molecules as response predictive markers for gefitinib (Iressa, ZD1839) in chemotherapy-resistant non-small cell lung cancerInt J Cancer200511310911515386420
- TracySMukoharaTHansenMMeyersonMJohnsonBEJännePAGefitinib induces apoptosis in the EGFRL858R non-small-cell lung cancer cell line H3255Cancer Res2004647241724415492241
- WangYKreisbergJIGhoshPMCross-talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancerCurr Cancer Drug Targets20077591604Review.17896924
- NelsonECEvansCPMackPCDevere-WhiteRWLaraPNInhibition of Akt pathways in the treatment of prostate cancerProstate Cancer and Prostatic Diseases200719